

Abstract
We performed hypothesis-free linkage analysis and exome sequencing in a family with two siblings who had neuronal ceroid lipofuscinosis (NCL). Two linkage peaks with maximum LOD scores of 3.07 and 2.97 were found on chromosomes 7 and 17, respectively. Unexpectedly, we found these siblings to be homozygous for a c.813_816del (p.Thr272Serfs∗10) mutation in the progranulin gene (GRN, granulin precursor) in the latter peak. Heterozygous mutations in GRN are a major cause of frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP), the second most common early-onset dementia. Reexamination of progranulin-deficient mice revealed rectilinear profiles typical of NCL. The age-at-onset and neuropathology of FTLD-TDP and NCL are markedly different. Our findings reveal an unanticipated link between a rare and a common neurological disorder and illustrate pleiotropic effects of a mutation in the heterozygous or homozygous states.
Main Text
The neuronal ceroid lipofuscinoses (NCLs) are neurodegenerative diseases characterized by storage of abnormal lipopigment in lysosomes. The more common childhood-onset forms, which are usually associated with visual failure, are due to recessive mutations in a group of genes believed to be involved in lysosomal processing.1 Adult-onset cases are rarer and can present with or without retinopathy; the genetic causes are now being unraveled.2,3
We employed genetic linkage analysis and massively parallel sequencing to identify the genetic cause of NCL with retinopathy in a family with apparent recessive inheritance for whom screening of likely known genes (PPT1 [MIM 600722], CLN3 [MIM 607042], and CLN6 [MIM 606725]) was unrewarding. Two siblings with apparently unrelated, healthy parents were affected (Figure 1A). The 28-year-old proband presented with rapidly progressive visual failure at 22 years, followed by major convulsions at 25 years, and myoclonic seizures at 26 years. Clinical examination showed mild cerebellar ataxia, early cognitive deterioration, and retinal dystrophy (Figure 1B). Electroencephalogram (EEG) results showed generalized polyspike wave discharges, electroretinogram results showed severe attenuation of both rod and cone responses, and MRI results showed cerebellar atrophy. Electron microscopic examination of a skin biopsy demonstrated numerous fingerprint profiles in membrane-bound structures in eccrine-secretory cells and in endothelium (Figure 1C). The proband's 26-year-old sister began having recurrent convulsions at 23 years, sometimes preceded by visual distortions. Her vision was initially normal but deteriorated when she was 25 years old. Examination revealed cerebellar ataxia and retinal dystrophy. EEG results showed polyspike wave discharges with a posterior emphasis, and MRI results revealed cerebellar atrophy.
Figure 1.
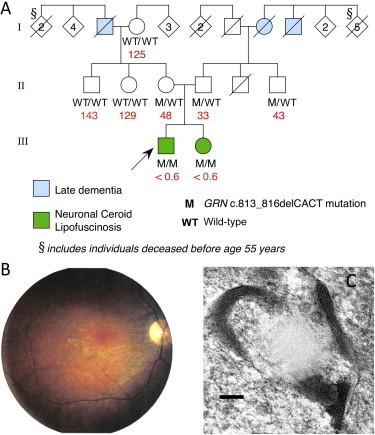
Family Pedigree and Clinical Features of NCL in Proband
(A) Pedigree of family (arrow indicates proband) indicating mutation status for GRN (c.813_816del). Plasma progranulin values are shown in red (ng/ml; median normal value is 126 ng/ml).
(B) Retinal photograph of proband showing optic atrophy, arteriolar attenuation, and irregular retinal pigmentation.
(C) Electron micrograph of skin biopsy from the proband. Typical fingerprint profiles within a membrane-bound structure in an eccrine pale cell are shown. The bar indicates 100 nm.
Clinical studies were approved by the Human Research Ethics Committee of Austin Health, Melbourne, Australia, and written informed consent was obtained from participating family members. DNA isolated from blood samples of the two affected siblings and their parents was genotyped through the use of Illumina Infinium HumanHap610W-Quad BeadChip genotyping arrays at the Australian Genome Research Facility (Melbourne). We analyzed genotypes for a subset of 11,572 SNP markers that have high heterozygosity and are in approximate linkage equilibrium (one SNP was chosen per 0.3 cM). Marker selection was performed by the Perl script linkdatagen.pl.4
Although the parents were not known to be related, they came from nearby small villages in Lombardy, Italy. To determine whether consanguinity was present, we estimated the inbreeding coefficients (F) of the affected siblings using FEstim,5 obtaining F = 0.000 for the affected son and F = 0.006 for the affected daughter. This suggests that the parents are distantly related (approximately second cousins once removed; expected F = 0.0078).
To ascertain the chromosomal locus, we performed an initial multipoint parametric linkage analysis that assumed no relationship between the two parents using MERLIN.6 We specified a fully penetrant recessive genetic model, a disease allele frequency of 0.0001, and allele frequencies from the Centre d'Etude du Polymorphisme Humain (CEPH; Utah residents with ancestry from northern and western Europe) HapMap population. FEstim was then used to adjust the LOD scores produced by MERLIN to account for the estimated inbreeding.7
This analysis revealed two linkage peaks located on chromosomes 7 and 17 (Figure 2). These peaks do not overlap any of the following genes previously implicated in NCL: PPT1, TPP1, CLN3, CLN5, CLN6, MFSD8, CLN8, CTSD, or DNAJC5. The linkage peak on chromosome 7 achieved a maximum adjusted LOD score of 3.07 and was located between rs1981701 and rs318572 (50.54 to 54.14 cM via deCODE), spanning 3.19 Mb and encompassing 25 genes. The linkage peak on chromosome 17 achieved a maximum adjusted LOD score of 2.97 and was located between rs2019231 and rs3809854 (70.07 to 73.75 cM); it spanned 5.42 Mb and encompassed 186 genes.
Figure 2.
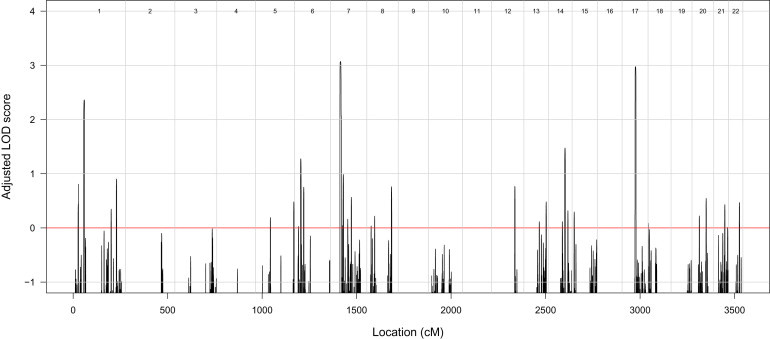
Results of Linkage Analysis
Autosomal genome-wide LOD scores, adjusted for inbreeding, for the family under a fully penetrant recessive genetic model.
To efficiently identify a potential causative mutation, we sequenced the exomes of the two parents and their affected son. Exome capture and sequencing was performed by Axeq Technologies (Rockville, MD, USA). DNA samples were enriched for approximately 62 Mb of the genome identified as being “exomic” with the use of Illumina TruSeq capture and sequenced using an Illumina HiSeq 2000 machine, generating 110 bp paired-end reads.
Reads were aligned to the hg19 reference genome with the use of Novoalign, with quality score calibration performed. Multimapping reads were removed; potential PCR duplicates were removed with Picard. Variants were called with the mpileup and bcftools view utilities from the SAMtools package8,9 and filtered with the vcfutils.pl script from the same package. Variants were annotated with ANNOVAR.10 We used HumVar-trained PolyPhen-2 (v2.1.0r367) to predict the biological impact of nonsynonymous variants.11
Table S1, available online, summarizes the number of reads mapped to each exome, and Table S2 summarizes the distribution of coverage for targeted bases.
We detected 325,946 variants from the reference sequence in at least one member of the trio. No rare (not reported or with frequencies of less than 1% in the 1000 Genomes May 2011 release12) exonic or splice-site variants were detected in PPT1, TPP1, CLN3, CLN5, CLN6, MFSD8, CLN8, CTSD, or DNAJC5. A total of 1,688 variants (0.52%) were located within the two linkage regions, of which 352 were rare and 20 were predicted to affect exonic regions or splice sites. All of these 20 variants were located within the linkage peak on chromosome 17. We discarded three variants that resulted in synonymous changes not predicted to affect splicing. Of the remaining 17 variants, three had inferred genotypes following the pattern expected under the consanguineous recessive disease model (i.e., the affected son was homozygous and unaffected parents were heterozygous; missing genotypes were permitted).
The three candidate disease-causing variants are described in Table S3. All three variants were present in dbSNP 132. Two variants (in DBF4B [MIM 611661] and ADAM11 [MIM 155120]) were present in the 1000 Genomes data set, both with an alternate allele frequency of 0.006. The third variant was not present in the 1000 Genomes data set, but was a known pathogenic mutation: a 4 bp deletion in GRN (MIM 138945; NM_002087.2), which encodes progranulin (also known as the granulin precursor). The c.813_816del mutation (rs63749877) results in a frameshift and premature termination of translation 10 residues downstream (p.Thr272Serfs∗10). Sanger sequencing confirmed that the variant was genuine and segregated as expected in the family, with the mother and father heterozygous for the variant and both affected children homozygous.
Heterozygous c.813_816del GRN mutations are known to cause frontotemporal lobar degeneration13–16 (FTLD-TDP; MIM 607485). This presents in later life, so we reevaluated the family for features of frontal lobe dementia. There was a history of late dementia (approximately seventh decade) in the maternal grandfather, paternal grandmother, and great uncle; as all three individuals are deceased, pathological diagnoses could not be made, and DNA could not be obtained. A number of older relatives had died before 55 years of age, so a more obvious pattern of autosomal-dominant dementia may have been masked. The parents, both heterozygous for the mutation, were in their fifties and healthy. DNA from all available family members was screened. The results (Figure 1) indicate that the mother must have inherited the c.813_816del mutation from her father, who had dementia, and that an additional family member in the parents' generation is a c.813_816del heterozygote. Plasma progranulin was measured in available family members with a commercial kit (Human Progranulin ELISA Kit, AdipoGen, Seoul, South Korea).17 Plasma progranulin correlated with the genotypes; values were 125–143 ng/ml in three individuals with no copies of the deletion (median normal value: 126 ng/ml17), 33–48 ng/ml in the three heterozygous subjects, which is below the cutoff value (62 ng/ml) for predicting heterozygous null mutations,17 and undetectable (<0.6 ng/ml) in the two homozygous affected siblings (Figure 1A).
Subsequently, we sequenced 20 unrelated cases of proven or suspected adult NCL wherein known NCL genes had been excluded. All 13 exons, exon-intron boundaries, and untranslated regions of GRN were PCR-amplified, sequenced with the BigDye Terminator v3.1 Cycle Sequencing protocol, and run on an ABI 3730XL DNA Analyzer (Life Technologies, Carlsbad, CA, USA). We did not find homozygous or compound-heterozygous mutations in any of these individuals, although one was found to be heterozygous for the most common GRN mutation, c.1477C>T (p.Arg493∗; rs63751294).18 This patient presented with limb dystonia, spasticity, gait disturbance, apraxia, and dementia at the age of 46. Death occurred around the age of 48–49. We excluded the possibility that this individual had a partial deletion of the second GRN allele by performing a multiplex ligation-dependent probe amplification assay. We were unable to reevaluate the original diagnosis of NCL as the original electron micrographs and tissue samples were unavailable. This diagnosis was made on the basis of lymphocyte inclusions, which may be unreliable in adult NCL; we suspect that the individual may have had FTLD-TDP misdiagnosed as adult NCL. We also sequenced the same regions of GRN for 188 anonymous blood-bank controls without detecting any known mutations or novel variants.
Ahmed et al. previously generated mice deficient in the orthologous Grn (Grn−/−) and reported accumulation of abnormal intraneuronal ubiquitin-positive autofluorescent lipofuscin detected by light microscopy.19 This finding has since been replicated with the use of independently generated Grn-deficient mice.20 Electron microscopic examination of fixed brain tissue from the Ahmed et al. mice revealed abundant rectilinear profiles diagnostic of NCL (Figure 3). Animal studies were performed in accordance with the Mayo Clinic Institutional Animal Care and Use Committee.
Figure 3.
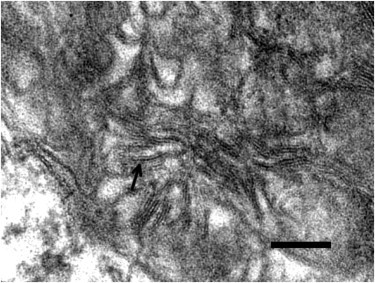
NCL Pathology in Progranulin-Deficient Mice
Electron micrograph showing a high-power view of one of the storage granules in a neuron from the habenula of a Grn−/− mouse. The pattern corresponds to the rectilinear complex, which, along with fingerprint profiles, is characteristic of most types of NCL. The arrow points to a pentalaminar profile. The bar indicates 100 nm.
We report that homozygous c.813_816del mutations in GRN cause adult-onset NCL and suggest that this locus be designated CLN11. This result is remarkable, given that mutations in this gene are an important cause of FTLD-TDP in the heterozygous state and that the two clinically distinct neurological disorders have very different pathologies.
Clinically, mutations in specific genes usually determine important phenotypes in either the heterozygous or homozygous state, but not both. In classical recessive disorders, heterozygous individuals are usually healthy, asymptomatic carriers, although there may be a clinically trivial or biochemical phenotype or sometimes even a survival advantage, such as for sickle cell trait (MIM 603903).21 In classical dominant disorders, the population allele frequency is low, so homozygous individuals are exceptional, except in inbred communities.22 When observed, such rare homozygous cases may differ little from heterozygous family members (e.g., in Huntington disease [MIM 143100] and epidermolysis bullosa simplex [MIM 131900]) or have a more severe form of the same phenotype, suggesting an additive effect on the gene product dysfunction (e.g., in Marfan syndrome [MIM 154700], achondroplasia [MIM 100800], and Charcot-Marie-Tooth disease [MIM 118200]).22
One example of different diseases with homozygous versus heterozygous mutations involves the gene that encodes acid beta-glucosidase (GBA [MIM 606463]). Homozygous GBA mutations have been long known to cause the various forms of Gaucher disease (MIMs 608013, 230800, 230900, 231000, and 231005), where there is storage of glucocerebroside. More recently, various heterozygous GBA mutations have been shown to increase the risk of late-onset Parkinson disease (MIM 168600), not as a Mendelian trait but rather as a susceptibility factor.23,24
Although the present result was unexpected and we did not identify additional cases, the finding of similar and diagnostic electron-microscopic features of NCL in the human material and in the progranulin-deficient mouse supports our conclusion of strikingly different phenotypes for this GRN mutation depending on whether it is homozygous or heterozygous. The sibling pair had early-adult onset of severe retinal disease with seizures, ataxia, and cognitive change. Although brain biopsies were not available from the siblings, the mouse brain pathology mirrored the known human pathology of NCL in its storage of abnormal autofluorescent lipopigment with characteristic ultrastructural features in neurons.25,26 This contrasts with FTLD-TDP due to heterozygous GRN mutation; the disorder presents in later life with behavioral and cognitive impairment, but seizures are unusual, and ataxia and retinal involvement are absent. The pathology does not reveal lipopigment storage material but rather neurons with TDP-43-immunoreactive neuronal and glial inclusions.27,28
To the best of our knowledge, homozygous mutations have not previously been reported in GRN. Indeed, Bruni et al.29 found no homozygotes in a highly consanguineous Calabrian family in which 19 individuals were found to be heterozygous for a c.1144dup (p.Thr382Asnfs∗32) mutation in GRN. The authors speculated that the loss of both GRN alleles might cause embryonic lethality,29 but our data showing homozygotes with undetectable progranulin levels and the viability of the Grn−/− mouse show that progranulin deficiency is compatible with life.
Progranulin has multiple physiological roles, and it influences inflammation, early embryogenesis, tumorigenesis, and adult tissue repair.30 How haploinsufficiency causes FTLD-TDP remains uncertain. In Caenorhabditis elegans, progranulin deficiency causes more rapid clearance of apoptotic cells and inadvertently results in the clearance of neurons that would otherwise recover from initiation of apoptosis.31 This observation, however, does not explain how progranulin deficiency would result in NCL. Given the link between NCL and proteins that affect lysosomal processing, the association between GRN mutation and NCL suggests that progranulin has a lysosomal function. There is good evidence that progranulin is neurotrophic or neuroprotective,30 potentially through the modulation of survival signaling. Disruption of cell signaling processes could conceivably result in rapid “aging” of unprotected progranulin-deficient cells with respect to lipofuscinosis. Indeed, homozygous GRN deficiency in mice results in microglial activation, increased ubiquitination, and excess lipopigment deposition.19
The history of dementia in this family was not regarded as remarkable when first assessed. Early death in many older family members masked the likely autosomal-dominant pattern on both sides of the family (Figure 1A). It has been estimated that 50% of c.813_816del heterozygotes show symptoms by the age of 63 and that 97.3% are symptomatic by the age of 73.14 Thus, the discovery of homozygous mutation in children presents an unexpected ethical dilemma in terms of new knowledge regarding the likelihood of late-life dementia in the parents and other healthy heterozygotes.
With the recent widespread implementation of massively parallel sequencing to uncover causes of hitherto obscure early-onset recessive phenotypes,32 even in single families as here, further examples of pleiotropic effects of homozygous and heterozygous mutations may emerge. This will create opportunities for better pathophysiological understanding, but may also result in unexpected ethical quandaries upon the emergence of age-dependent heterozygous phenotypes.
Acknowledgments
K.R.S. is supported by a PhD scholarship funded by the Pratt Foundation. S.F.B. is supported by a National Health and Medical Research Council (NHMRC) Australia Fellowship and an NHMRC Program Grant. M.B. is supported by an Australian Research Council (ARC) Future Fellowship and an NHMRC Program Grant. S.E.M. is supported by the Medical Research Council. The Batten Disease Support and Research Association supported S.E.M., J.S., and K.S. We thank Pawan Mann and Olivia Galante for technical assistance; Danya Vears, Robyn Ferguson, and Karen Oliver for technical support; the Rare NCL Gene Consortium for encouraging collaborative research on variant NCL cases; and the families and physicians for providing samples.
Contributor Information
Melanie Bahlo, Email: bahlo@wehi.edu.au.
Samuel F. Berkovic, Email: s.berkovic@unimelb.edu.au.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Novoalign, www.novocraft.com
Picard, http://picard.sourceforge.net/
References
- 1.Mole, S.E., and Williams, R.E. (2010). Neuronal Ceroid-Lipofuscinoses. In GeneReviews, R.A. Pagon, T.D. Bird, and C.R. Dolan, et al., eds. (Seattle, WA: University of Washington), http://www.ncbi.nlm.nih.gov/books/NBK1428/.
- 2.Arsov T., Smith K.R., Damiano J., Franceschetti S., Canafoglia L., Bromhead C.J., Andermann E., Vears D.F., Cossette P., Rajagopalan S. Kufs disease, the major adult form of neuronal ceroid lipofuscinosis, caused by mutations in CLN6. Am. J. Hum. Genet. 2011;88:566–573. doi: 10.1016/j.ajhg.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nosková L., Stránecký V., Hartmannová H., Přistoupilová A., Barešová V., Ivánek R., Hůlková H., Jahnová H., van der Zee J., Staropoli J.F. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet. 2011;89:241–252. doi: 10.1016/j.ajhg.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bahlo M., Bromhead C.J. Generating linkage mapping files from Affymetrix SNP chip data. Bioinformatics. 2009;25:1961–1962. doi: 10.1093/bioinformatics/btp313. [DOI] [PubMed] [Google Scholar]
- 5.Leutenegger A.L., Prum B., Génin E., Verny C., Lemainque A., Clerget-Darpoux F., Thompson E.A. Estimation of the inbreeding coefficient through use of genomic data. Am. J. Hum. Genet. 2003;73:516–523. doi: 10.1086/378207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 7.Leutenegger A.L., Labalme A., Genin E., Toutain A., Steichen E., Clerget-Darpoux F., Edery P. Using genomic inbreeding coefficient estimates for homozygosity mapping of rare recessive traits: application to Taybi-Linder syndrome. Am. J. Hum. Genet. 2006;79:62–66. doi: 10.1086/504640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durbin R.M., Abecasis G.R., Altshuler D.L., Auton A., Brooks L.D., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benussi L., Binetti G., Sina E., Gigola L., Bettecken T., Meitinger T., Ghidoni R. A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol. Aging. 2008;29:427–435. doi: 10.1016/j.neurobiolaging.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 14.Benussi L., Ghidoni R., Pegoiani E., Moretti D.V., Zanetti O., Binetti G. Progranulin Leu271LeufsX10 is one of the most common FTLD and CBS associated mutations worldwide. Neurobiol. Dis. 2009;33:379–385. doi: 10.1016/j.nbd.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 15.Le Ber I., Camuzat A., Hannequin D., Pasquier F., Guedj E., Rovelet-Lecrux A., Hahn-Barma V., van der Zee J., Clot F., Bakchine S., French research network on FTD/FTD-MND Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain. 2008;131:732–746. doi: 10.1093/brain/awn012. [DOI] [PubMed] [Google Scholar]
- 16.Yu C.-E., Bird T.D., Bekris L.M., Montine T.J., Leverenz J.B., Steinbart E., Galloway N.M., Feldman H., Woltjer R., Miller C.A. The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch. Neurol. 2010;67:161–170. doi: 10.1001/archneurol.2009.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghidoni R., Stoppani E., Rossi G., Piccoli E., Albertini V., Paterlini A., Glionna M., Pegoiani E., Agnati L.F., Fenoglio C. Optimal plasma progranulin cutoff value for predicting null progranulin mutations in neurodegenerative diseases: a multicenter Italian study. Neurodegener. Dis. 2012;9:121–127. doi: 10.1159/000333132. [DOI] [PubMed] [Google Scholar]
- 18.Rademakers R., Baker M., Gass J., Adamson J., Huey E.D., Momeni P., Spina S., Coppola G., Karydas A.M., Stewart H. Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C—>T (Arg493X) mutation: an international initiative. Lancet Neurol. 2007;6:857–868. doi: 10.1016/S1474-4422(07)70221-1. [DOI] [PubMed] [Google Scholar]
- 19.Ahmed Z., Sheng H., Xu Y.F., Lin W.-L., Innes A.E., Gass J., Yu X., Wuertzer C.A., Hou H., Chiba S. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 2010;177:311–324. doi: 10.2353/ajpath.2010.090915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petkau T.L., Neal S.J., Milnerwood A., Mew A., Hill A.M., Orban P., Gregg J., Lu G., Feldman H.H., Mackenzie I.R.A. Synaptic dysfunction in progranulin-deficient mice. Neurobiol. Dis. 2012;45:711–722. doi: 10.1016/j.nbd.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 21.Pasvol G., Weatherall D.J., Wilson R.J.M. Cellular mechanism for the protective effect of haemoglobin S against P. falciparum malaria. Nature. 1978;274:701–703. doi: 10.1038/274701a0. [DOI] [PubMed] [Google Scholar]
- 22.Zlotogora J. Dominance and homozygosity. Am. J. Med. Genet. 1997;68:412–416. doi: 10.1002/(sici)1096-8628(19970211)68:4<412::aid-ajmg8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 23.Goker-Alpan O., Giasson B.I., Eblan M.J., Nguyen J., Hurtig H.I., Lee V.M.-Y., Trojanowski J.Q., Sidransky E. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology. 2006;67:908–910. doi: 10.1212/01.wnl.0000230215.41296.18. [DOI] [PubMed] [Google Scholar]
- 24.Goker-Alpan O., Schiffmann R., LaMarca M.E., Nussbaum R.L., McInerney-Leo A., Sidransky E. Parkinsonism among Gaucher disease carriers. J. Med. Genet. 2004;41:937–940. doi: 10.1136/jmg.2004.024455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson G., Elleder M., Goebel H.H. Morphological diagnostic and pathological considerations. In: Mole S.E., Williams R.E., Goebel H.H., editors. The neuronal ceroid lipofuscinoses (Batten disease) Oxford University Press; Oxford, UK: 2011. pp. 35–49. [Google Scholar]
- 26.Carpenter S., Karpati G., Andermann F., Jacob J.C., Andermann E. The ultrastructural characteristics of the abnormal cytosomes in Batten-Kufs' disease. Brain. 1977;100:137–156. doi: 10.1093/brain/100.1.137. [DOI] [PubMed] [Google Scholar]
- 27.Josephs K.A., Ahmed Z., Katsuse O., Parisi J.F., Boeve B.F., Knopman D.S., Petersen R.C., Davies P., Duara R., Graff-Radford N.R. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J. Neuropathol. Exp. Neurol. 2007;66:142–151. doi: 10.1097/nen.0b013e31803020cf. [DOI] [PubMed] [Google Scholar]
- 28.Mackenzie I.R.A., Baker M., Pickering-Brown S., Hsiung G.-Y.R., Lindholm C., Dwosh E., Gass J., Cannon A., Rademakers R., Hutton M., Feldman H.H. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006;129:3081–3090. doi: 10.1093/brain/awl271. [DOI] [PubMed] [Google Scholar]
- 29.Bruni A.C., Momeni P., Bernardi L., Tomaino C., Frangipane F., Elder J., Kawarai T., Sato C., Pradella S., Wakutani Y. Heterogeneity within a large kindred with frontotemporal dementia: a novel progranulin mutation. Neurology. 2007;69:140–147. doi: 10.1212/01.wnl.0000265220.64396.b4. [DOI] [PubMed] [Google Scholar]
- 30.Bateman A., Bennett H.P.J. The granulin gene family: from cancer to dementia. Bioessays. 2009;31:1245–1254. doi: 10.1002/bies.200900086. [DOI] [PubMed] [Google Scholar]
- 31.Kao A.W., Eisenhut R.J., Martens L.H., Nakamura A., Huang A., Bagley J.A., Zhou P., de Luis A., Neukomm L.J., Cabello J. A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc. Natl. Acad. Sci. USA. 2011;108:4441–4446. doi: 10.1073/pnas.1100650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Najmabadi H., Hu H., Garshasbi M., Zemojtel T., Abedini S.S., Chen W., Hosseini M., Behjati F., Haas S., Jamali P. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.