

Abstract
Williams-Beuren syndrome is a rare contiguous gene syndrome, characterized by intellectual disability, facial dysmorphisms, connective-tissue abnormalities, cardiac defects, structural brain abnormalities, and transient infantile hypercalcemia. Genes lying telomeric to RFC2, including CLIP2, GTF2I and GTF2IRD1, are currently thought to be the most likely major contributors to the typical Williams syndrome cognitive profile, characterized by a better-than-expected auditory rote-memory ability, a relative sparing of language capabilities, and a severe visual-spatial constructive impairment. Atypical deletions in the region have helped to establish genotype-phenotype correlations. So far, however, hardly any deletions affecting only a single gene in the disease region have been described. We present here two healthy siblings with a pure, hemizygous deletion of CLIP2. A putative role in the cognitive and behavioral abnormalities seen in Williams-Beuren patients has been suggested for this gene on the basis of observations in a knock-out mouse model. The presented siblings did not show any of the clinical features associated with the syndrome. Cognitive testing showed an average IQ for both and no indication of the Williams syndrome cognitive profile. This shows that CLIP2 haploinsufficiency by itself does not lead to the physical or cognitive characteristics of the Williams-Beuren syndrome, nor does it lead to the Williams syndrome cognitive profile. Although contribution of CLIP2 to the phenotype cannot be excluded when it is deleted in combination with other genes, our results support the hypothesis that GTF2IRD1 and GTF2I are the main genes causing the cognitive defects associated with Williams-Beuren syndrome.
Main Text
Williams-Beuren syndrome (WBS [MIM 194050]) is a rare neurodevelopmental disorder with an estimated frequency of 1/7,500 to 1/15,000. The WBS phenotype includes recognizable facial dysmorphisms, connective tissue abnormalities, cardiac defects (SVAS/PPS), structural brain abnormalities, transient infantile hypercalcemia, and a specific cognitive profile.1–3 Independent of the global IQ level, which is commonly in the mild range of intellectual disability, the Williams syndrome cognitive profile (WSCP) is globally characterized by a better-than-expected auditory rote memory ability, relatively spared language capabilities, and a severe visual-spatial constructive impairment.4 The WSCP has been divided into sub-profiles involving severe impairments in visual-spatial processing and better skills in the verbal domain. It has further been demonstrated that specific defects exist in the working memory of WBS patients; these defects relate to both the verbal and spatial domains when patients need to process stored information.5 Although the four indices of the Wechsler intelligence testing (verbal comprehension, working memory, perceptual organization, and processing speed) specifically query discrete cognitive domains, cognitive profiling of WBS patients typically reveals sub-index differences on the individual subtest scores. Specifically, the WSCP translates to significantly higher scores on the vocabulary and similarities subtests of the verbal comprehension index, as well as to significantly lower scores on arithmetic, digit-symbol, and block-design subtests from the working-memory, processing-speed, and perceptual-organization tests, respectively. Despite the global impairment in working memory, WBS patients show a significant strength in the working-memory digit-span subtest.4–8
The causative microdeletion is commonly 1.45 Mb and comprises 28 genes on 7q11.23. The deleted region is delineated by low-copy repeats of high sequence similarity, facilitating genomic rearrangement by means of nonallelic homologous recombination (NAHR).9,10 The reciprocal 7q11.23 microduplication causes the Sommerville-Van der Aa syndrome (MIM 609757), characterized by mild but distinct facial dysmorphisms and cognitive abnormalities, the most notable of which is a severe language delay.11–13 Because the phenotype of the microduplication mirrors that of the microdeletion in some aspects, it has been speculated that specific genes in the 7q11.23 region are very sensitive to dosage alterations that might affect human language and visual-spatial abilities.12
The study of rare atypical deletion patients suggested that genes telomeric to RFC2 (MIM 600404), including CLIP2 (MIM 603432), GTF2I (MIM 601679) and GTF2IRD1 (MIM 604318), are most likely responsible for the typical WSCP and social behavior of WBS patients.1,14–19 Based on studies in mouse and fruit fly models, hypotheses of association between specific aspects of the WBS phenotype and individual genes, such as ELN (MIM 130160) for the cardiac and connective-tissue defects,20 GTF2IRD1 and BAZ1B (MIM 605681) for craniofacial features,15,21 and STX1A (MIM 186590) and MLXIPL (MIM 605678) for diabetes mellitus,22,23 were formulated. Both GTF2IRD1 and CLIP2 were linked to the neurological and cognitive symptoms.24–26 Of these genes, ELN is the only one whose role has been confirmed so far by detection of point mutations and single-gene deletions in patients with isolated SVAS.27–29
We present a pure, hemizygous CLIP2 deletion in two unaffected siblings. The intragenic deletion was initially identified as a supplementary finding in a patient referred to our department on indication of global developmental delay and microcephaly. As a toddler, he was diagnosed with autism on the basis of repetitive behavior, poor interaction skills, and hand flapping. Psycho-diagnostic evaluation at the age of 15 years showed a total IQ of 69 (WISC-R). In late adolescence he developed visual as well as auditory hallucinations and was diagnosed with schizophrenia. Dysmorphic features, apart from microcephaly, included long palpebral fissures, a high nasal bridge, and a prominent inverted lower lip. We detected a 580 kb microduplication at 16p11.2 (MIM 611913) by using an Illumina HumanCNV370-Quad BeadChip. The clinical presentation of this patient fits well within the described phenotype of carriers of a 16p11.2 microduplication. The phenotype of the patient is thus fully explained by the de novo 16p11.2 microduplication.30,31
With the approval of the institutional ethics committee, the patients' DNA was included in a follow-up research cohort. Refined data analysis with CNV-WebStore identified an additional deletion, ranging from position 73,760,704 to 73,810,096, of five consecutive probes in CLIP2 on chromosome 7q11.23.32 The deletion was confirmed by reanalysis on an Illumina HumanCyto12-v1.0 BeadChip. On the basis of the higher coverage of 19 consecutive probes in this disease-associated region, the deletion was predicted to range from position 73,734,542 to 73,815,967. MLPA analysis showed that the CLIP2 deletion was also present in the unaffected mother and her brother, referred to as “healthy siblings,” whereas the duplication of 16p11.2 in the autistic patient appeared de novo.33 PCR primers spanning the deletion were designed, allowing characterization of the breakpoints at the nucleotide level. The deleted region spanned 83 kb, ranging from position 73,740,492 to 73,823,520 (NCBI GRCh37). The proximal breakpoint is located in intron 2 of CLIP2, and the distal breakpoint maps in the intergenic region between CLIP2 and GTF2IRD1 (Figure 1). Both breakpoints were located in AluSz repeats as identified by RepeatMasker (Figure 2).34 Locally aligning these elements by using LALIGN showed more than 82% similarity between the flanking repeats and 26 base pairs of perfect identity at the breakpoints, indicating Alu-mediated, unequal homologous recombination as the causative mechanism for the rearrangement.35,36
Figure 1.

Well-Characterized, Reported Atypical Deletions in the WBS Critical Region
Deletions (in red) relevant to this study are shown. DNA from the individuals presented here was isolated from peripheral blood after informed consent was obtained. Segmental duplications flanking the WBS critical region are indicated by black arrows. Affected genes and cognitive details, if available, are listed on the right.
Figure 2.

AluSz Alignment wth the LALIGN Program
Breakpoint position is indicated by a red rectangle around the flanking nucleotides. Proximal AluSz coordinates: chr7:73,740,287–73,740,582 with the breakpoint at position 73,740,492. Distal AluSx coordinates: chr7:73,823,314–73,823,618 with the breakpoint at position 73,823,520.
Expression of the CLIP2, CLIP1, GTF2IRD1, GTF2I, LAT2, RFC2, STX1A, and WBSCR22 genes was examined by qPCR on lymphoblastoid cell lines with GAPDH, YWAZ, and HPRT as reference genes (Figure 3). A set of 15 cognitively tested individuals of normal intelligence was used as a control. Because no lymphoblastoid material was available from the male sibling, two replicate cDNA syntheses from two independent cell lines of the female sibling were used as test samples. A significant reduction of CLIP2 expression to about 40% residual expression was detected (p < 0.001). No evidence of a compensation effect by CLIP1 overexpression was found. No significant alterations in the expression of GTF2I, LAT2, RFC2, STX1A, or WBSCR22 was detected, but the expression of GTF2IRD1, located 48 kb 3′ of CLIP2, was significantly increased (p < 0.001) in comparison to the normal controls. These results are in line with studies in full-deletion WBS patients, demonstrating that the expression of most candidate genes, including CLIP2, is reduced to 50% of the level observed in controls when this expression is analyzed in lymphoblast cell lines.37–39 In contrast, no reduction in GTF2IRD1 expression is observed in this cell type for typical WBS patients, presumably as a consequence of a negative autoregulation mechanism.18,19,40 In fibroblast cell lines, however, the expression of GTF2IRD1 is reduced, indicating tissue-specific regulation.39 Due to this tissue-dependent regulation, the relevance of the observed overexpression of GTF2IRD1 in blood cells of the female sibling is unclear, and expression might be differently affected in the central nervous system.
Figure 3.
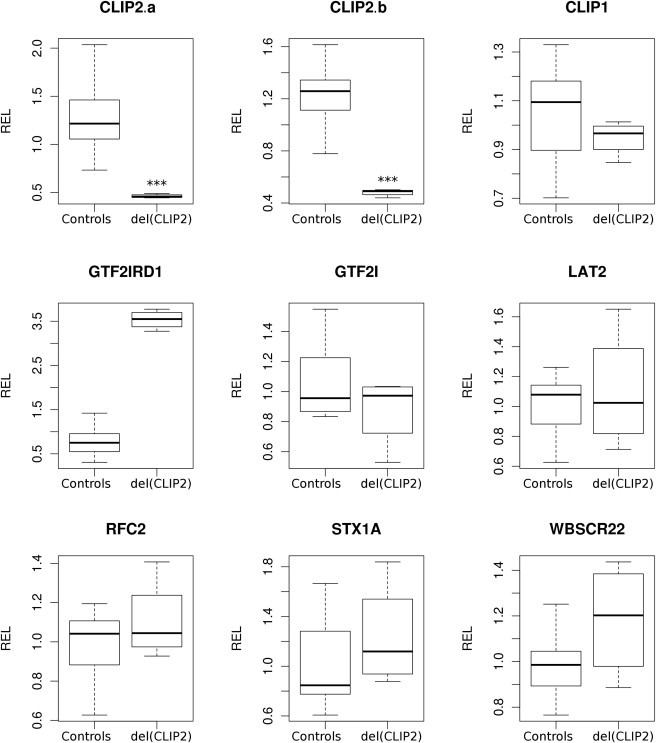
Results of Gene-Expression Study
Relative gene-expression levels (RELs) of CLIP2, CLIP1, GTF2IRD1, GTF2I, STX1A, RFC2, WBSCR22, and LAT2 in lymphoblastoid cell lines from 15 independent control samples and four samples from the female carrier. Isolation of total RNA, cDNA synthesis, and RT-PCR normalization and analysis were performed as described before.1 CLIP2a and CLIP2b represent two distinct probesets targeting different exons of CLIP2 within the deleted region. The four carrier samples represent two cDNA syntheses from RNA isolated from two independent cell cultures of the same individual. ∗∗∗p < 0.001, Mann-Whitney U test.
Both siblings with the pure CLIP2 deletion are healthy adults of European descent in their fifties with an unremarkable medical, developmental, and academic history. The woman has worked as a solicitor's secretary for the last 25 years, and her brother is a librarian. Both of them are married and have children. Contrary to adults with the WBS, they have a normal body habitus and an average proportional stature.41 There is no hearing impairment, there are no signs of cardiovascular disease or hypertension, and there is no history of gastrointestinal problems or diabetes. On physical examination, there is no scoliosis and no stiffness or hypermobility in the joints. There is no premature greying of the hair or sagging of the cheeks, as seen in adults with WBS. The typical dysmorphic features of WBS in adults are a wide mouth and prominent lips together with prominence of the supra orbital ridges and a narrow nasal root, and these features were not present in the siblings. Neurological examination in the female sibling showed slight divergent strabismus of the right eye and a weakness in focalization, leading to a suspicion of amblyopia. This was not confirmed by an ophthalmologist. Examination of ocular motility was otherwise normal. Cranial nerve evaluation showed no abnormalities. Peripheral motor and sensibility evaluation as well as reflexes were all normal. Coordination, gait, and equilibrium were normal. She has no history of psychiatric signs or symptoms. Psychiatric evaluation did not reveal any signs of comorbid behavioral or the mental-health problems, such as generalized anxiety, attention-deficit hyperactivity disorder, depression, and phobias and obsessions, that are seen in WBS patients.41,42 The brother has no obvious neurological or psychiatric abnormalities, but thorough examination by a neurologist was not performed.
Both siblings were subjected to formal cognitive testing with Wechsler Adult Intelligence Scales (WAIS-III, The Psychological Corporation, 1997). Verbal IQ was scored by seven tests: vocabulary, similarities, calculating, series of numbers, information, understanding, and repeating of numbers and letters. Five tests were used for performance IQ testing: symbol substitution, picture arrangement, matrix reasoning, object assembly, and block design. The majority of adult WBS patients score in the mild range of intellectual disability (55 to 69 IQ points) on standardized intelligence tests and have a slightly higher VIQ than PIQ score, although this difference is not significant in most cases.7 It can be appreciated from Figure 4 and Table 1 that the siblings' obtained IQ scores closely resemble a normal cognitive profile. Both siblings performed on an average to above-average level on the different indices. The brother's verbal IQ was 5 points higher than performance IQ, and the sister's verbal IQ was 6 points lower. Thus, the siblings showed small but non-significant and reciprocal differences between verbal and performance IQ. The four secondary indices of the WAIS-III (verbal comprehension, working memory, perceptual organization, and processing speed) were also scored so that maximum information on the cognitive strengths and weaknesses of the patients would be available. No evidence for a relative verbal strength was present at this level either. The brother did not show any significant differences between the individual index scores, whereas the sister showed significant relative strengths in working memory and processing speed, contributing to verbal and performance IQ scores, respectively.
Figure 4.

Cognitive Profiles Associated with the Reported Deletions in Figure 1, for those Cases where FSIQ, VIQ, and PIQ Was Available
Howlin et al. (2010) represents typical WBS patients who were at least 40 years old and were tested with WAIS-III. Expected IQ values in control populations are 100. ∗CLIP2 was not deleted but had lowered expression.
Table 1.
WAIS-III Results of the Cognitive Profiling in Both Deletion Carriers
| Female Carrier | Male Carrier | |
|---|---|---|
| Global IQ | 107 | 105 |
| Performal IQ | 111 | 103 |
| Verbal IQ | 105 | 107 |
| Verbal comprehension index (VCI) | 103 | 105 |
| Perceptual organisationindex (POI) | 99 | 107 |
| Working memory index (WMI) | 122 | 114 |
| Processing speed index (PSI) | 123 | 103 |
| Significant differences | WMI and PSI versus VCI and PRI | none |
Formal cognitive testing was performed by a trained psychologist. The significance threshold was taken from the WAIS III NL Technische Handleiding (Pearson, Amsterdam, The Netherlands). Significant deviations between secondary indices and verbal or performance IQ are defined as deviations seen in less than 5% from the standardized sample used for calibration of the test.
In addition to a mild global intellectual disability and a slightly elevated VIQ/PIQ ratio, significant differences between individual subtests are highly indicative of the WSCP. WBS patients typically have low arithmetic, digit-symbol, and block-design scores, whereas scores on vocabulary similarities and digit span are higher. A tendency to higher object-assembly and matrix-reasoning scores has also been observed.4,6–8 Individual subtest scores for these siblings (see Figure 5 and Table 2) did not reveal the described common pattern of strengths and weaknesses as seen in WBS patients. Both siblings had normal to high scores on all subtests and no significant deviations from the reference values. The highest score for both was on digit span. Although performance on this subtest is typically a relative strength of WBS patients, we feel we cannot attribute this to the CLIP2 deletion because the strength in digit span might be merely a consequence of training during the siblings' daily habits and occupation. None of the other significant deviations in subtest scores, where these are shared by both siblings, are indicative of the WSCP. Furthermore, the sister has a significant relative strength in digit symbol coding, and her brother scores poorly in vocabulary, both contrary to the results typical for the WSCP. Finally, no evidence for the visual-spatial impairment seen in WBS patients was present in the scores for the block-design subtest.
Figure 5.

Individual WAIS-III Subtest Scores
Howlin et al. (2010), 40+ represents typical WBS patients who were at least 40 years old when they were tested with WAIS-III. All scores are age-corrected scaled scores. Normal results are indicated by a gray area (mean 10, sd 3).
Table 2.
Summary of Results from WAIS-III Individual Subtests for both CLIP2-Deletion Carriers
|
Female Carrier |
Male Carrier |
WSCP4,6–8 |
|||||
|---|---|---|---|---|---|---|---|
| Age-Corrected Scaled Score | Deviation from Own Average∗∗ | Strengths and Weaknesses | Age-Corrected Scaled Score | Deviation from Own Average∗∗∗ | Strengths and Weaknesses | Strengths and Weaknesses | |
| Verbal IQ Subtests | |||||||
| Vocabulary | 10 | −1.86 | 9 | −2.29 | weakness | strength | |
| Similarities | 10 | −1.86 | 12 | +0.71 | strength | ||
| Arithmetic | 8 | −3.86 | weakness | 10 | −1.29 | weakness | |
| Digit span | 16 | +4.14 | strength | 16 | +4.71 | strength | strength |
| Information | 12 | +0.14 | 12 | +0.71 | |||
| Comprehension | 10 | −1.86 | 9 | −2.29 | |||
| Letter-number Sequencing | 17 | +5.14 | strength | 11 | −0.29 | ||
| Performance IQ Subtest | |||||||
| Picture completion | 8 | −3.8 | weakness | 9 | −1.6 | ||
| Digit symbol | 15 | +3.2 | strength | 9 | −1.6 | weakness | |
| Block design | 10 | −1.8 | 11 | +0.4 | weakness | ||
| Matrix reasoning | 12 | +0.2 | 14 | +3.4 | strength | ||
| Picture arrangement | 14 | +2.2 | 10 | −0.6 | |||
| Symbol search | 13 | +1.2 | 12 | +1.94 | |||
∗∗Averages of verbal and performance subtests are 11.86 and 11.8. ∗∗∗Averages of verbal and performance subtests are 11.29 and 10.06. Full results are available in sections 1 and 2 of the Supplemental Data. Testing was performed as summarized in the legend of Table 1.
These results demonstrate that CLIP2 haploinsufficiency by itself might not be critical to the pathology and cognitive profile of WBS. This contrasts with the suggested involvement of CLIP2 in the clinical manifestation of the syndrome in earlier functional and animal studies. CLIP2 codes for the CAP-GLY-domain-containing linker protein 2 (CLIP-115). The protein colocalizes with CLIP-170, encoded by CLIP2 paralog CLIP1, at the plus ends of growing microtubules along with other so-called microtubule-plus-end tracking proteins (+TIPS). These proteins regulate the polymerization of the microtubule network to form the actin skeleton, and it has been hypothesized that deregulation of this process might underlie the clinical presentation of Williams-Beuren Syndrome.43,44 Two lines of evidence support this hypothesis. First, mutations in +TIPs other than CLIP-115 and CLIP-170 cause neurological disorders such as lissencephaly (PAFAH1B1 [MIM 601545]) and amyotrophic lateral sclerosis (DCTN1 [MIM 105400]).45,46 Second, mice heterozygous for Clip2 showed phenotypic traits compatible with the characteristic abnormalities seen in WBS patients, including mild structural brain abnormalities and growth retardation. Furthermore, disturbed hippocampal functioning, correlating with the visual-spatial impairment of WBS patients,25,26 was observed.
However, the role of CLIP2 in the WBS phenotype was only partially supported by studies of atypical deletions in patients because these studies provided evidence both for and against a role for CLIP2 in various aspects of the phenotype on the basis of cognitive and physical characteristics (see Figure 1). Antonell et al.18 presented two families whose members had deletions in the WBS region, slightly lower cognitive performance, and mild facial dysmorphisms but who lacked the WSCP. CLIP2 expression was affected in both families, whereas GTF2I expression was not. It was thus hypothesized that CLIP2 might contribute to some of the general aspects of WBS but that hemizygosity of GTF2I and GTF2IRD1 was necessary for the typical WSCP and also greatly affected the complete phenotype. Two atypical deletions previously described by Hirota et al.47 support this hypothesis. The first deletion included CLIP2, and the second spanned only EIF4H and just spared CLIP2. Both deletions shared the typical centromeric breakpoint. Both carriers had a similar full-scale IQ in the mild intellectual-disability range (FSIQ = 64/65) and partially lacked the WSCP characteristics, thus implicating GTF2I and GTF2IRD1 haploinsufficiency in the WSCP. It was noted by the authors that the VIQ/PIQ ratio of 64/72 for the first case is exceptional and in fact opposed to what is expected for the WSCP. Tassabehji et al.15 reported a patient with a deletion that went from the typical centromeric breakpoint to GTF2IRD1 and included CLIP2. This patient had typical but mild syndrome-typical features, such as spatial-cognition defects and facial characteristics suggestive of WBS. In combination with results from GTF2IRD1 and CLIP2 mouse models, these findings led to the postulation that CLIP2 was involved in the observed mild neurological phenotype in this patient, and the craniofacial abnormalities were attributed to GTF2IRD1. Haploinsufficiency of GTF2I was proposed to be necessary for the full WBS phenotype. Dai et al.48 reported on an individual who had an atypical deletion sparing only GTF2I and presented with a milder phenotype but had some of the typical WSCP traits, including visual-spatial impairment and a high VIQ/PIQ ratio. The authors postulated that GTF2I is most likely implicated in the hypersocial phenotype and general cognitive defects of WBS, whereas GTF2IRD1 is responsible for the visual-spatial defects of the WSCP. This hypothesis was further supported by the family presented by Morris et al.49 because the carriers of the deletion, affecting GTF2IRD1 but sparing GTF2I, had normal intelligence levels and fit the WSCP. However, Ferrero et al.17 recently reported on a patient with a deletion spanning a region from BAZ1B to CLIP2. The patient showed a normal full-scale IQ, signs of the WSCP, and a high VIQ/PIQ ratio, indicating a role for CLIP2, rather than GTF2IRD1, in the WSCP. These findings are compatible with the deletions reported by Dai et al. and Morris et al., both including CLIP2 and both found in individuals who showed signs of the WSCP.
In combination with previous results from atypical deletions, our observations that CLIP2 haploinsufficiency by itself does not have apparent clinical consequences support a pivotal role of GTF2I and GTF2IRD1 in the visual-spatial and cognitive aspects of the WBS phenotype. Described deletions including these two genes are always associated with a cognitive deficit. However, it cannot be excluded that CLIP2 contributes to the clinical manifestation of the disorder when deleted in combination with one or more additional genes. Defects in these and other candidate genes will provide more insight in the genotype-phenotype correlation of the WBS microdeletion.
Acknowledgments
We are grateful to the patient and his family for participation in this study. We thank Sien Braat for help with the real-time experiments and our tissue-culture laboratory for providing cell cultures. Our work is supported by the Belgian National Fund for Scientific Research – Flanders (FWO) and the Marguerite-Marie Delacroix foundation.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Tassabehji M. Williams-Beuren syndrome: A challenge for genotype-phenotype correlations. Hum. Mol. Genet. 2003;12(Spec No 2):R229–R237. doi: 10.1093/hmg/ddg299. [DOI] [PubMed] [Google Scholar]
- 2.Jackowski A.P., Rando K., Maria de Araújo C., Del Cole C.G., Silva I., Tavares de Lacerda A.L. Brain abnormalities in Williams syndrome: a review of structural and functional magnetic resonance imaging findings. Eur. J. Paediatr. Neurol. 2009;13:305–316. doi: 10.1016/j.ejpn.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Pober B.R. Williams-Beuren syndrome. N. Engl. J. Med. 2010;362:239–252. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- 4.Mervis C.B., Robinson B.F., Bertrand J., Morris C.A., Klein-Tasman B.P., Armstrong S.C. The Williams syndrome cognitive profile. Brain Cogn. 2000;44:604–628. doi: 10.1006/brcg.2000.1232. [DOI] [PubMed] [Google Scholar]
- 5.Rhodes S.M., Riby D.M., Fraser E., Campbell L.E. The extent of working memory deficits associated with Williams syndrome: exploration of verbal and spatial domains and executively controlled processes. Brain Cogn. 2011;77:208–214. doi: 10.1016/j.bandc.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 6.Bellugi U., Lichtenberger L., Jones W., Lai Z., St George M. I. The neurocognitive profile of Williams Syndrome: A complex pattern of strengths and weaknesses. J. Cogn. Neurosci. 2000;12(Suppl 1):7–29. doi: 10.1162/089892900561959. [DOI] [PubMed] [Google Scholar]
- 7.Howlin P., Elison S., Udwin O., Stinton C. Cognitive, linguistic and adaptive functioning in Williams Syndrome: Trajectories from early to middle adulthood. J. Appl. Res. Intellect. Disabil. 2010;23:322–336. [Google Scholar]
- 8.Howlin P., Davies M., Udwin O. Cognitive functioning in adults with Williams syndrome. J. Child Psychol. Psychiatry. 1998;39:183–189. [PubMed] [Google Scholar]
- 9.Merla G., Brunetti-Pierri N., Micale L., Fusco C. Copy number variants at Williams-Beuren syndrome 7q11.23 region. Hum. Genet. 2010;128:3–26. doi: 10.1007/s00439-010-0827-2. [DOI] [PubMed] [Google Scholar]
- 10.Cuscó I., Corominas R., Bayés M., Flores R., Rivera-Brugués N., Campuzano V., Pérez-Jurado L.A. Copy number variation at the 7q11.23 segmental duplications is a susceptibility factor for the Williams-Beuren syndrome deletion. Genome Res. 2008;18:683–694. doi: 10.1101/gr.073197.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van der Aa N., Rooms L., Vandeweyer G., van den Ende J., Reyniers E., Fichera M., Romano C., Delle Chiaie B., Mortier G., Menten B. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur. J. Med. Genet. 2009;52:94–100. doi: 10.1016/j.ejmg.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Somerville M.J., Mervis C.B., Young E.J., Seo E.-J., del Campo M., Bamforth S., Peregrine E., Loo W., Lilley M., Pérez-Jurado L.A. Severe expressive-language delay related to duplication of the Williams-Beuren locus. N. Engl. J. Med. 2005;353:1694–1701. doi: 10.1056/NEJMoa051962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gagliardi C., Bonaglia M.C., Selicorni A., Borgatti R., Giorda R. Unusual cognitive and behavioural profile in a Williams syndrome patient with atypical 7q11.23 deletion. J. Med. Genet. 2003;40:526–530. doi: 10.1136/jmg.40.7.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tassabehji M., Hammond P., Karmiloff-Smith A., Thompson P., Thorgeirsson S.S., Durkin M.E., Popescu N.C., Hutton T., Metcalfe K., Rucka A. GTF2IRD1 in craniofacial development of humans and mice. Science. 2005;310:1184–1187. doi: 10.1126/science.1116142. [DOI] [PubMed] [Google Scholar]
- 16.Howald C., Merla G., Digilio M.C., Amenta S., Lyle R., Deutsch S., Choudhury U., Bottani A., Antonarakis S.E., Fryssira H. Two high throughput technologies to detect segmental aneuploidies identify new Williams-Beuren syndrome patients with atypical deletions. J. Med. Genet. 2006;43:266–273. doi: 10.1136/jmg.2005.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrero G.B., Howald C., Micale L., Biamino E., Augello B., Fusco C., Turturo M.G., Forzano S., Reymond A., Merla G. An atypical 7q11.23 deletion in a normal IQ Williams-Beuren syndrome patient. Eur. J. Hum. Genet. 2010;18:33–38. doi: 10.1038/ejhg.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antonell A., Del Campo M., Magano L.F., Kaufmann L., de la Iglesia J.M., Gallastegui F., Flores R., Schweigmann U., Fauth C., Kotzot D., Pérez-Jurado L.A. Partial 7q11.23 deletions further implicate GTF2I and GTF2IRD1 as the main genes responsible for the Williams-Beuren syndrome neurocognitive profile. J. Med. Genet. 2010;47:312–320. doi: 10.1136/jmg.2009.071712. [DOI] [PubMed] [Google Scholar]
- 19.Edelmann L., Prosnitz A., Pardo S., Bhatt J., Cohen N., Lauriat T., Ouchanov L., González P.J., Manghi E.R., Bondy P. An atypical deletion of the Williams-Beuren syndrome interval implicates genes associated with defective visuospatial processing and autism. J. Med. Genet. 2007;44:136–143. doi: 10.1136/jmg.2006.044537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li D.Y., Faury G., Taylor D.G., Davis E.C., Boyle W.A., Mecham R.P., Stenzel P., Boak B., Keating M.T. Novel arterial pathology in mice and humans hemizygous for elastin. J. Clin. Invest. 1998;102:1783–1787. doi: 10.1172/JCI4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashe A., Morgan D.K., Whitelaw N.C., Bruxner T.J., Vickaryous N.K., Cox L.L., Butterfield N.C., Wicking C., Blewitt M.E., Wilkins S.J. A genome-wide screen for modifiers of transgene variegation identifies genes with critical roles in development. Genome Biol. 2008;9:R182. doi: 10.1186/gb-2008-9-12-r182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iizuka K., Horikawa Y. ChREBP: a glucose-activated transcription factor involved in the development of metabolic syndrome. Endocr. J. 2008;55:617–624. doi: 10.1507/endocrj.k07e-110. [DOI] [PubMed] [Google Scholar]
- 23.Lam P.P., Leung Y.M., Sheu L., Ellis J., Tsushima R.G., Osborne L.R., Gaisano H.Y. Transgenic mouse overexpressing syntaxin-1A as a diabetes model. Diabetes. 2005;54:2744–2754. doi: 10.2337/diabetes.54.9.2744. [DOI] [PubMed] [Google Scholar]
- 24.Osborne L.R. Animal models of Williams syndrome. Am. J. Med. Genet. C. Semin. Med. Genet. 2010;154C:209–219. doi: 10.1002/ajmg.c.30257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Hagen J.M., van der Geest J.N., van der Giessen R.S., Lagers-van Haselen G.C., Eussen H.J., Gille J.J., Govaerts L.C., Wouters C.H., de Coo I.F., Hoogenraad C.C. Contribution of CYLN2 and GTF2IRD1 to neurological and cognitive symptoms in Williams Syndrome. Neurobiol. Dis. 2007;26:112–124. doi: 10.1016/j.nbd.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 26.Hoogenraad C.C., Koekkoek B., Akhmanova A., Krugers H., Dortland B., Miedema M., van Alphen A., Kistler W.M., Jaegle M., Koutsourakis M. Targeted mutation of Cyln2 in the Williams syndrome critical region links CLIP-115 haploinsufficiency to neurodevelopmental abnormalities in mice. Nat. Genet. 2002;32:116–127. doi: 10.1038/ng954. [DOI] [PubMed] [Google Scholar]
- 27.Curran M.E., Atkinson D.L., Ewart A.K., Morris C.A., Leppert M.F., Keating M.T. The elastin gene is disrupted by a translocation associated with supravalvular aortic stenosis. Cell. 1993;73:159–168. doi: 10.1016/0092-8674(93)90168-p. [DOI] [PubMed] [Google Scholar]
- 28.Li D.Y., Toland A.E., Boak B.B., Atkinson D.L., Ensing G.J., Morris C.A., Keating M.T. Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis. Hum. Mol. Genet. 1997;6:1021–1028. doi: 10.1093/hmg/6.7.1021. [DOI] [PubMed] [Google Scholar]
- 29.Olson T.M., Michels V.V., Urban Z., Csiszar K., Christiano A.M., Driscoll D.J., Feldt R.H., Boyd C.D., Thibodeau S.N. A 30 kb deletion within the elastin gene results in familial supravalvular aortic stenosis. Hum. Mol. Genet. 1995;4:1677–1679. doi: 10.1093/hmg/4.9.1677. [DOI] [PubMed] [Google Scholar]
- 30.Shinawi M., Liu P., Kang S.H., Shen J., Belmont J.W., Scott D.A., Probst F.J., Craigen W.J., Graham B.H., Pursley A. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet. 2010;47:332–341. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacquemont S., Reymond A., Zufferey F., Harewood L., Walters R.G., Kutalik Z., Martinet D., Shen Y., Valsesia A., Beckmann N.D. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011;478:97–102. doi: 10.1038/nature10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vandeweyer G., Reyniers E., Wuyts W., Rooms L., Kooy R.F. CNV-WebStore: Online CNV analysis, storage and interpretation. BMC Bioinformatics. 2011;12:4. doi: 10.1186/1471-2105-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rooms L., Vandeweyer G., Reyniers E., van Mol K., de Canck I., Van der Aa N., Rossau R., Kooy R.F. Array-based MLPA to detect recurrent copy number variations in patients with idiopathic mental retardation. Am. J. Med. Genet. A. 2011;155A:343–348. doi: 10.1002/ajmg.a.33810. [DOI] [PubMed] [Google Scholar]
- 34.Smit, A.F.A., Hubley, R., Green, P. (1996) RepeatMasker Open-3.0.
- 35.Pearson W.R., Wood T., Zhang Z., Miller W. Comparison of DNA sequences with protein sequences. Genomics. 1997;46:24–36. doi: 10.1006/geno.1997.4995. [DOI] [PubMed] [Google Scholar]
- 36.Batzer M.A., Deininger P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- 37.Gao M.C., Bellugi U., Dai L., Mills D.L., Sobel E.M., Lange K., Korenberg J.R. Intelligence in Williams Syndrome is related to STX1A, which encodes a component of the presynaptic SNARE complex. PLoS ONE. 2010;5:e10292. doi: 10.1371/journal.pone.0010292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henrichsen C.N., Csárdi G., Zabot M.T., Fusco C., Bergmann S., Merla G., Reymond A. Using transcription modules to identify expression clusters perturbed in Williams-Beuren syndrome. PLoS Comput. Biol. 2011;7:e1001054. doi: 10.1371/journal.pcbi.1001054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Merla G., Howald C., Henrichsen C.N., Lyle R., Wyss C., Zabot M.T., Antonarakis S.E., Reymond A. Submicroscopic deletion in patients with Williams-Beuren syndrome influences expression levels of the nonhemizygous flanking genes. Am. J. Hum. Genet. 2006;79:332–341. doi: 10.1086/506371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palmer S.J., Santucci N., Widagdo J., Bontempo S.J., Taylor K.M., Tay E.S., Hook J., Lemckert F., Gunning P.W., Hardeman E.C. Negative autoregulation of GTF2IRD1 in Williams-Beuren syndrome via a novel DNA binding mechanism. J. Biol. Chem. 2010;285:4715–4724. doi: 10.1074/jbc.M109.086660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pober B.R., Morris C.A. Diagnosis and management of medical problems in adults with Williams-Beuren syndrome. Am. J. Med. Genet. C. Semin. Med. Genet. 2007;145C:280–290. doi: 10.1002/ajmg.c.30139. [DOI] [PubMed] [Google Scholar]
- 42.Elison S., Stinton C., Howlin P. Health and social outcomes in adults with Williams syndrome: Findings from cross-sectional and longitudinal cohorts. Res. Dev. Disabil. 2010;31:587–599. doi: 10.1016/j.ridd.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 43.Jaworski J., Hoogenraad C.C., Akhmanova A. Microtubule plus-end tracking proteins in differentiated mammalian cells. Int. J. Biochem. Cell Biol. 2008;40:619–637. doi: 10.1016/j.biocel.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 44.Hoogenraad C.C., Akhmanova A., Galjart N., De Zeeuw C.I. LIMK1 and CLIP-115: linking cytoskeletal defects to Williams syndrome. Bioessays. 2004;26:141–150. doi: 10.1002/bies.10402. [DOI] [PubMed] [Google Scholar]
- 45.Reiner O., Carrozzo R., Shen Y., Wehnert M., Faustinella F., Dobyns W.B., Caskey C.T., Ledbetter D.H. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 46.Münch C., Sedlmeier R., Meyer T., Homberg V., Sperfeld A.D., Kurt A., Prudlo J., Peraus G., Hanemann C.O., Stumm G., Ludolph A.C. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63:724–726. doi: 10.1212/01.wnl.0000134608.83927.b1. [DOI] [PubMed] [Google Scholar]
- 47.Hirota H., Matsuoka R., Chen X.N., Salandanan L.S., Lincoln A., Rose F.E., Sunahara M., Osawa M., Bellugi U., Korenberg J.R. Williams syndrome deficits in visual spatial processing linked to GTF2IRD1 and GTF2I on chromosome 7q11.23. Genet. Med. 2003;5:311–321. doi: 10.1097/01.GIM.0000076975.10224.67. [DOI] [PubMed] [Google Scholar]
- 48.Dai L., Bellugi U., Chen X.N., Pulst-Korenberg A.M., Järvinen-Pasley A., Tirosh-Wagner T., Eis P.S., Graham J., Mills D., Searcy Y., Korenberg J.R. Is it Williams syndrome? GTF2IRD1 implicated in visual-spatial construction and GTF2I in sociability revealed by high resolution arrays. Am. J. Med. Genet. A. 2009;149A:302–314. doi: 10.1002/ajmg.a.32652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morris C.A., Mervis C.B., Hobart H.H., Gregg R.G., Bertrand J., Ensing G.J., Sommer A., Moore C.A., Hopkin R.J., Spallone P.A. GTF2I hemizygosity implicated in mental retardation in Williams syndrome: Genotype-phenotype analysis of five families with deletions in the Williams syndrome region. Am. J. Med. Genet. A. 2003;123A:45–59. doi: 10.1002/ajmg.a.20496. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.