

Abstract
Autosomal-recessive inheritance, severe to profound sensorineural hearing loss, and partial agenesis of the corpus callosum are hallmarks of the clinically well-established Chudley-McCullough syndrome (CMS). Although not always reported in the literature, frontal polymicrogyria and gray matter heterotopia are uniformly present, whereas cerebellar dysplasia, ventriculomegaly, and arachnoid cysts are nearly invariant. Despite these striking brain malformations, individuals with CMS generally do not present with significant neurodevelopmental abnormalities, except for hearing loss. Homozygosity mapping and whole-exome sequencing of DNA from affected individuals in eight families (including the family in the first report of CMS) revealed four molecular variations (two single-base deletions, a nonsense mutation, and a canonical splice-site mutation) in the G protein-signaling modulator 2 gene, GPSM2, that underlie CMS. Mutations in GPSM2 have been previously identified in people with profound congenital nonsyndromic hearing loss (NSHL). Subsequent brain imaging of these individuals revealed frontal polymicrogyria, abnormal corpus callosum, and gray matter heterotopia, consistent with a CMS diagnosis, but no ventriculomegaly. The gene product, GPSM2, is required for orienting the mitotic spindle during cell division in multiple tissues, suggesting that the sensorineural hearing loss and characteristic brain malformations of CMS are due to defects in asymmetric cell divisions during development.
Main Text
The autosomal-recessively inherited disorder, Chudley-McCullough Syndrome (CMS [MIM 604213]), was first described1 in Canadian siblings of Dutch-German Mennonite (sometimes referred to as Old Colony or Chortitza Mennonite) ancestry, who presented with hydrocephalus and profound sensorineural hearing loss. Several subsequent reports2–8 have expanded the clinical phenotype to include partial agenesis of the corpus callosum, frontal polymicrogyria, gray matter heterotopia, cerebellar dysplasia, and arachnoid cysts. This combination of brain malformations is highly distinctive and not seen in any other genetic syndrome.
In an effort to identify mutations that cause CMS, we recruited individuals with CMS from centers in Canada and the United States. Study subjects were enrolled with informed consent under protocols approved by the health research ethics boards of the participating academic institutions. All affected individuals had severe or profound sensorineural hearing loss and ventriculomegaly (Table 1). Brain imaging revealed additional findings characteristic of the syndrome, including posterior agenesis of the corpus callosum, frontal polymicrogyria, frontal heterotopia, cerebellar dysplasia, and arachnoid cysts (Figure 1, Table 2). The affected individuals were nondysmorphic, except for 5B (Table 1), who had downslanting palpebral fissures and low-set, posteriorly rotated ears.1 Only subject 3B had developmental issues beyond what is typically seen in individuals with severe hearing loss (Table 1). Perhaps most surprising given the polymicrogyria and heterotopia in all individuals, seizures were present in only two subjects (3B and 7B), and they were well controlled with medication.
Table 1.
Clinical Features in Subjects with GPSM2-Related Chudley-McCullough Syndrome
| Indiv.a | Sex | Age | DNA Change | Protein Change | Ethnicity | Hearing Loss | Aids/Implants | Motor Delay | Comm. Delayb | Cognitive Impairment | Other Features |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1A | M | 1 yr | c.1471delG | p.G491GfsX6 | Mennonite | severe | cochlear implant | mild | mild | no | none |
| 2A | F | 25 yr | c.1471delG | p.G491GfsX6 | Mennonite | severe | cochlear implant | no | no | no | none |
| 3A | M | 12 yr | c.1471delG | p.G491GfsX6 | Mennonite | severe | none | no | yes | mild | none |
| 3B | F | 15 yr | c.1471delG | p.G491GfsX6 | Mennonite | severe | none | yes | yes | mild to moderate ID | h/o seizures, now off medications |
| 4A | F | 4 yr | c.1471delG | p.G491GfsX6 | Mennonite | profound | cochlear implant | mild | mild | mild | none |
| 5A | F | 17 yr | c.741delC | p.N247NfsX34 | Mennonite | severe-profound | cochlear implant | no | no | no | none |
| 5B | M | 21 yr | c.741delC | p.N247NfsX34 | Mennonite | profound | cochlear implant | no | mild, resolved | no | downslanting palpebral fissures, rotated ears, nasal voice |
| 6A | F | 2 yr | c.741delC | p.N247NfsX34 | European-American | profound | cochlear implant | mild | no | no | none |
| 7A | F | 10 yr | c.741delC | p.N247NfsX34 | Dutch | profound | cochlear implant | no | no | mild, resolved | none |
| c.1661C>A | p.S554X | ||||||||||
| 7B | F | 4 yr | c.741delC | p.N247NfsX34 | Dutch | severe-profound | cochlear implant | mild, mildly increased tone | mild, resolving | mild, resolved | controlled seizures, breath holding |
| c.1661C>A | p.S554X | ||||||||||
| 8A | F | 7 yr | c.1062+1G>T | p.R318RfsX8 | Mexican-American | profound | cochlear implant | mild | no | no | none |
| 8B | M | 6 yr | c.1062+1G>T | p.R318RfsX8 | Mexican-American | profound | cochlear implant | mild | no | no | none |
| Previously Published Subjects with GPSM2 Mutations9,10 | |||||||||||
| 9A, Walsh9 CG6 | M | 26 yr | c.379C>T | p.R127X | Palestinian | severe-profound | none | no | no | no | none |
| 10A, Yariz10 IV-1 | F | 16 yr | c.1684C>T | p.Q562X | Turkish | severe-profound | none | no | no | no | none |
| 10B, Yariz10 IV-2 | M | 12 yr | c.1684C>T | p.Q562X | Turkish | severe-profound | none | no | no | no | none |
| 11A,c Yariz10 IV-3 | M | 16 yr | c.1684C>T | p.Q562X | Turkish | severe-profound | none | no | no | no | none |
Indiv., individual; Comm., communication; M, male; F, female; yr, years of age; ID, intellectual disability; h/o, “history of.”
A and B indicate siblings.
More than expected for severe to profound hearing loss.
11A is a first cousin of 10A and 10B.
Figure 1.
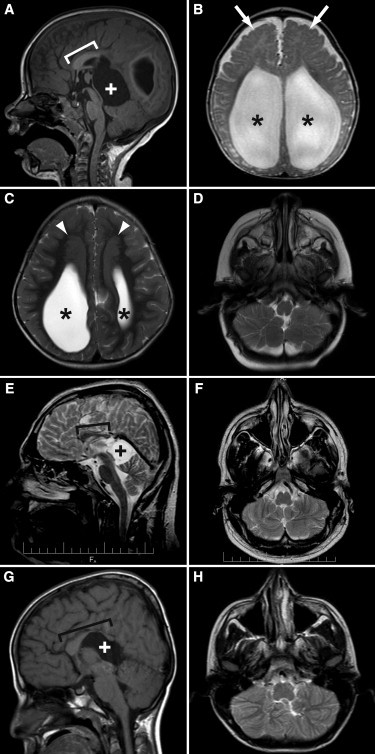
Characteristic Neuroimaging Features of GPSM2-related Chudley-McCullough Syndrome
(A) illustrates posterior agenesis of the corpus callosum (bracket indicates remaining corpus callosum) and a quadrigeminal plate cistern cyst (white plus sign) causing mass effect on the cerebellum and tectum in individual 8A at 3 years of age.
(B) illustrates severe ventriculomegaly (black asterisks) and frontal polymicrogyria (white arrows) in individual 8B at 6 months of age.
(C) illustrates large frontal gray matter heterotopia (white arrowheads) located superior and medial to the enlarged lateral ventricles (black asterisks) in individual 8A at 3 years of age.
(D) illustrates inferior cerebellar hemisphere dysplasia in individual 8B at 6 months of age.
(E) and (F) illustrate a short corpus callosum (bracket indicates remaining corpus callosum) and a quadrigeminal plate cistern cyst (black plus sign) causing mass effect on the cerebellum and tectum as well as cerebellar hemisphere dysplasia in individual 9A (CG6 from Walsh et al.9) at 26 years of age.
(G) and (H) illustrate similar findings in individual 10B (IV-2 from Yariz et al.10) at 12 years of age, although the corpus callosum is thinned posteriorly and dysplastic anteriorly, rather than short.
(A) and (G) are sagittal T1-weighted images; (B)–(D), (F), and (H) are axial T2-weighted images, and (E) is a sagittal T2-weighted image.
Table 2.
Neuroimaging Features in Subjects with GPSM2-Related Chudley-McCullough Syndrome
| Individuala | Ventriculomegaly | CC | Heterotopia | Frontal PMG | Cerebellar Dysplasia | Arachnoid Cyst |
|---|---|---|---|---|---|---|
| 1A | yes | posterior agenesis | small | moderate | unable to score due to mass effect | large bilateral CPA cysts |
| 2A | yes, right > left | posterior agenesis | moderate | presentb | yes | ND |
| 3A | shunted HC | posterior agenesis | small | extensive | yes | no, right ventricle herniation into midline |
| 3B | shunted HC | posterior agenesis | small | moderate | yes | left CPA |
| 4A | yes | posterior agenesis | extensive | extensive | no | right CPA |
| 5Ac | HC, foramen of Monro fenestration | probable partial agenesis | ND | ND | ND | ND |
| 5Bc | shunted HC | probable partial agenesis | ND | ND | ND | ND |
| 6A | shunted HC | posterior agenesis | small | extensive | yes | large interhemispheric and small left CPA cysts |
| 7A | yes | posterior agenesis | small | moderate | yes | moderate interhemispheric cyst |
| 7B | shunted HC | posterior agenesis | small | moderate | yes | small pineal cyst |
| 8A | yes | posterior agenesis | large | extensive | yes | large interhemispheric and small bilateral CPA cysts |
| 8B | shunted HC | posterior agenesis | moderate | extensive | yes | moderate interhemispheric and small right CPA cysts |
| Previously Published Subjects with GPSM2 Mutations9,10 | ||||||
| 9A, Walsh9 CG6 | no | short and thin | moderate | moderate | yes | moderate interhemispheric cyst |
| 10A, Yariz10 IV-1 | no | short and thin | small | subtle | no | moderate interhemispheric cyst |
| 10B, Yariz10 IV-2 | no | short, severely thin posteriorly | moderate | subtle | yes | small interhemispheric cyst |
| 11A,d Yariz10 IV-3 | no | short and thin | small | subtle | mild on right | moderate interhemispheric cyst |
CC, corpus callosum; CPA, cerebellopontine angle; HC, hydrocephalus; ND, no data; PMG, polymicrogyria.
A and B indicate siblings.
Unable to determine the extent due to technical limitations of the available images.
Only computed tomography scans available.
11A is a first cousin of 10A and 10B.
In four Mennonite families, genomic DNA from six affected individuals and their unaffected relatives was genotyped with the Affymetrix GeneChip Human Mapping 250K NspI SNP array. Loss of heterozygosity on chromosome 1p was observed in all six affected individuals, but not in any member of their extended families. Four of the six individuals studied were identically homozygous for a 5.8 Mb region (range: 8.3 Mb to 76.5 Mb). The remaining two individuals (a sister and brother) also shared a homozygous interval on 1p, but their haplotype differed from the other four affected individuals. Overlap between the two unique haplotypes was approximately 2.9 Mb (from rs2863991 to rs402684), a chromosomal segment within 1p13.3 containing 42 known and putative genes (Genome Reference Consortium human genome build 37 [GRCh37]/hg19).
As part of a cross-Canada initiative known as FORGE (Finding of Rare Disease Genes), genomic DNA from two Mennonite individuals (one of each unique haplotype described above) and four non-Mennonite affected individuals from other parts of Canada and the United States were subjected to whole-exome sequencing. Details of exome-capture-library preparation, sequencing, and bioinformatics analysis can be found in Supplemental Data (available online). In brief, the span of the human genome covered by at least one qualified aligned read (total sequence yield) averaged 1.77 Gb per subject (Table S1). More than 23,000 sequence variants were identified in each subject with > 12,000 of these being nonsynonymous variants. Although more than 2,500 identified variants per subject were not cataloged in dbSNP129 or dbSNP130, only about 200 per subject were novel; that is, not listed in the 1000 Genomes Project database or the noncancer genome database compiled at the Michael Smith Genome Sciences Centre (Vancouver, BC, Canada). Of these 200 novel variants, the only gene within the identified homozygous SNP interval that carried biallelic mutations in all six sequenced subjects was the G protein-signaling modulator 2 gene (GPSM2 [MIM 609245]). The mutations identified with exome sequencing were verified with Sanger sequencing in all affected subjects and, when available, in the extended family of each proband. The first mutation, a homozygous single-base deletion (c.1471delG) predicted to cause a frameshift (p.Gly491GlyfsX6), was identified in all four Mennonite subjects (1A, 2A, 3A, and 3B) who displayed the 5.8 Mb SNP haplotype (Figure 2A) and was also detected in subject 4A, who was not analyzed for SNPs. The second mutation, also a homozygous single-base deletion (c.741delC) predicted to cause a frameshift (p.Asn247AsnfsX34), was identified in the Mennonite sister and brother (5A and 5B) of the second haplotype and in an unrelated subject (6A) of European ancestry from the southern United States (Figure 2B). Affected siblings (7A and 7B) of Dutch ancestry were heterozygous for the c.741delC mutation (paternally transmitted) and also for a maternally transmitted c.1661C>A (p.Ser554X) mutation (Figure 2C). The final two subjects were siblings of Mexican ancestry (8A and 8B) who were found to be homozygous for a mutation in the donor splice site for exon 9, c.1062+1G>T (Figure 2D). This mutation results in a transcript that is missing exon 9 (Figure 2E) and is predicted to generate a truncated protein, p.Arg318ArgfsX8.
Figure 2.
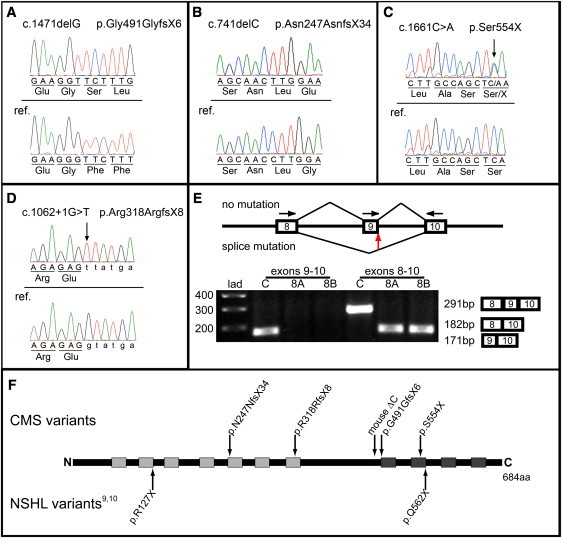
GPSM2 Sequence Analysis and GPSM2 Schematic
With the use of the primers specified in Table S2, genomic DNA from each subject was PCR-amplified, and the products were isolated and purified, as described in Supplemental Data. Sanger sequencing was conducted on both the forward and reverse strand; only the forward strand is shown here. The reference (ref.) sequence (NM_013296.4) is depicted directly below the variant sequence in each panel.
(A) depicts the c.1471delG mutation identified in five subjects.
(B) depicts the c.741delC homozygous mutation identified in three subjects. Two other study subjects were heterozygous for this mutation (sequence not shown).
(C) depicts the c.1661C>A heterozygous mutation from one of the two subjects who was also heterozygous for c.741delC.
(D) depicts the homozygous splice-site mutation identified in siblings 8A and 8B.
(E) depicts the effect of the splice-site mutation on the splicing of exon 9. Primers in exons 8 and 9 generated a ∼200 base-pair (bp) product in the unaffected control, but no product in subjects 8A and 8B. Primers in exons 8 and 10 generated a ∼300 bp fragment in the control and a ∼200 bp product in subjects 8A and 8B, consistent with the loss of exon 9. Sequencing of this product revealed that exon 8 is spliced to exon 10 in the affected siblings (data not shown). The horizontal arrows indicate the primer positions; the vertical red arrow indicates the location of the splice-site mutation. “lad” indicates the ladder lane and “C” indicates the control lanes. The expected exon composition and size of the various products are indicated to the right of the gel.
(F) depicts the positions of amino-acid variants that account for NSHL and CMS, and includes the mouse-engineered variant ΔC. Three of the variants occur within the seven tetratricopeptide repeat domains of GPSM2, and three within the four GoLoco motifs.
Two of the four mutations were defined in individuals of Mennonite ancestry and probably represent founder mutations of European origin. Although the c.1661C>A mutation was not in dbSNP129 or dbSNP130, it is now listed as rs145191476, having been identified (as heterozygous) in one of 3,510 European-American subjects who participated in the National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project (NHLBI GO ESP; see Web Resources). We identified this change in CMS-affected siblings of Dutch ancestry (7A and 7B), confirming that this allele is also of European origin. The origin of the identified splice-site mutation (c.1062+1G>T) in the affected siblings of Mexican ancestry is unknown, and it was absent from 3,510 European-American subjects in the NHLBI GO ESP data set.
Recently, a GPSM2 nonsense mutation, c.875C>T (p.Arg127X), was reported as the cause of recessively inherited nonsyndromic hearing loss (NSHL) (DFNB82) in a large Palestinian family.9 Subsequently, a second nonsense mutation, c.1684C>T (p.Gln562X), was identified in a Turkish family whose members also displayed profound hearing loss,10 confirming that GPSM2 is one of more than 50 genes in which mutations are known to cause recessive deafness.11 Given the identification of GPSM2 mutations in individuals with CMS and in individuals with profound, apparently nonsyndromic hearing loss, we investigated whether the latter group might have asymptomatic brain malformations. Brain imaging was performed on one affected individual from the Palestinian family and all three affected individuals from the Turkish family. Despite the fact that study subjects 9A, 10A, 10B, and 11A did not exhibit any neurological deficits (Table 1), all four individuals displayed imaging features consistent with CMS (Table 2 and Figure 1E–1H). In contrast to the CMS subjects, none of the individuals studied solely because of hearing loss had ventriculomegaly, and the corpus callosum abnormalities tended to be less severe. Genotype-phenotype correlations were not apparent, in that the individuals (5A, 5B, 6A, and 9A) with GPSM2 mutations predicted to result in the shortest proteins (p.Asn247AsnfsX34 and p.Arg127X) did not have more severe brain malformations or clinical features than the other subjects studied.
The GPSM2 mutations identified in individuals with CMS highlight the role of GPSM2 in normal brain development and in mechanisms that underlie common brain malformations, such as partial agenesis of the corpus callosum, heterotopia, and polymicrogyria. During early neurogenesis in the mouse cerebral cortex, Gpsm2 is required for planar orientation of the mitotic spindle in apical progenitor cells (radial glia).12,13 In mice, an engineered variant (ΔC) very similar to the human p.Gly491GlyfsX6 variant (Figure 2F) results in abnormally localized apical progenitors but does not seem to radically affect the number or organization of cortical neurons, although phenotypes in mature mouse brain have not been published.13,14 It is tempting to speculate that the ectopic neuronal precursors could result in heterotopic neurons analogous to the heterotopia observed in individuals with CMS. GPSM2 is also required for correct spindle orientation in keratinocyte progenitors,15 T cells,16 oocytes,17 and epithelial cells,18 as well as for neurotransmitter localization in mature neurons.19 Given this central role of GPSM2 (also known as LGN [Leu-Gly-Asn repeat-enriched protein] and Pins [Partner of Inscuteable, homolog of Drosophila]) in cell division, it is surprising that truncating mutations do not cause more widespread defects in individuals with CMS and in the mouse model.
In conclusion, we provide compelling evidence that GPSM2 mutations account for CMS in most, if not all, affected individuals, confirming a role for GPSM2 in human brain development. In contrast to others affected with hydrocephalus, agenesis of the corpus callosum, polymicrogyria, and heterotopia, individuals with CMS usually do not have significant cognitive impairment or seizures. Therefore, GPSM2 sequencing should be performed both in individuals with brain-imaging findings of CMS and in individuals with sensorineural hearing loss who have not undergone brain imaging. Identification of GPSM2 mutations in fetuses and infants with agenesis of the corpus callosum and heterotopia would greatly alter prognostic counseling and allow for early detection and treatment of hearing loss after birth. Although it seems probable that GPSM2-related defects in asymmetric cell division underlie the hearing loss and abnormal brain development in CMS, the mechanistic details remain to be established through future investigations.
Acknowledgments
We are grateful to the family members who participated in this study, to Sunita Khatkar for conducting candidate gene analysis, to Kirk McManus for preparing Figure 2, and to Tom Walsh and Mary-Claire King for critically reading the manuscript. The research was supported by grants from the National Institutes of Health, KL2-RR025015 (to D.D.), R01DC009645 (to M.T.), and R01DC011835 (to M.K.); the Manitoba Institute of Child Health (to A.E.C. and B.T.R.); and the Winnipeg Rh Institute Foundation (to T.Z.). FORGE (Finding of Rare Disease Genes) Canada funding was provided by the government of Canada through Genome Canada, the Canadian Institutes of Health Research, and the Ontario Genomics Institute (OGI-049). Additional funding was provided by Genome Quebec and Genome British Columbia.
FORGE Canada Consortium Steering Committee: Kym Boycott (leader; University of Ottawa), Jan Friedman (coleader; University of British Columbia), Jacques Michaud (coleader; Université de Montréal), Francois Bernier (University of Calgary), Michael Brudno (University of Toronto), Bridget Fernandez (Memorial University), Bartha Knoppers (McGill University), Mark Samuels (Université de Montréal), Steve Scherer (University of Toronto).
Contributor Information
Dan Doherty, Email: ddoher@u.washington.edu.
Teresa Zelinski, Email: zelinski@cc.umanitoba.ca.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NHLBI Grand Opportunity Exome Sequencing Project (NHLBI GO ESP) Exome Variant Server (EVS), http://evs.gs.washington.edu/EVS
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Chudley A.E., McCullough C., McCullough D.W. Bilateral sensorineural deafness and hydrocephalus due to foramen of Monro obstruction in sibs: a newly described autosomal recessive disorder. Am. J. Med. Genet. 1997;68:350–356. doi: 10.1002/(sici)1096-8628(19970131)68:3<350::aid-ajmg19>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 2.Hendriks Y.M.C., Laan L.A.E.M., Vielvoye G.J., van Haeringen A. Bilateral sensorineural deafness, partial agenesis of the corpus callosum, and arachnoid cysts in two sisters. Am. J. Med. Genet. 1999;86:183–186. [PubMed] [Google Scholar]
- 3.Lemire E.G., Stoeber G.P. Chudley-McCullough syndrome: bilateral sensorineural deafness, hydrocephalus, and other structural brain abnormalities. Am. J. Med. Genet. 2000;90:127–130. doi: 10.1002/(sici)1096-8628(20000117)90:2<127::aid-ajmg8>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 4.Welch K.O., Tekin M., Nance W.E., Blanton S.H., Arnos K.S., Pandya A. Chudley-McCullough syndrome: expanded phenotype and review of the literature. Am. J. Med. Genet. A. 2003;119A:71–76. doi: 10.1002/ajmg.a.10180. [DOI] [PubMed] [Google Scholar]
- 5.Østergaard E., Pedersen V.F., Skriver E.B., Brøndum-Nielsen K. Brothers with Chudley-McCullough syndrome: sensorineural deafness, agenesis of the corpus callosum, and other structural brain abnormalities. Am. J. Med. Genet. A. 2004;124A:74–78. doi: 10.1002/ajmg.a.20380. [DOI] [PubMed] [Google Scholar]
- 6.Matteucci F., Tarantino E., Bianchi M.C., Cingolani C., Fattori B., Nacci A., Ursino F. Sensorineural deafness, hydrocephalus and structural brain abnormalities in two sisters: the Chudley-McCullough syndrome. Am. J. Med. Genet. A. 2006;140:1183–1188. doi: 10.1002/ajmg.a.31178. [DOI] [PubMed] [Google Scholar]
- 7.Alrashdi I., Barker R., Patton M.A. Chudley-McCullough syndrome: another report and a brief review of the literature. Clin. Dysmorphol. 2011;20:107–110. doi: 10.1097/MCD.0b013e328341d007. [DOI] [PubMed] [Google Scholar]
- 8.Kau T., Veraguth D., Schiegl H., Scheer I., Boltshauser E. Chudley-McCullough syndrome: case report and review of the neuroimaging spectrum. Neuropediatrics. 2012;43:44–47. doi: 10.1055/s-0032-1307451. [DOI] [PubMed] [Google Scholar]
- 9.Walsh T., Shahin H., Elkan-Miller T., Lee M.K., Thornton A.M., Roeb W., Abu Rayyan A., Loulus S., Avraham K.B., King M.-C., Kanaan M. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am. J. Hum. Genet. 2010;87:90–94. doi: 10.1016/j.ajhg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yariz K.O., Walsh T., Akay H., Duman D., Akkaynak A.C., King M.-C., Tekin M. A truncating mutation in GPSM2 is associated with recessive non-syndromic hearing loss. Clin. Genet. 2012;81:289–293. doi: 10.1111/j.1399-0004.2011.01654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dror A.A., Avraham K.B. Hearing loss: mechanisms revealed by genetics and cell biology. Annu. Rev. Genet. 2009;43:411–437. doi: 10.1146/annurev-genet-102108-134135. [DOI] [PubMed] [Google Scholar]
- 12.Morin X., Jaouen F., Durbec P. Control of planar divisions by the G-protein regulator LGN maintains progenitors in the chick neuroepithelium. Nat. Neurosci. 2007;10:1440–1448. doi: 10.1038/nn1984. [DOI] [PubMed] [Google Scholar]
- 13.Konno D., Shioi G., Shitamukai A., Mori A., Kiyonari H., Miyata T., Matsuzaki F. Neuroepithelial progenitors undergo LGN-dependent planar divisions to maintain self-renewability during mammalian neurogenesis. Nat. Cell Biol. 2008;10:93–101. doi: 10.1038/ncb1673. [DOI] [PubMed] [Google Scholar]
- 14.Shioi G., Konno D., Shitamukai A., Matsuzaki F. Structural basis for self-renewal of neural progenitors in cortical neurogenesis. Cereb. Cortex. 2009;19(Suppl 1):i55–i61. doi: 10.1093/cercor/bhp042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams S.E., Beronja S., Pasolli H.A., Fuchs E. Asymmetric cell divisions promote Notch-dependent epidermal differentiation. Nature. 2011;470:353–358. doi: 10.1038/nature09793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oliaro J., Van Ham V., Sacirbegovic F., Pasam A., Bomzon Z., Pham K., Ludford-Menting M.J., Waterhouse N.J., Bots M., Hawkins E.D. Asymmetric cell division of T cells upon antigen presentation uses multiple conserved mechanisms. J. Immunol. 2010;185:367–375. doi: 10.4049/jimmunol.0903627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo X., Gao S. Pins homolog LGN regulates meiotic spindle organization in mouse oocytes. Cell Res. 2009;19:838–848. doi: 10.1038/cr.2009.54. [DOI] [PubMed] [Google Scholar]
- 18.Zheng Z., Zhu H., Wan Q., Liu J., Xiao Z., Siderovski D.P., Du Q. LGN regulates mitotic spindle orientation during epithelial morphogenesis. J. Cell Biol. 2010;189:275–288. doi: 10.1083/jcb.200910021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sans N., Wang P.Y., Du Q., Petralia R.S., Wang Y.X., Nakka S., Blumer J.B., Macara I.G., Wenthold R.J. mPins modulates PSD-95 and SAP102 trafficking and influences NMDA receptor surface expression. Nat. Cell Biol. 2005;7:1179–1190. doi: 10.1038/ncb1325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.