

Abstract
The evolutionarily conserved cohesin complex was originally described for its role in regulating sister-chromatid cohesion during mitosis and meiosis. Cohesin and its regulatory proteins have been implicated in several human developmental disorders, including Cornelia de Lange (CdLS) and Roberts syndromes. Here we show that human mutations in the integral cohesin structural protein RAD21 result in a congenital phenotype consistent with a “cohesinopathy.” Children with RAD21 mutations display growth retardation, minor skeletal anomalies, and facial features that overlap findings in individuals with CdLS. Notably, unlike children with mutations in NIPBL, SMC1A, or SMC3, these individuals have much milder cognitive impairment than those with classical CdLS. Mechanistically, these mutations act at the RAD21 interface with the other cohesin proteins STAG2 and SMC1A, impair cellular DNA damage response, and disrupt transcription in a zebrafish model. Our data suggest that, compared to loss-of-function mutations, dominant missense mutations result in more severe functional defects and cause worse structural and cognitive clinical findings. These results underscore the essential role of RAD21 in eukaryotes and emphasize the need for further understanding of the role of cohesin in human development.
Introduction
In eukaryotic cells, sister chromatids remain physically connected from replication until their separation during anaphase. This phenomenon, called sister-chromatid cohesion, is essential for the proper segregation of the duplicated genome. Sister-chromatid cohesion is mediated by cohesin, a multimeric complex that consists of at least four core proteins: a heterodimer of SMC1 and SMC3 that forms a hinged ring-like structure, plus two “clasp” proteins, STAG (stromal antigen, or Stromalin, also known as SA) and RAD21, that play a central structural role.1 A number of regulatory proteins, including NIPBL and ESCO2, have also been found to have roles in cohesin's function.2–4 In addition to regulating sister-chromatid cohesion, the cohesin complex has been implicated in global transcriptional regulation5–10 and DNA double-strand break (DSB) repair.11–15
Mutations in NIPBL (MIM 608667) have been identified in ∼60% of individuals with classical Cornelia de Lange syndrome (CdLS [MIM 122470]), also termed Brachmann-de Lange syndrome (BdLS), a dominantly inherited developmental disorder with characteristic facial features including synophrys, arched eyebrows, a short anteverted nose, micrognathia, hirsutism, upper-extremity abnormalities ranging from small hands to severe deficiencies of the limbs, cardiac defects, growth retardation, and moderate to severe neurodevelopmental delay.16,17 Subsequently, mutations in SMC1A18,19 (MIM 300040) and SMC319 (MIM 606062) were identified in ∼5% of children with milder variants of CdLS; these children demonstrate fuller eyebrows, a prominence of the nasal bridge, predominant cognitive involvement, and few structural anomalies.20 In addition, mutations in the gene encoding cohesin-regulatory protein ESCO2 (MIM 609353) cause Roberts Syndrome and SC Phocomelia21,22 (RS/SCP [MIM 268300]), recessive disorders that, like CdLS, involve growth failure and mental retardation, although the facial features and limb anomalies are distinct from characteristics of CdLS.
The RAD21 subunit of the cohesin complex plays important structural and functional roles, in that it serves as the only physical link between the SMC1/SMC3 heterodimer and the STAG subunit and that its integrity regulates the association or disassociation of functional cohesin with chromatin.1 Although human RAD21 (MIM 606462) was initially cloned in 1996,23 in this work we identify RAD21 mutations that cause an additional clinically overlapping disorder of cohesin. These findings thus expand our understanding of the pervasive roles of the cohesin complex in human development.
Subjects and Methods
Human Subjects
All individuals were enrolled in the study under an Institutional Review Board-approved protocol of informed consent at The Children's Hospital of Philadelphia, the Institut für Humangenetik Lübeck, or the Institut für Humangenetik Essen.
Genome-wide Copy-Number Analysis
Whole-genome SNP genotyping was performed with Illumina (San Diego, CA) Infinium HumanHap550 Beadchip or Affymetrix (Fremont, CA) Genome-Wide Human SNP 6.0 arrays according to the manufacturers' protocols. Copy-number calling was performed with custom algorithms24 and PennCNV.25 Inspection of copy-number variants was performed for 8q24 by analysis of allele frequency and log R ratios with Illumina BeadStudio (ver. 3.1.3) or Affymetrix Chromosome Analysis Suite (version 1.0) software as described.26
Mutation Identification
Genomic DNA was screened for mutations in the coding exons and intron-exon boundaries by PCR of genomic DNA followed by high-resolution melt-curve analysis27 and sequencing. Primers were designed with ExonPrimer. Primer sequences and PCR conditions for RAD21 are available upon request. Amplimers were analyzed in duplicate with a LightScanner (Idaho Technology, Salt Lake City, Utah). Any variants identified by high-resolution melt-curve analysis were subsequently sequenced. Sequencing was performed with BigDye Terminator v3.1 cycle sequencing and analyzed on an ABI 3730 (Applied Biosystems, Carlsbad, CA). All probands were pre-screened and found to be negative for mutations in NIPBL, SMC1A, and SMC3. Sequence analysis was performed with Sequence Pilot (JSI Medical Systems, Kippenheim, Germany), Sequencher v4.8 (Gene Codes Corporation, Ann Arbor, MI) and MacVector v10.5.1 (Accelrys Corp, San Diego, CA).
Reference Sequences and RAD21 Conservation Analysis
RefSeq ID numbers for mRNAs and proteins are as follows: human RAD21 (NM_006265.2); human RAD21 (EAW91965); mouse Rad21 (AF332086_1); Xenopus laevis rad21 (AAC26809); zebrafish Rad21 (NP_955889); Drosophila melanogaster Rad21 (EAA46289); C. elegans SCC-1 (NP_494836); and Saccharomyces cerevisiae Scc1 (NP_011321). Sequences were aligned by the ClustalW method28 with MacVector software (Accelrys Corp, San Diego, CA).
Modeling of the p.Cys585Arg Mutation on RAD21-SMC1 Interaction
A wild-type model (wt) and p.Cys585Arg (c.1753T>C) model (mut) of human RAD21 in complex with human SMC1A were built with the YASARA v10.8.16 software package.29,30 For the homology models, running PSI-BLAST against UniProt generated a position-specific scoring matrix (PSSM). The PSSM was used in a Protein Data Bank search for potential modeling templates. The templates were ranked by an alignment score and WHAT_CHECK.31 The highest-scoring template for human RAD21 complexed with human SMC1A was the X-ray structure of the yeast SCC1-SMC1complex32 (PDB code 1W1W). It was used for creating the 3D structure of the wt and mut models with default settings. A hybrid model derived from 20 initial models was used for further analysis. All models were energy minimized by a YAMBER3 force field so that bumps were removed, and the covalent geometry was corrected.33,34 After removal of conformational stress by a short, steep descent minimization, the procedure continued by simulated annealing until convergence was reached. After validation with WHAT_CHECK, the average-quality Z score for the wt and mut models was −1.4, which is better than that for the template (−2.1). A structure file of the models and alignments is available upon request.
After homology modeling, a molecular dynamics (MD) simulation was performed on the wt and mut human RAD21-SMC1A complexes. At first, a cubic cell was created around the atoms of the homology model and was filled with water to a density of 0.997 g/liter. Counter ions were placed, and minimizations, first with the water solvent and then with the whole system, were done. After this, a short equilibration procedure calibrated the models to 298K. Resulting models were used for a 4 ns MD simulation (time step 2.5 fs).
Tissue Culture
Lymphoblastoid cell lines (LCLs) were cultured in RPMI 1640 medium supplemented to 20% fetal bovine serum (FBS) and 2% 20 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37°C in 5% CO2. HeLa cells were cultured at 37°C and 5% CO2 in DMEM supplemented to 10% FBS. Nonsense-mediated decay was inhibited with 1 mg/ml cycloheximide (Sigma-Aldrich, St. Louis, MO) in culture media for 6 hr prior to RNA harvesting.
RT-PCR
LCLs were lysed and RNA was harvested with RNeasy Plus Mini Kit (QIAGEN, Valencia, CA). cDNA was synthesized with the SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA) and random hexamers. TaqMan qRT-PCR gene-expression assays were performed on the SDS-7900HT system (Applied Biosystems, Carlsbad, CA. Samples were run in triplicate for RAD21 (ABI Hs01085854_mH) and normalized to endogenous MAPK1 (ABI Hs01052196_m1) levels.6 Analysis was performed with RQ Manager 2.0 software (Applied Biosystems).
Immunoblotting
Cells were lysed in a Nonidet P-40 lysis buffer (0.15M NaCl, 1% NP-40, and 0.05M Tris-HCl [pH 8.0]) treated with a mixture of protease inhibitors (0.25 mM PMSF, 10 μg/ml aprotinin and leupeptin, and 1mM DTT). Protein was separated via 7.5% SDS-PAGE and transferred to a nitrocellulose membrane (Whatman, London, UK). Membranes were incubated with a 1:200 dilution of anti-Rad21 (Abcam, Cambridge, UK), a 1:500 dilution of anti-GAL4-DBD/AD (Santa Cruz), or a 1:250 dilution of anti-actin (Sigma) in 5% BLOTTO overnight at 4°C. Secondary antibody exposure was performed for 1 hr with 1:2000 anti-rabbit HRP (Amersham Biosciences). Blots were visualized with the ECL Western Blotting Analysis System (GE Healthcare). Quantitative chemiluminescent analysis was performed with a VersaDoc 5000 (Bio-Rad Laboratories, Philadelphia, PA).
Constructs
The human RAD21 (IMAGE 6044010; GenBank BC050381) and zebrafish rad21 (NM_199595) open-reading frames, with the addition of BglII and XhoI sites, were amplified by PCR. They were subsequently digested and ligated into BamHI- and XhoI-digested pCS2+.35,36 Mutations were introduced with the QuikChange Site-Directed Mutagenesis Kit (Stratagene/Agilent, Santa Clara, CA). Oligonucleotide sequences used are listed in Table S5.
Mammalian Two-Hybrid Quantitative Reporter Assays
The full-length open-reading frame of human RAD21 was inserted into the pCMV-BD expression plasmid (Stratagene, La Jolla, CA). Mutant constructs were generated by site-directed in vitro mutagenesis as above. The full-length open-reading frames of STAG1, STAG2 and STAG3 (accession numbers BC064699, BX640970, and BC047490, respectively) were cloned in-frame into the pCMV-AD plasmid. HeLa cells were transiently transfected in 24-well plates (Sarstedt, Nümbrecht, Germany) with FuGene-HD (Roche, Mannheim, Germany) according to the manufacturer's instructions, and the expression of fusion proteins was verified by immunoblotting. The phRG-TK Renilla expression vector (Promega, Mannheim, Germany) was used as a transfection control. After incubations of 24 hr, the activities of firefly and Renilla luciferase were measured with the Dual Luciferase Reporter Assay System (Promega) in a Berthold Luminometer (Berthold, Bad Wildbad, Germany). All measurements were performed in at least six independent experiments in triplicate. Relative luciferase activity was determined as the average firefly:renilla luciferase activity ratio.
Cell-Survival Assays
LCLs from affected individuals and controls were irradiated on ice with graded doses of gamma radiation (0, 1, 2, 3, 4, and 5 Gy) from a 137Cs source at a dose rate of 0.56 Gy per minute. Cells were cultured for 96 hr, and cell viability was determined with a tetrazolinium salt WST-1 [2-(4-Iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium] (Roche Applied Science, Indianapolis, IN) according to the manufacturer's instructions. WST-1 accumulates in proportion to mitochondrial density and therefore is an indirect measure of cell number. The fraction of cell survival was expressed as a ratio of surviving cells to unirradiated cells (0 Gy). Each data point represents the mean of four replicates. Three independent experiments were performed for each assay. Survival curves were fitted by nonlinear regression analysis with the exponential model (Prism version 5.01, GraphPad Software, San Diego, California USA).
Micronucleus Assays
The cytokinesis-block micronucleus assay was performed essentially as described elsewhere.37 In brief, LCLs were subjected to 5 Gy of gamma radiation as described above. Cells were placed at 37°C for 4 hr to allow recovery. Unirradiated cells were used as controls for spontaneous micronucleus frequencies. Cytochalasin-B (Sigma-Aldrich, St. Louis, MO) was added at a final concentration of 2 μg/ml so that cells that had undergone a single karyokinesis but not cytokinesis could be identified; i.e., micronuclei from previous cell-cycle transits were excluded. Cells were harvested 44 hr after the addition of Cytochalasin-B, fixed in three parts methanol:one part acetic acid, and dropped onto poly-L slides. Slides were air-dried for 10 min and stained with Diff-Quick (Lab Aids Pty Ltd, North Narrabeen, NSW, Australia). Slides were scored for the number of binucleated cells (BNCs) containing (1) micronuclei (MNi), (2) nucleoplasmic bridges (NPBs), and (3) both MNi and NPBs according to described criteria.38 Two hundred BNCs were scored for each cell line.
Single-Cell Gel Electrophoresis, or Comet Assays
This assay quantifies DNA fragmentation as a result of DNA DSBs, the main ionizing radiation (IR)-induced lesion correlating with cell death. LCLs were irradiated on ice with 8 Gy of gamma irradiation as above. Cells were harvested 0, 1 and 4 hr after irradiation. Unirradiated cells were used as a control for the basal level of DNA damage. The comet assay for the detection of DSBs was performed essentially as described.39 In brief, cell lysis was performed in neutral lysis buffer (2M NaCl, 30 mM Na2EDTA, 10 mM Tris-HCl, 1% Sarkosyl, 1% Triton X-100, and 10% DMSO [pH 8.3]), and electrophoresis was carried out in TBE buffer (2 mM Na2EDTA, 90 mM Tris-HCl and 90 mM Boric acid [pH 8.3]). DNA damage was measured as the tail moment (tail moment = [(tail length × tail DNA intensity)/entire cell DNA intensity (head and tail)] × 100) with CASP image analysis software (http://casp.sourceforge.net). For each data point, eight to ten images were captured. Tail moments were measured for all cells, and doublets in each image were excluded. The mean of tail moment was calculated with a minimum of 50 cells per data point.
Sister-Chromatid-Cohesion Assays
Metaphase spreads were performed on LCLs. Sister-chromatid cohesion was evaluated in two independent laboratories via a semiquantitative method where cells were arrested in 0.15 μg/ml Colcemid (Sigma-Aldrich, St. Louis, MO) for 2 hr at 37°C. After a wash in PBS, cells were incubated in 1 ml 0.075M KCl hypotonic buffer for 10 min. Cells were then fixed in three parts methanol:one part acetic acid and dropped onto Poly-L slides. Slides were air dried and stained with Diff-Quick (Lab Aids Pty Ltd, North Narrabeen, NSW, Australia). The frequencies of cells with arms open (chromosome arms showed a clear separation), partially open (chromosome arms were easily distinguishable), and closed (chromosome arms had no separation) were scored. From 36 to 53 metaphase chromosome spreads were counted per cell line.
Cell-Cycle Analysis
LCLs in exponential growth were seeded at a density of 1 million cells per ml. Bromodeoxyuridine (BrdU), an analog of the DNA precursor thymidine, was added to a final concentration of 10 μM, and cells were incubated at 37°C with 5% CO2. BrdU is incorporated into newly synthesized DNA by cells in the S phase of the cell cycle. After 1 hr incubation in the presence of BrdU, cells were washed twice in the culture medium so that unincorporated BrdU would be removed, and they were then cultured in 20 ml fresh medium. At various time points (0, 6, and 21 hr after BrdU labeling), 5 ml of cells were removed and washed twice with PBS. Cells were then fixed and processed for BrdU staining and flow cytometry with the BrdU Flow Kit (BD PharMingen, San Diego, CA, USA) according to the manufacturer's instructions. BrdU-positive cells were detected by FITC-conjugated anti-BrdU antibody in combination with total DNA content via propidium iodide (PI) staining. Cell-cycle analysis was performed with FCS Express3 software.
Zebrafish Lines
Zebrafish were maintained as described previously.40 Embryos for experiments were grown at 28°C or 22°C in E3 medium to the required developmental stages. All zebrafish research was approved by the University of Otago Animal Ethics Committee.
RNA Synthesis and Microinjection
Capped mRNAs corresponding to the human and zebrafish RAD21 variants and controls were synthesized with an SP6 mMessage mMachine transcription kit (Ambion) according to the manufacturer's instructions. All mRNAs were resuspended in water. For microinjection, 400 pg of each mRNA was injected into the cell of 1 cell embryos from rad21nz171 heterozygous intercrosses.
Zebrafish whole-mount in situ hybridization was performed as described previously.41
Statistical Analyses
Statistical analyses of crystal modeling are noted above and in the Supplemental Data. For mammalian two-hybrid assays and unirradiated-basal-DNA-damage comet assays, significance was calculated with unpaired two-tailed t tests. Analysis of significance for grouped data sets in the comet assays for sister-chromatid separation, micronucleus, and DNA-damage recovery was performed by chi-square analysis. Fisher's exact test was used for cell-cycle fraction analysis. Analysis of ionizing radiation survival was performed with two-term linear regression. Graphing and statistical analyses were performed with Prism v. 5.0 (GraphPad Software, La Jolla, CA) or Stata 11 (StataCorp, College Station, TX). Significance values are indicated in the text and figure legends.
Results
Identification of Children with Mutations in RAD21
To identify additional potential causal loci for CdLS, we performed genome-wide array-based copy-number analysis of 101 individuals with typical CdLS and 189 with overlapping features. All individuals were negative for mutations in NIPBL, SMC1A, and SMC3, genes previously identified as having mutations in CdLS. We identified a single boy (P1) with a chromosomal alteration containing a known cohesin gene, specifically, a de novo 8q24.1 interstitial microdeletion that includes RAD21, which encodes the kleisin subunit that binds to the SMC1 and SMC3 components of the cohesin complex as well as to other regulatory proteins, including the STAG/Scc3 proteins1 (Figures 1A and 1B). This boy's phenotypic features included synophrys with highly arched eyebrows, micrognathia, short stature, and minor radial-bone anomalies (Figure 1C and Figure S1), but unlike CdLS individuals with NIPBL, SMC1A, or SMC3 mutations, he displayed variant features that included sparse temporal scalp hair and, most remarkably, normal cognitive development. He was diagnosed as an infant on the basis of his facial and growth characteristics, which overlapped with those of CdLS.
Figure 1.
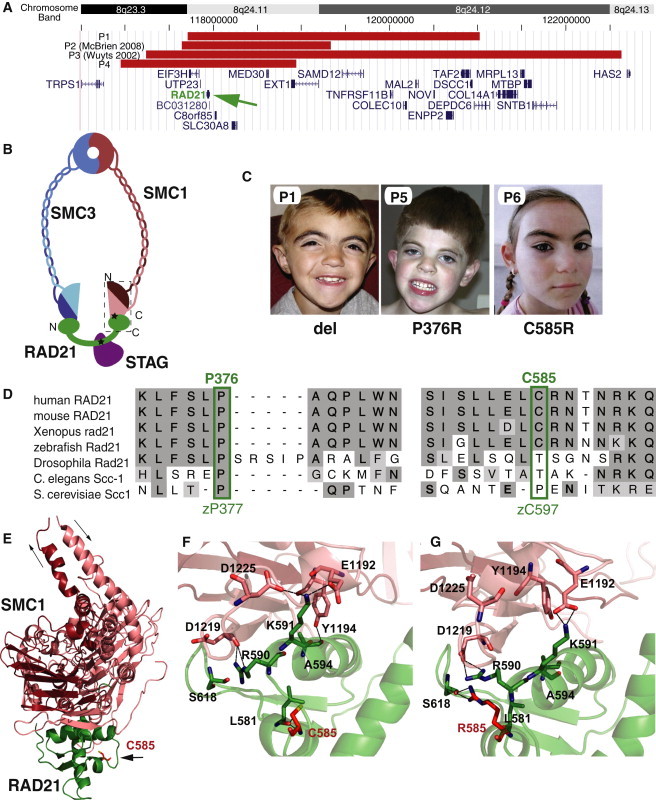
Mutations in RAD21
(A) Deletions including RAD21. Localization of deletions (red bars) on 8q24 is indicated by chromosome band and position (hg 18). Gene locations are indicated in blue with gene names. RAD21 is indicated by a green arrow.
(B) Schematic representation of the cohesin complex. SMC1A (red) and SMC3 (blue) N- (lighter) and C- (darker) termini are indicated. Relative positions of RAD21 binding to SMC1A via its C terminus and to STAGs via its central region are noted. Black stars indicate relative positions of missense mutations. A dashed box indicates the region represented in the crystal structure in (D).
(C). Facial features of children with RAD21 mutations. del = RAD21 deletion, P376R = p.Pro376Arg, and C585R = p.Cys585Arg mutations.
(D) Conservation of RAD21 sequence and position of missense mutations.
(E) Localization of the RAD21 p.Cys585Arg (C585) mutation near the interface with SMC1.
(F) Detail of SMC1 wild-type RAD21 molecular interface.
(G) Modeled effect of RAD21 p.Cys585Arg mutation on SMC1 interaction.
To further investigate the effect of 8q24.1 deletions, we compared the features of two previously reported individuals (P242 and P343) and one newly identified individual (P4) with overlapping deletions. All four children (Figures 1A and 1C) had features consistent with those of other cohesin disorders; such features included short stature, microcephaly, highly arched or thick eyebrows with synophrys, and palatal and vertebral anomalies. They also displayed minor cognitive delays, typically less significant than those seen in even “mild” CdLS (Figure S1 and Table S1). Of those genes in the minimal overlapping interval, RAD21 is the only one known to function in the cohesin complex, and therefore we hypothesized that it is responsible for this cohesinopathy-like phenotype. Sequencing of the remaining non-deleted allele for probands P1, P3, and P4 revealed no additional mutations in RAD21.
Subsequently, we screened the exons of RAD21 for mutations in 258 individuals who had CdLS or overlapping features and who were previously found to be negative for mutations in NIPBL, SMC1A, and SMC3. This analysis identified two additional probands who had atypical clinical features and who demonstrated heterozygous de novo missense mutations (Figure 1C and Table S1). These children (P5 and P6) had c.1127C>G (p.Pro376Arg) and c.1753T>C (p.Cys585Arg) mutations, respectively. Both individuals had mild neurodevelopmental deficits and highly arched eyebrows, although P5 had more substantial cognitive dysfunction along with a cleft palate, tetralogy of Fallot, and minor radial head and vertebral anomalies. All of these features are observed in individuals with mutations in other cohesin genes.44 Importantly, neither of these mutations was observed in any of the more than 600 control chromosomes from people of European descent (Table S2).
Expression of RAD21 in Cells Carrying RAD21 Mutations
To understand how these mutations could result in altered RAD21 activity, we first measured RNA and protein expression levels from the two available LCLs, one with a contiguous gene deletion (P1) and one with the c.1127C>G (p.Pro376Arg) missense mutation (P5). The cell line with the RAD21 deletion expressed approximately half the normal level of RAD21 RNA, but the line with the c.1127C>G (p.Pro376Arg) mutation expressed a slightly higher total level of RAD21 RNA, while maintaining expression of both alleles (Figures S2A an S2B). The RAD21-deleted cell line also demonstrated reduced protein levels, whereas the p.Pro376Arg mutation was expressed at wild-type levels with no altered mobility in immunoblotting (Figures S2C and S2D). These data support the hypotheses that the deletion mutation results in haploinsufficiency and that another mechanism of pathogenicity probably explains the effect of the p.Pro376Arg mutation.
RAD21 Mutations Alter Conserved Amino Acids
To determine whether the RAD21 missense mutations were pathogenic in these children, we performed a series of additional analyses. Computational analysis of the identified missense mutations demonstrated that the substituted proline of the p.Pro376Arg mutation is identically conserved through S. cerevisiae and that the substituted cysteine of the p.Cys585Arg mutation is conserved in vertebrates (Figure 1D). Furthermore, SIFT45 and Polyphen46 analyses suggest that each of these mutations is likely to be pathogenic (Table S1). Note that for experiments described later in this work, the conserved residues for these mutations in zebrafish are Pro377 and Cys597, as represented in Figure 1D.
Structural Modeling of the RAD21 p.Cys585Arg Mutation-SMC1A Complex
We next assessed the potential impact of these mutations on cohesin structure. To perform its crucial role in regulating sister-chromatid cohesion, RAD21 binds to both SMC1 and SMC3.1 Specifically, it is thought that the C terminus of RAD21 initially binds to the C terminus of SMC1 to incorporate RAD21 into the cohesin ring. This is followed by interaction of the N terminus with the SMC3 head domain to close the cohesin ring32 (Figure 1B). To understand the effects of RAD21 mutations in the structure of the cohesin complex, we modeled the human SMC1A-RAD21 structure on the basis of available data from the S. cerevisiae Scc1 cocrystallized with the Smc1 head domain32 (Figures S3 and S4). Although the p.Pro376Arg mutation lies outside of this resolved crystal structure, the p.Cys585Arg mutation is located within the resolved structure of the C terminus of RAD21 (Figures 1B and 1E–1G) and is positioned near the interface of RAD21 and the C-terminal amino acids of SMC1 (Figure 1E), suggesting that it might be involved in the interaction of these two proteins. Additional structural modeling suggests that the p.Cys585Arg mutation alters hydrogen bonding of the 585 residue from RAD21 Leu581 to Ser618. This shifts the conformation of the RAD21 alpha helix that interacts with SMC1 and predicts decreased hydrogen bonding between the two proteins at this interface (Figures 1F and 1G and Figures S5 and S6). In summary, the structural modeling data strongly suggest altered interactions at the RAD21-SMC1A interface for the p.Cys585Arg mutation, and such altered interactions would predict altered functionality of the cohesin complex. Unfortunately, despite numerous strategies, we have been unable to model this mutation effectively in an in vitro assay, and we have no cell line for this girl and thus cannot assay in vivo interactions.
The p.Pro376Arg Mutation Alters RAD21-STAG Interaction
In addition to binding to both SMC1 and SMC3, RAD21 binds to STAG, the fourth core cohesin subunit (Figure 1B). This interaction is also integral in the regulation of sister-chromatid cohesion.2,47 Importantly, the p.Pro376Arg mutation resides in a region of conserved homology (amino acids 372–392) that has been shown to be essential for interaction of RAD21 with the STAG proteins.2 Thus, we hypothesized that the p.Pro376Arg mutation might disrupt RAD21's interaction with the STAG proteins.
To test RAD21-STAG interaction in a quantitative manner, we performed a mammalian two-hybrid assay. In these experiments, wild-type or mutant human RAD21 was tested for binding to STAG1, STAG2, or STAG3. Strikingly, the p.Pro376Arg mutation resulted in an increased interaction of RAD21 with STAG1 and STAG2 (120% and 125%, respectively), whereas in that with STAG3, the meiotic stromalin was unchanged (Figure 2A). As expected, the p.Cys585Arg mutation that lies outside the STAG binding region does not alter STAG binding affinity (Figure 2A).
Figure 2.
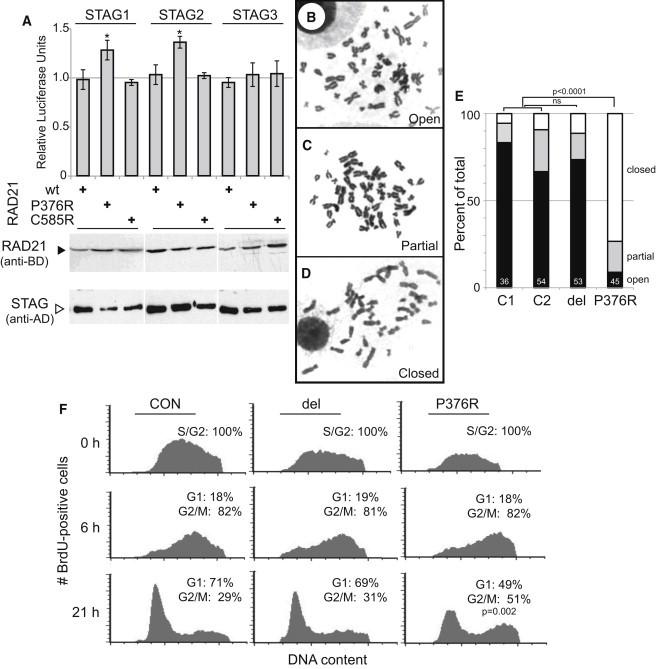
RAD21 Alteration Effects on the Cohesin Complex
(A) RAD21 p.Pro376Arg increases binding affinity to mitotic STAG proteins. Immunoblotting of similarly expressed hybrid proteins is indicated below. Significant p values (<0.05) from two-tailed t tests are indicated (∗). Open (B), partially open (C) and closed (D) sister-chromatid phenotypes are demonstrated. (E) Increased prevalence of closed sister chromatids in LCLs from the boy with the p.Pro376Arg mutation and in normal control LCLs (C1, C2). The RAD21-deleted LCLs (del) show no difference from controls. The number of metaphases analyzed for each cell line is indicated in white at the base. Chi-square calculations of significance in difference of all cell phenotypes of RAD21 mutant cell lines compared with the averaged controls are indicated above the columns. (F) Flow cytometry smoothened histogram plots of BrdU-positive cells versus DNA content showing the proportions of G1, S, and G2/M cells 0, 6, and 21 hr after BrdU labeling. The proportion of cells in each cell cycle phase is expressed as the percentage of BrdU-positive cells. Two-sided Fisher's exact test is shown. Error bars indicate standard deviations. del = RAD21 deletion, P376R = p.Pro376Arg, C585R = p.Cys585Arg mutations, and CON = control cells.
These data suggest that the RAD21 p.Pro376Arg mutation might interfere with cohesin activity by increasing the binding of STAG1 and STAG2 to RAD21, an activity which has been shown to mediate sister-chromatid cohesion along the length of chromosome arms.48 This altered RAD21-STAG interaction might be predicted to lead to consequences for sister chromatids in metaphase analyses.
Tighter Sister-Chromosome Cohesion is Observed in Cells with the p.Pro376Arg Mutation
Because the role of RAD21 has been well described in sister-chromatid cohesion, and because the above experiments indicated altered STAG binding, we assessed whether LCLs from two available probands (P1 [RAD21del] and P5 [p.Pro376Arg]) demonstrated alterations in sister-chromatid cohesion. Analysis of metaphase spreads (Figures 2B–2E) showed a substantially higher percentage of sister chromatids with a closed-arm phenotype in cells with the p.Pro376Arg mutation (76%) than in controls (5%). Correspondingly, there was a marked decrease in the percentage of metaphase spreads showing an open-arm configuration in p.Pro376Arg (9%) versus control (83%) LCLs. Additional examination of chromosome numbers in these LCLs also revealed an increase in aneuploidy; metaphase spreads demonstrated the gain or loss of one or more chromosome in 39% (17/44) and 29% (10/34) of RAD21 p.Pro376Arg and deletion mutant LCLs, respectively. This is in contrast to control cells, where aneuploidy was observed in7% (3/45) of LCLs. This finding was not mirrored in DNA obtained from peripheral blood of either child, as analyzed by genome-wide SNP array under a lower limit of mosaic aneuploidy detection of ∼5%.
Cell-Cycle Progression Is Altered in Cells with the p.Pro376Arg Mutation
This appearance of closely bound sister chromatids suggests that these cells might be impeded in their progression through mitosis. To assess this, we used BrdU to pulse labeled S-phase cells and monitored their progression through the cell cycle at 0, 6, and 21 hr time points after labeling. These analyses of BrdU-positive cells showed that, 6 hr after the BrdU pulse, there is no effect on progression from replication to mitosis, and the cell-cycle profiles of both the deletion and p.Pro376Arg mutants are similar to those of control cells; more than 80% are BrdU positive at the G2/M phase (Figure 2F). However, there appears to be a delay during mitosis, and by 21 hr after the BrdU pulse, when the G1 population has progressed to 71% in control cells and 69% in RAD21-deletion cells, only 49% of p.Pro376Arg-mutated cells are in G1 (Figure 2F). These data suggest that increased sister-chromatid cohesin in the p.Pro376Arg cells does indeed lead to a delay in progression from the G2/M to G1 phase.
Cellular Response to IR Is Compromised by RAD21 Mutations
In addition to its role in sister-chromatid cohesion, RAD21 and its homologs have been shown to play central roles in mediating IR response and DNA-damage repair in yeast49,50 and vertebrates.14,51 To determine whether the mutations we identified resulted in enhanced sensitivity to IR, a potent inducer of DNA DSBs, we tested the LCLs in several assays. In a cell-survival assay after IR, we noted that both RAD21-heterozygote cell lines exhibited significantly lower post-IR cell survival than control cells, but this reduction was not as great as that noted in cells derived from a homozygous individual with the classical IR-sensitivity disorder, ataxia telangiectasia52 (AT, Figure 3A).
Figure 3.
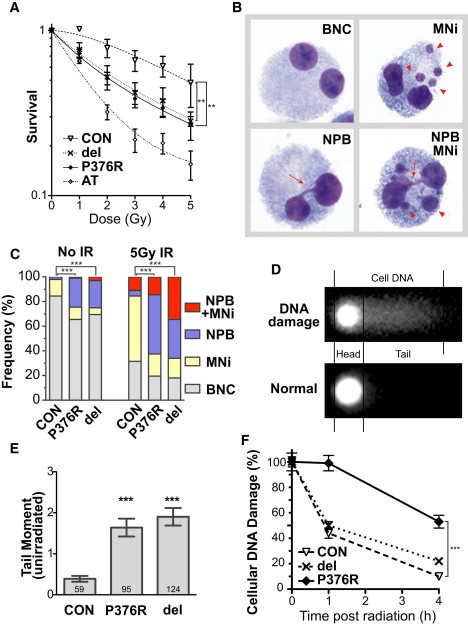
RAD21 Mutations Increase Susceptibility to DNA Damage
(A) Cell survival after a graded dose of ionizing radiation (IR). Radiation doses are indicated beneath the samples. Fractional survival was expressed relative to unirradiated cells. Control (CON), ATM-mutated (AT), RAD21-deleted (del), and RAD21 p.Pro376Arg-mutated (P376R) lymphoblastoid cell lines are indicated. Each data point represents the mean of three independent experiments. Error bars represent SEM.
(B) Representative images of binucleated cells (BNCs, top left) with micronuclei (MNi, top right, arrowheads); nucleoplasmic bridges (NPBs, bottom left, arrows), and NPBs accompanying MNi (bottom right panel).
(C) The frequencies of spontaneous and radiation-induced MNi, NPBs, and NPBs accompanying MNi, as shown in (B). The frequencies were calculated per 200 binucleated cells.
(D) A diagram of the Comet assay, indicating the head and tail dimensions used for assessments.
(E) The basal level of DNA damage, as measured by the tail moment in unirradiated cells via the Comet assay. A significant difference was observed between the control cell line (CON) and the RAD21 mutant cell lines. Numbers at base of column indicate number of nuclei assayed.
(F) DNA damage repair kinetics after IR at 8 Gy. Cellular DNA damage was measured as the tail moment and expressed relative to 0 hr after irradiation. Also see Figure S7. Significant p values for p < 0.0001 (∗∗∗) and p < 0.01 (∗∗) are noted. del = RAD21 deletion, P376R = p.Pro376Arg, and C585R = p.Cys585Arg mutations.
It has been shown that, in addition to showing decreased survival, mouse embryonic fibroblast cell lines with heterozygous null Rad21 mutations exhibit enhanced chromosomal rearrangements after IR.14 To test whether the human RAD21 mutant cell lines were similarly affected, we used a cytokinesis-block micronucleus assay53 to examine the level of chromosomal damage induced by IR. In this assay, one can observe binucleated normal cells (BNCs); micronuclei (MNi), which result from chromosomal breakage; and nucleoplasmic bridges (NPBs), which result from chromosomal structural rearrangements such as dicentric chromosomes and ring chromosomes (Figure 3B). Our results showed that both RAD21 mutant cell lines exhibited elevated levels of NPBs in unirradiated cells, suggesting an increased level of spontaneous aberrations (Figure 3C, left). After IR, an increase in the overall number of chromosomal abnormalities was observed in all lines (Figure 3C). Unlike in control cells, where MNi were prominent, NPBs and NPBs accompanying MNi accounted for the majority of chromosomal aberrations in the two cell lines with RAD21 mutations (Figure 3C right). These data suggest that spontaneous and IR-induced chromosomal damage in RAD21 mutant cells leads to increased complex chromosomal aberrations.
To further investigate whether the increased chromosomal damage in RAD21 mutant cell lines is due to a defect in DSB repair, we performed a comet assay to evaluate the kinetics of IR-induced DSB repair in RAD21 mutant and control LCLs. We determined the tail moment, which provides an estimate of DNA damage in the form of fragmentation (Figure 3D), in both unirradiated and irradiated cells to assess DNA damage in basal and IR-induced states, respectively.54 Our results showed that the basal level of DNA damage, as measured by the tail moment of unirradiated cells, is significantly higher in both RAD21 mutant cell lines than in the control cell lines (Figure 3E and Figure S7). To determine the repair kinetics of IR-induced DNA damage, we compared the tail moments 1 and 4 hr after IR to those at 0 hr (Figure 3F and Figure S7). In the control cell line, approximately 45% of DNA damage remained 1 hr after IR, and by 4 hr after IR only ∼10% of damage remained. In contrast, the p.Pro376Arg mutant cell line showed significantly slower DNA damage-repair kinetics; approximately 98% and 53% of residual DNA damage remained 1 and 4 hr, respectively, after IR. The RAD21-deletion mutant cell line also showed impaired repair kinetics, but to a lesser extent; approximately 50% and 21% of residual DNA damage remained 1 and 4 hr, respectively, after IR. These results further corroborate an impaired IR-induced DNA-damage repair in RAD21 mutant cells and, in conjunction with the higher basal-DNA-damage rate, increased cytogenetic damage, and impaired repair of that damage, strongly suggest that the RAD21 mutations identified in these individuals result in a susceptibility to the processing of both endogenous and exogenous induced DNA damage.
In summary, these data show that both the RAD21-deletion and p.Pro376Arg-mutated LCLs are deficient in their response to IR, as manifested by decreased survival and increased chromosomal rearrangements and DSBs. Furthermore, the p.Pro376Arg mutation causes deficient repair of cellular DNA damage. These observations all support the hypothesis that these mutations disrupt RAD21 function.
RAD21 Mutations Alter Transcription in a Zebrafish Model
Significant evidence has demonstrated that, in addition to functioning in sister-chromatid cohesion and DNA stability, the cohesin complex plays a key role in transcriptional regulation.5,6,8,9,55–57 One of the best examples of this activity has been observed in zebrafish, where Rad21 is required for transcription of runx1 in the posterior lateral plate mesoderm (PLM) and for the transcription of runx3 in neuronal tissues.8 runx1 is required for both hematopoietic and neuronal cell development (Figure 4A). Remarkably, expression of runx1 is absent in the PLM of mutants carrying homozygous rad21nz171-null alleles, whereas normal expression is retained in neuronal cells from the 4–15 somite stage (11–15 hr post-fertilization [hpf]), indicating a tissue-specific effect of Rad21 loss on runx1 expression8 (Figure 4C and Table S3). Expression of runx1 could be rescued by microinjection of either human RAD21 or zebrafish rad21 mRNA into rad21-null embryos8 (Figure 4D), whereas a transcript containing the rad21nz171-null mutation did not rescue expression (Tables S3 and S4). Therefore, we used the biological readout of rescued runx gene-expression pattern in zebrafish rad21 mutants to determine whether the identified human RAD21 mutations are competent to facilitate transcription.
Figure 4.
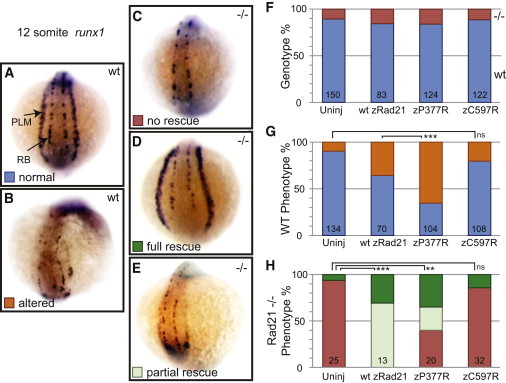
RAD21 Mutations Alter Transcriptional Activity
Rescue of lateral-plate-mesoderm expression of runx1 in Rad21-null zebrafish embryos with zebrafish rad21 mRNA that are either wild-type (wt zRad21) or carrying the equivalent human mutations (see Figure 1D).
(A) Wild-type, uninjected 12 somite embryo viewed from the posterior. PLM = posterior lateral-plate mesoderm and RB = Rohon-Beard cells.
(B) Wild-type embryo with altered runx1 expression.
(C) Rad21 homozygous null embryo with loss of PLM runx1 expression.
(D and E) Rad21 homozygous null embryos with full and partial rescues by mRNA injection.
(F) Genotype fractions of embryos analyzed for each injection cohort. Red = Rad21-null, blue = wild-type, uninj = uninjected.
(G) Frequency of altered runx1 expression in wild-type embryos. Orange = altered expression and blue = normal expression.
(H) Frequency of runx1 LPM rescue. Green = full rescue, light green = partial rescue, and red = no rescue. Error bars indicate standard deviations. Significant p values for p < 0.0001 (∗∗∗) and p < 0.01 (∗∗) are noted.
Because the wild-type human RAD21 mRNAs were moderately toxic to developing zebrafish embryos, the missense mutations were introduced into the equivalent positions of the zebrafish Rad21 (see Figure 1D), and in-vitro-transcribed mRNA encoding each of these mutations was injected into 1 cell zebrafish embryos. The distribution of runx1 or runx3 transcripts was examined at the 10–12 somite stage. After being scored, embryos were genotyped for the rad21nz171-null allele.
Zebrafish p.Pro377Arg rescued expression of runx1 in 65% of rad21-null zebrafish embryos (Figures 4D and 4H; Table S3). However, this variant also caused altered transcription in wild-type embryos. It resulted in ∼50% more atypical runx1 expression than wild-type Rad21 (Figures 4B and 4G). Remarkably, the human p.Pro376Arg RAD21 mutation altered spatiotemporal expression of runx1 in nearly all surviving embryos, suggesting that the human p.Pro376Arg protein has additional activity when compared with wild-type RAD21. Analysis of runx3 expression yielded similar results (Table S4). These data reflect the greater disruption of normal features in the boy carrying the RAD21 p.Pro376Arg mutation.
In contrast, the zebrafish p.Cys597Arg Rad21 mutation failed to rescue runx1 expression in rad21-null embryos and exhibited only background levels of activity in wild-type embryos. Analysis of runx3 expression yielded similar results (Table S4), and the activity of the zebrafish p.Cys597Arg mutation was equivalent to that of the rad21nz171-null mRNA, which contains a nonsense mutation and does not produce Rad21 protein.8 Furthermore, these data are consistent with the compromised human p.Cys585Arg RAD21 function predicted by the structural analysis and with the clinical features of the boy carrying this mutation, who demonstrates mild features similar to those of the patients with deletion alleles.
Discussion
Using a strategy of genome-wide copy-number analysis of individuals with presumed CdLS, we identified a single proband with a de novo deletion of 8q24.1 that includes RAD21. We subsequently assessed three additional individuals with deletions that include RAD21 and identified two with de novo missense mutations in RAD21 (Figure 1, Figure S1, and Table S1). Quite remarkably, although these children have some overlap with CdLS, they clearly have some divergence in the facial features and, most notably, have extremely mild cognitive and physical abnormalities. Common features in these patients include short stature, synophrys, micrognathia, brachydactyly, mild radioulnar differences, vertebral anomalies, and very mild cognitive involvement. The boy with the p.Pro376Arg mutation has more severe features, including mild mental retardation, tetralogy of Fallot, and hearing loss.
Of note, RAD21 lies between TRPS1 (MIM 604386) and EXT1 (MIM 608177) and would be deleted in persons with the Langer-Giedion/Trichorhinophalangeal syndrome, type II (TRPS II [MIM 150230]) deletion.58 It is likely that the mild facial and cognitive involvement seen in these individuals with RAD21 heterozygous loss-of-function mutations causes many individuals to go clinically unnoticed, and in the context of TRPS II, these features would be overshadowed by the distinctive facial and hand features caused by disruption of TRPS1.59
To confirm the pathogenicity of individual RAD21 mutations, we employed a number of assessments of RAD21 function. In silico analyses revealed conservation of identity of the mutated residues in vertebrate lineages, and they revealed conservation of the p.Pro376Arg mutation through yeast (Figure 1D). Structural modeling suggests that the p.Cys585Arg mutation would cause significant alterations in hydrogen bonding and thus disrupt the RAD21/SMC1 interface (Figures 1F and 1G). Because the SMC1-RAD21 interaction probably initiates RAD21 incorporation into the SMC1/SMC3 complex,32 disruption of this interaction would probably cause a defect in formation of functional cohesin complexes. Efforts to obtain a functional readout of the interaction between the RAD21 p.Cys585Arg mutation and SMC1A via two-hybrid or coimmunoprecipitation assays of overexpressed or tagged proteins (data not shown) were unfortunately unsuccessful. Furthermore, it is not possible to assess this interaction in vivo with endogenous proteins because a cell line for this girl is unavailable.
Because the RAD21 p.Pro376Arg mutation resides in the amino acid domain that interacts with the STAG proteins,2 we tested the interaction of STAG1, -2, and -3 with wild-type and mutant RAD21. As expected, the p.Cys585Arg mutation, which resides outside the interaction region, does not affect RAD21-STAG binding. Strikingly, the p.Pro376Arg mutation increases binding to both STAG1 and STAG2 (Figure 2A). This suggests that pathology associated with p.Pro376Arg could be due to retained association of RAD21 with STAG through anaphase;60 such retained association could lead to illegitimate sister-chromosome interactions, such as the increased nucleoplasmic bridging that we noted in our cytokinesis block assays (Figures 3B and 3C). Alternatively, it could result from reduced dissociation of the cohesin complex from STAG1 and -2 in prophase,60 causing delayed sister-chromatid separation or altered transcription.10 Finally, tighter association of RAD21 with STAG could cause cohesin to be resistant to removal by the WAPAL/PDS5 complex in interphase cells. This would lead to longer cohesin residency times on chromosomes and in turn lead to disregulation of genes normally controlled by cohesin.
Cued by the structural data and previous work demonstrating that alterations in the cohesin complex disrupt transcription,6–8 we used the rescue of rad21-null zebrafish embryos, which lack expression of runx1 and runx3, as a biological readout for transcriptional competency of RAD21 mutations. Unlike wild-type Rad21, the p.Cys585Arg/zebrafish p.Cys597Arg mutation had little ability to rescue runx gene expression, confirming a loss of function for this mutation. In contrast, the p.Pro376Arg/zebrafish p.Pro377Arg mutation was able to partially rescue runx1/3 expression in rad21-null fish, but it also surprisingly affected expression in wild-type embryos, suggesting an altered activity rather than a loss of function. Inappropriate activation of runx1 expression in zebrafish embryos expressing the p.Pro376Arg mutation is consistent with structural data indicating tighter binding to STAG.
To test whether the RAD21 mutations altered sister-chromatid cohesion,61 we assessed sister-chromatid separation in metaphase spreads of lymphoblastoid cells, which demonstrated decreased separation in those carrying the p.Pro376Arg mutation. These data are consistent with the increased binding of the p.Pro376Arg mutant protein to STAG and suggest that this binding might be improperly retained, resulting in tighter binding of sister chromatids as a result of the improper removal of cohesin via the prophase pathway of cohesin dissociation.60,62
Because RAD21 is involved in radiation damage response and DNA repair,12,14,50,63–65 we assessed the radiation sensitivity of LCLs from the individuals with RAD21-deletion and p.Pro376Arg mutations. These cell lines demonstrated an increased IR sensitivity, resulting in reduced survival and increased chromosomal aberrations (Figure 3). Further analysis revealed that RAD21 mutant cells showed slower DNA DSB repair, suggesting that a deficiency in DSB repair is probably the underlying cause of enhanced cellular IR sensitivity and chromosomal aberrations in RAD21 mutant cells. Enhanced cellular sensitivity to IR has also been observed in CdLS cell lines with NIPBL, SMC1A, and SMC3 mutations, concordant with the RAD21 mutations and consistent with the idea that cellular hypersensitivity to DNA-damaging agents might be a general feature of CdLS.11,65 These data are consistent with this notion.
Although short-term radiation sensitivity is elevated in cells derived from individuals with CdLS, the clinical implications of such findings are less clear. RAD21 variants were found in radiosensitive (RS) cancer patients, suggesting altered RAD21 function associates with radiation sensitivity.66 Additionally, Rad21 heterozygous mice demonstrated global enhancement of radiosensitivity after whole-body irradiation.14 Our findings of cellular radiosensitivity in cells with the heterozygous RAD21-deletion mutation (P1) and the p.Pro376Arg mutation (P5) raises the possibility that these individuals might sustain greater than expected side effects upon exposure to DNA-damaging agents such as radiotherapy. Significantly, no tumors or malignancies have been noted in these children, nor in other people with CdLS.67,68 Although cells from patients with many human radiation-sensitivity syndromes, such as ataxia telangiectasia (AT [MIM 208900]), Nijmegen breakage syndrome, (NBS [MIM 251260]), and ataxia-telangiectasia-like disorder (ATLD [MIM 604391]), have genomic instability and the individuals are prone to cancer, these two features do not invariably coassociate. For example, individuals with trichothiodystrophy (TTDP [MIM 601675]) and Cockayne syndrome (MIM 216400) display elevated radiation sensitivity but no apparent cancer predisposition. It is also possible that defects in cohesin protect individuals from cancer given that MYC is often downregulated when there is a loss of cohesin function.69
Overall, this combination of phenotypic, molecular, and cellular data emphasizes a role for RAD21 mutations as a cause of a human congenital disorder. Although only six individuals have been studied in this work, we would speculate that the extremely mild nature of this cohesinopathy leads to underascertainment. Thus, we would expect that in the coming years, with the advent of whole-exome and -genome sequencing for clinical diagnosis, additional persons will be identified.
Acknowledgments
We are exceptionally grateful to the children and families who participated in this study as well as to the referring physicians and colleagues who have contributed samples and clinical information. We thank Maninder Kaur and Melanie Hullings for technical assistance and Pavel Lobachevsky and Goli Mostoufi-Moab for assistance with statistical analysis of the radiation-survival experiments. We are indebted to the continued support of the USA and International Cornelia de Lange Syndrome Foundations. This work was supported by National Institutes of Health grants K08HD055488 (from the National Institute of Child Health and Human Development [NICHD] to M.A.D.), P01 HD052860 (from the NICHD to I.D.K.), research grants from the USA CdLS Foundation, institutional funds from the Children's Hospital of Philadelphia, Cancer Council Victoria Australia grant 081219 (to H.X. and M.J.M.), Australian National Health and Medical Research Council project grant 1007659 (to R.G.R., H.X., and M.J.M.), Marsden grant UOO0713 (to E.D., M.M., and J.H.), funds from New Zealand Lottery Health Research (to E.D., M.M., and J.H.), Spanish Ministerio de Sanidad grant PI091422 (to M.C.G.-R.), and intramural funding of the University of Lübeck, Germany (“Schwerpunktprogramm: Medizinische Genetik,” to F.J.K. and G.G.-K.).
Contributor Information
Matthew A. Deardorff, Email: deardorff@email.chop.edu.
Frank J. Kaiser, Email: frank.kaiser@uk-sh.de.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ExonPrimer, http://ihg.gsf.de/ihg/ExonPrimer
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
SIFT (Sorting Intolerant from Tolerant) Analysis, http://sift.jcvi.org
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Nasmyth K., Haering C.H. Cohesin: Its roles and mechanisms. Annu. Rev. Genet. 2009;43:525–558. doi: 10.1146/annurev-genet-102108-134233. [DOI] [PubMed] [Google Scholar]
- 2.Shintomi K., Hirano T. Releasing cohesin from chromosome arms in early mitosis: Opposing actions of Wapl-Pds5 and Sgo1. Genes Dev. 2009;23:2224–2236. doi: 10.1101/gad.1844309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watrin E., Schleiffer A., Tanaka K., Eisenhaber F., Nasmyth K., Peters J.M. Human Scc4 is required for cohesin binding to chromatin, sister-chromatid cohesion, and mitotic progression. Curr. Biol. 2006;16:863–874. doi: 10.1016/j.cub.2006.03.049. [DOI] [PubMed] [Google Scholar]
- 4.Hou F., Zou H. Two human orthologues of Eco1/Ctf7 acetyltransferases are both required for proper sister-chromatid cohesion. Mol. Biol. Cell. 2005;16:3908–3918. doi: 10.1091/mbc.E04-12-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hadjur S., Williams L.M., Ryan N.K., Cobb B.S., Sexton T., Fraser P., Fisher A.G., Merkenschlager M. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009;460:410–413. doi: 10.1038/nature08079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J., Zhang Z., Bando M., Itoh T., Deardorff M.A., Clark D., Kaur M., Tandy S., Kondoh T., Rappaport E. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. 2009;7:e1000119. doi: 10.1371/journal.pbio.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Misulovin Z., Schwartz Y.B., Li X.Y., Kahn T.G., Gause M., MacArthur S., Fay J.C., Eisen M.B., Pirrotta V., Biggin M.D. Association of cohesin and Nipped-B with transcriptionally active regions of the Drosophila melanogaster genome. Chromosoma. 2008;117:89–102. doi: 10.1007/s00412-007-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horsfield J.A., Anagnostou S.H., Hu J.K., Cho K.H., Geisler R., Lieschke G., Crosier K.E., Crosier P.S. Cohesin-dependent regulation of Runx genes. Development. 2007;134:2639–2649. doi: 10.1242/dev.002485. [DOI] [PubMed] [Google Scholar]
- 9.Wendt K.S., Yoshida K., Itoh T., Bando M., Koch B., Schirghuber E., Tsutsumi S., Nagae G., Ishihara K., Mishiro T. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 10.Dorsett D. Cohesin, gene expression and development: Lessons from Drosophila. Chromosome Res. 2009;17:185–200. doi: 10.1007/s10577-009-9022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vrouwe M.G., Elghalbzouri-Maghrani E., Meijers M., Schouten P., Godthelp B.C., Bhuiyan Z.A., Redeker E.J., Mannens M.M., Mullenders L.H., Pastink A. Increased DNA damage sensitivity of Cornelia de Lange syndrome cells: Evidence for impaired recombinational repair. Hum. Mol. Genet. 2007;16:1478–1487. doi: 10.1093/hmg/ddm098. [DOI] [PubMed] [Google Scholar]
- 12.Watrin E., Peters J.M. The cohesin complex is required for the DNA damage-induced G2/M checkpoint in mammalian cells. EMBO J. 2009;28:2625–2635. doi: 10.1038/emboj.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dodson H., Morrison C.G. Increased sister chromatid cohesion and DNA damage response factor localization at an enzyme-induced DNA double-strand break in vertebrate cells. Nucleic Acids Res. 2009;37:6054–6063. doi: 10.1093/nar/gkp684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu H., Balakrishnan K., Malaterre J., Beasley M., Yan Y., Essers J., Appeldoorn E., Tomaszewski J.M., Vazquez M., Verschoor S. Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS ONE. 2010;5:e12112. doi: 10.1371/journal.pone.0012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Musio A., Montagna C., Mariani T., Tilenni M., Focarelli M.L., Brait L., Indino E., Benedetti P.A., Chessa L., Albertini A. SMC1 involvement in fragile site expression. Hum. Mol. Genet. 2005;14:525–533. doi: 10.1093/hmg/ddi049. [DOI] [PubMed] [Google Scholar]
- 16.Krantz I.D., McCallum J., DeScipio C., Kaur M., Gillis L.A., Yaeger D., Jukofsky L., Wasserman N., Bottani A., Morris C.A. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 2004;36:631–635. doi: 10.1038/ng1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tonkin E.T., Wang T.J., Lisgo S., Bamshad M.J., Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet. 2004;36:636–641. doi: 10.1038/ng1363. [DOI] [PubMed] [Google Scholar]
- 18.Musio A., Selicorni A., Focarelli M.L., Gervasini C., Milani D., Russo S., Vezzoni P., Larizza L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet. 2006;38:528–530. doi: 10.1038/ng1779. [DOI] [PubMed] [Google Scholar]
- 19.Deardorff M.A., Kaur M., Yaeger D., Rampuria A., Korolev S., Pie J., Gil-Rodriguez C., Arnedo M., Loeys B., Kline A.D. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. 2007;80:485–494. doi: 10.1086/511888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohatgi S., Clark D., Kline A.D., Jackson L.G., Pie J., Siu V., Ramos F.J., Krantz I.D., Deardorff M.A. Facial diagnosis of mild and variant CdLS: Insights from a dysmorphologist survey. Am. J. Med. Genet. A. 2010;152A:1641–1653. doi: 10.1002/ajmg.a.33441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schüle B., Oviedo A., Johnston K., Pai S., Francke U. Inactivating mutations in ESCO2 cause SC phocomelia and Roberts syndrome: no phenotype-genotype correlation. Am. J. Hum. Genet. 2005;77:1117–1128. doi: 10.1086/498695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vega H., Waisfisz Q., Gordillo M., Sakai N., Yanagihara I., Yamada M., van Gosliga D., Kayserili H., Xu C., Ozono K. Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat. Genet. 2005;37:468–470. doi: 10.1038/ng1548. [DOI] [PubMed] [Google Scholar]
- 23.McKay M.J., Troelstra C., van der Spek P., Kanaar R., Smit B., Hagemeijer A., Bootsma D., Hoeijmakers J.H. Sequence conservation of the rad21 Schizosaccharomyces pombe DNA double-strand break repair gene in human and mouse. Genomics. 1996;36:305–315. doi: 10.1006/geno.1996.0466. [DOI] [PubMed] [Google Scholar]
- 24.Shaikh T.H., Gai X., Perin J.C., Glessner J.T., Xie H., Murphy K., O'Hara R., Casalunovo T., Conlin L.K., D'Arcy M. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang K., Li M., Hadley D., Liu R., Glessner J., Grant S.F., Hakonarson H., Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peiffer D.A., Le J.M., Steemers F.J., Chang W., Jenniges T., Garcia F., Haden K., Li J., Shaw C.A., Belmont J. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–1148. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wittwer C.T., Reed G.H., Gundry C.N., Vandersteen J.G., Pryor R.J. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin. Chem. 2003;49:853–860. doi: 10.1373/49.6.853. [DOI] [PubMed] [Google Scholar]
- 28.Eddy S.R. Multiple alignment using hidden Markov models. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1995;3:114–120. [PubMed] [Google Scholar]
- 29.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krieger E., Joo K., Lee J., Raman S., Thompson J., Tyka M., Baker D., Karplus K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009;77(Suppl 9):114–122. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hooft R.W., Vriend G., Sander C., Abola E.E. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 32.Haering C.H., Schoffnegger D., Nishino T., Helmhart W., Nasmyth K., Lowe J. Structure and stability of cohesin's Smc1-kleisin interaction. Mol. Cell. 2004;15:951–964. doi: 10.1016/j.molcel.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 33.Krieger E., Darden T., Nabuurs S.B., Finkelstein A., Vriend G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins. 2004;57:678–683. doi: 10.1002/prot.20251. [DOI] [PubMed] [Google Scholar]
- 34.Krieger E., Nielsen J.E., Spronk C.A., Vriend G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006;25:481–486. doi: 10.1016/j.jmgm.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 35.Rupp R.A., Snider L., Weintraub H. Xenopus embryos regulate the nuclear localization of XMyoD. Genes Dev. 1994;8:1311–1323. doi: 10.1101/gad.8.11.1311. [DOI] [PubMed] [Google Scholar]
- 36.Turner D.L., Weintraub H. Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev. 1994;8:1434–1447. doi: 10.1101/gad.8.12.1434. [DOI] [PubMed] [Google Scholar]
- 37.Lovelock P.K., Wong E.M., Sprung C.N., Marsh A., Hobson K., French J.D., Southey M., Sculley T., Pandeya N., Brown M.A. Prediction of BRCA1 and BRCA2 mutation status using post-irradiation assays of lymphoblastoid cell lines is compromised by inter-cell-line phenotypic variability. Breast Cancer Res. Treat. 2007;104:257–266. doi: 10.1007/s10549-006-9415-5. [DOI] [PubMed] [Google Scholar]
- 38.Fenech M. The in vitro micronucleus technique. Mutat. Res. 2000;455:81–95. doi: 10.1016/s0027-5107(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 39.Wojewódzka M., Buraczewska I., Kruszewski M. A modified neutral comet assay: elimination of lysis at high temperature and validation of the assay with anti-single-stranded DNA antibody. Mutat. Res. 2002;518:9–20. doi: 10.1016/s1383-5718(02)00070-0. [DOI] [PubMed] [Google Scholar]
- 40.Westerfield M. University of Oregon Press; Eugene, OR: 1995. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio) [Google Scholar]
- 41.Kalev-Zylinska M.L., Horsfield J.A., Flores M.V., Postlethwait J.H., Vitas M.R., Baas A.M., Crosier P.S., Crosier K.E. Runx1 is required for zebrafish blood and vessel development and expression of a human RUNX1-CBF2T1 transgene advances a model for studies of leukemogenesis. Development. 2002;129:2015–2030. doi: 10.1242/dev.129.8.2015. [DOI] [PubMed] [Google Scholar]
- 42.McBrien J., Crolla J.A., Huang S., Kelleher J., Gleeson J., Lynch S.A. Further case of microdeletion of 8q24 with phenotype overlapping Langer-Giedion without TRPS1 deletion. Am. J. Med. Genet. A. 2008;146A:1587–1592. doi: 10.1002/ajmg.a.32347. [DOI] [PubMed] [Google Scholar]
- 43.Wuyts W., Roland D., Ludecke H.J., Wauters J., Foulon M., Van Hul W., Van Maldergem L. Multiple exostoses, mental retardation, hypertrichosis, and brain abnormalities in a boy with a de novo 8q24 submicroscopic interstitial deletion. Am. J. Med. Genet. 2002;113:326–332. doi: 10.1002/ajmg.10845. [DOI] [PubMed] [Google Scholar]
- 44.Deardorff M.A., Clark D.M., Krantz I.D. Cornelia de Lange Syndrome. In: Pagon R.A., Bird T.D., Dolan C.R., editors. Gene Reviews. University of Washington; Seattle, WA: 2010. [Google Scholar]
- 45.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sunyaev S., Ramensky V., Koch I., Lathe W., 3rd, Kondrashov A.S., Bork P. Prediction of deleterious human alleles. Hum. Mol. Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 47.Losada A., Yokochi T., Kobayashi R., Hirano T. Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J. Cell Biol. 2000;150:405–416. doi: 10.1083/jcb.150.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Canudas S., Smith S. Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. J. Cell Biol. 2009;187:165–173. doi: 10.1083/jcb.200903096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Birkenbihl R.P., Subramani S. Cloning and characterization of rad21 an essential gene of Schizosaccharomyces pombe involved in DNA double-strand-break repair. Nucleic Acids Res. 1992;20:6605–6611. doi: 10.1093/nar/20.24.6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sjögren C., Nasmyth K. Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr. Biol. 2001;11:991–995. doi: 10.1016/s0960-9822(01)00271-8. [DOI] [PubMed] [Google Scholar]
- 51.Sonoda E., Matsusaka T., Morrison C., Vagnarelli P., Hoshi O., Ushiki T., Nojima K., Fukagawa T., Waizenegger I.C., Peters J.M. Scc1/Rad21/Mcd1 is required for sister chromatid cohesion and kinetochore function in vertebrate cells. Dev. Cell. 2001;1:759–770. doi: 10.1016/s1534-5807(01)00088-0. [DOI] [PubMed] [Google Scholar]
- 52.Sprung C.N., Chao M., Leong T., McKay M.J. Chromosomal radiosensitivity in two cell lineages derived from clinically radiosensitive cancer patients. Clin. Cancer Res. 2005;11:6352–6358. doi: 10.1158/1078-0432.CCR-04-1931. [DOI] [PubMed] [Google Scholar]
- 53.Thomas P., Umegaki K., Fenech M. Nucleoplasmic bridges are a sensitive measure of chromosome rearrangement in the cytokinesis-block micronucleus assay. Mutagenesis. 2003;18:187–194. doi: 10.1093/mutage/18.2.187. [DOI] [PubMed] [Google Scholar]
- 54.Dhawan A., Bajpayee M., Parmar D. Comet assay: a reliable tool for the assessment of DNA damage in different models. Cell Biol. Toxicol. 2009;25:5–32. doi: 10.1007/s10565-008-9072-z. [DOI] [PubMed] [Google Scholar]
- 55.Dorsett D. Cohesin: Genomic insights into controlling gene transcription and development. Curr. Opin. Genet. Dev. 2011;21:199–206. doi: 10.1016/j.gde.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kagey M.H., Newman J.J., Bilodeau S., Zhan Y., Orlando D.A., van Berkum N.L., Ebmeier C.C., Goossens J., Rahl P.B., Levine S.S. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawauchi S., Calof A.L., Santos R., Lopez-Burks M.E., Young C.M., Hoang M.P., Chua A., Lao T., Lechner M.S., Daniel J.A. Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/−) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet. 2009;5:e1000650. doi: 10.1371/journal.pgen.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lüdecke H.J., Johnson C., Wagner M.J., Wells D.E., Turleau C., Tommerup N., Latos-Bielenska A., Sandig K.R., Meinecke P., Zabel B. Molecular definition of the shortest region of deletion overlap in the Langer-Giedion syndrome. Am. J. Hum. Genet. 1991;49:1197–1206. [PMC free article] [PubMed] [Google Scholar]
- 59.Momeni P., Glockner G., Schmidt O., von Holtum D., Albrecht B., Gillessen-Kaesbach G., Hennekam R., Meinecke P., Zabel B., Rosenthal A. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat. Genet. 2000;24:71–74. doi: 10.1038/71717. [DOI] [PubMed] [Google Scholar]
- 60.Waizenegger I.C., Hauf S., Meinke A., Peters J.M. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell. 2000;103:399–410. doi: 10.1016/s0092-8674(00)00132-x. [DOI] [PubMed] [Google Scholar]
- 61.Vass S., Cotterill S., Valdeolmillos A.M., Barbero J.L., Lin E., Warren W.D., Heck M.M. Depletion of Drad21/Scc1 in Drosophila cells leads to instability of the cohesin complex and disruption of mitotic progression. Curr. Biol. 2003;13:208–218. doi: 10.1016/s0960-9822(03)00047-2. [DOI] [PubMed] [Google Scholar]
- 62.Hauf S., Roitinger E., Koch B., Dittrich C.M., Mechtler K., Peters J.M. Dissociation of cohesin from chromosome arms and loss of arm cohesion during early mitosis depends on phosphorylation of SA2. PLoS Biol. 2005;3:e69. doi: 10.1371/journal.pbio.0030069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bauerschmidt C., Arrichiello C., Burdak-Rothkamm S., Woodcock M., Hill M.A., Stevens D.L., Rothkamm K. Cohesin promotes the repair of ionizing radiation-induced DNA double-strand breaks in replicated chromatin. Nucleic Acids Res. 2010;38:477–487. doi: 10.1093/nar/gkp976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim H.S., Baek K.H., Ha G.H., Lee J.C., Kim Y.N., Lee J., Park H.Y., Lee N.R., Lee H., Cho Y. The hsSsu72 phosphatase is a cohesin-binding protein that regulates the resolution of sister chromatid arm cohesion. EMBO J. 2010;29:3544–3557. doi: 10.1038/emboj.2010.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Revenkova E., Focarelli M.L., Susani L., Paulis M., Bassi M.T., Mannini L., Frattini A., Delia D., Krantz I., Vezzoni P. Cornelia de Lange syndrome mutations in SMC1A or SMC3 affect binding to DNA. Hum. Mol. Genet. 2009;18:418–427. doi: 10.1093/hmg/ddn369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Severin D.M., Leong T., Cassidy B., Elsaleh H., Peters L., Venter D., Southey M., McKay M. Novel DNA sequence variants in the hHR21 DNA repair gene in radiosensitive cancer patients. Int. J. Radiat. Oncol. Biol. Phys. 2001;50:1323–1331. doi: 10.1016/s0360-3016(01)01608-x. [DOI] [PubMed] [Google Scholar]
- 67.Mannini L., Menga S., Musio A. The expanding universe of cohesin functions: A new genome stability caretaker involved in human disease and cancer. Hum. Mutat. 2010;31:623–630. doi: 10.1002/humu.21252. [DOI] [PubMed] [Google Scholar]
- 68.Xu H., Tomaszewski J.M., McKay M.J. Can corruption of chromosome cohesion create a conduit to cancer? Nat. Rev. Cancer. 2011;11:199–210. doi: 10.1038/nrc3018. [DOI] [PubMed] [Google Scholar]
- 69.Rhodes J.M., McEwan M., Horsfield J.A. Gene regulation by cohesin in cancer: is the ring an unexpected party to proliferation? Mol. Cancer Res. 2011;9:1587–1607. doi: 10.1158/1541-7786.MCR-11-0382. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.