

Abstract
Most autosomal genetic causes of childhood-onset hypogammaglobulinemia are currently not well understood. Most affected individuals are simplex cases, but both autosomal-dominant and autosomal-recessive inheritance have been described. We performed genetic linkage analysis in consanguineous families affected by hypogammaglobulinemia. Four consanguineous families with childhood-onset humoral immune deficiency and features of autoimmunity shared genotype evidence for a linkage interval on chromosome 4q. Sequencing of positional candidate genes revealed that in each family, affected individuals had a distinct homozygous mutation in LRBA (lipopolysaccharide responsive beige-like anchor protein). All LRBA mutations segregated with the disease because homozygous individuals showed hypogammaglobulinemia and autoimmunity, whereas heterozygous individuals were healthy. These mutations were absent in healthy controls. Individuals with homozygous LRBA mutations had no LRBA, had disturbed B cell development, defective in vitro B cell activation, plasmablast formation, and immunoglobulin secretion, and had low proliferative responses. We conclude that mutations in LRBA cause an immune deficiency characterized by defects in B cell activation and autophagy and by susceptibility to apoptosis, all of which are associated with a clinical phenotype of hypogammaglobulinemia and autoimmunity.
Introduction
In 86% of cases, childhood-onset agammaglobulinemia is an X-linked condition (XLA, [MIM 300755]) affecting male offspring.1 XLA is caused by mutations in BTK (MIM 300300), which encodes a signaling molecule downstream of the B cell antigen receptor,2 and is characterized by the lack of peripheral B cells (<1%).1 The remainder of agammaglobulinemias are rare autosomal-recessive (AR) traits; to date, six genes with mutations causing agammaglobulinemia have been described.3,4
In contrast to agammaglobulinemias, childhood-onset hypogammaglobulinemias are characterized by the presence of B cells in the periphery and by some residual immunoglobulin production. They might be transient or persistent and primary (inborn) or secondary as a result of, e.g., nephrosis, enteric protein loss, medication (immunosuppressive or antiepileptic drugs), or connatal infection, such as HIV or measles. In addition, primary T cell deficiencies combined with either the lack of peripheral B cells or a functional defect of persisting B cells (such as T-B+ severe combined immune deficiency or immunodeficiency, centromeric instability, and facial anomalies [ICF] syndrome [MIM 601457 and 242860, respectively]) might also include childhood-onset hypogammaglobulinemia as part of the phenotype.5,6
In adults, primary persistent hypogammaglobulinemia is either diagnosed as being caused by class-switch-recombination defects leading to various forms of hyper-IgM syndromes7 (MIM 308230, 605258, 606843, 608106, and 608184) or diagnosed as common variable immune deficiency (CVID), which is a diagnosis of exclusion. Hence, it is not surprising that CVID has a heterogeneous clinical and laboratory presentation.8 Affected individuals present with low IgG and IgA levels, whereas IgM levels can be low or normal.9 Most suffer from recurrent bacterial infections.10 Major complications include autoimmune, lymphoproliferative, and granulomatous diseases, which are each seen in 20%–30% of CVID-affected individuals.11
Fewer than 20% of adult individuals with a primary hypogammaglobulinemia have an affected relative.9 Reported families have either autosomal dominant (AD) or AR inheritance.12 Previously identified genetic defects associated with AR hypogammaglobulinemia are biallelic mutations in genes involved in B cell stimulation (such genes are CD19,13 MS4A1,14 CD81,15 CR2,16 and TNFRSF13C17 [MIM 613493, 613495, 613496, 610927, and 613494, respectively]) and a gene involved in T cell costimulation (ICOS [MIM 607594]).18 Interestingly, these individuals do not often have autoimmune phenomena, which are otherwise frequently seen in individuals with CVID. Polymorphisms in TNFRSF13B (encoding TACI, [MIM 240500]) and MSH5 (MIM 603382) have been associated with AD and simplex cases of CVID.19–22 For TNFRSF13B, most individuals with biallelic mutations develop CVID, whereas those with single-allele mutations are at a substantially increased risk of developing CVID and autoimmune phenomena.23 Large families with suggested dominant inheritance of CVID have been published. Genetic linkage studies in such families have found evidence of causative mutations on chromosome arms 4q24 and 16q,25 but disease-associated genes have not been identified.
In this study, we used a positional approach by seeking causative mutations that are found in individuals with severe, early-onset humoral immune deficiency and autoimmunity. By genome-wide SNP typing, genetic linkage analysis, and DNA sequencing of 16 families with putative recessive inheritance, we were able to find individuals with four distinct homozygous mutations in the gene encoding LRBA (lipopolysaccharide responsive beige-like anchor protein [MIM 606453]) in chromosomal region 4q31. Because LRBA expression has been found to be upregulated in cancer cells, it has been suggested that the protein acts as a positive regulator of cell survival by promoting proliferation and by preventing apoptosis.26 Here, we show that individuals with homozygous LRBA mutations have severe defects in B cell development and activation and in autophagy. B cell lines from these individuals show an increased susceptibility to apoptosis. Accordingly, LRBA has unanticipated functions in B cells, which are essential for normal development and humoral immune responses.
Material and Methods
Affected Individuals and Controls
Our study focused on five affected individuals from four consanguineous families for which genetic linkage evidence is the strongest. Family A is of Arab origin and was followed at the Meyer's Children Hospital, Rappaport School of Medicine (Haifa, Israel). Family B is of Sicilian origin and was under the care of the University Hospital Center of Liège (Liège, Belgium). Families C and D are from Iran and were followed at the Children Medical Center (Tehran, Iran). The affected individuals in families A, B, and C are alive, whereas the affected individual in family D is deceased. All affected individuals were diagnosed with childhood-onset CVID.
In addition, we analyzed 100 European, 145 Arabic (17 from the village of family A and 128 from elsewhere), and 233 Iranian healthy control samples to exclude the possibility that the detected mutations represent rare benign polymorphisms. For functional experiments, control samples from unrelated, healthy individuals were used unless otherwise specified.
All study participants gave their informed consent as appropriate under approved protocols from local institutional review boards. The research was conducted at University College London, Karolinska Institutet Stockholm, and the Universities of Freiburg, Brescia, and Oxford under approved protocols (#04/Q0501/119 for affected individuals, #07/H0720/182 for family members, and #08/H0720/46 for healthy controls).
Genotyping
Families A and B and the two Italian families were genome-wide typed with the Affymetrix (Santa Clara, CA, USA) GeneChip Human Mapping 250K Nsp Array (GEO Platform GPL3718) according to previously described methods.27 Fine mapping with selected microsatellite markers on chromosomes 4, 5, and 12 was performed in family A according to published protocols.24 Samples from 12 simplex Iranian cases, including the affected individuals from families C and D, had been previously genotyped with the older Affymetrix Human Mapping 10K Array Xba 142 (GEO Platform GPL2641).
Genetic Linkage Analysis
To identify perfect or near perfect intervals in families A and B, we used the in-house software “findhomoz” described previously.27 We used a modified version of “findhomoz” to reanalyze the SNP chip data from the 12 Iranian simplex cases to identify regions for which multiple individuals shared the same homozygous haplotype.
A marker or interval of markers segregates perfectly with recessive inheritance under consanguinity if the affected individuals are homozygous with the same genotype or haplotype, respectively, and the unaffected individuals have different genotypes or haplotypes. For the family structures of A, B, C, and D, the segregation is near perfect if there is one exception among the children. An exception could be an affected individual who does not share the same homozygous genotype (a “phenocopy”) or an unaffected individual who shares the same homozygous genotype (“nonpenetrant”). We limited our search for near-perfect intervals to the nonpenetrant type of exception.
Mutation Detection
Genomic DNA and cDNA were obtained from peripheral-blood mononuclear cells (PBMCs) of affected individuals, unaffected family members, and controls. We sequenced either gDNA or cDNA for 20 different genes with immune function in the linkage regions from chromosomes 4, 5, and 12 (Table S1, available online) by using standard protocols. We sequenced LRBA cDNA with 16 different overlapping PCR products. We sequenced LRBA gDNA with 61 pairs of oligonucleotides to amplify 58 exons of the chronologically first transcript (we used RefSeq NM_006726.2; the current version is NM_006726.4) with the longer coding region. Gene sequencing was according to the exon-intron structure suggested by the Ensembl record for transcript ENST00000357115, which has 100% sequence identity with NM_006726.4 except for the length of the poly(A) tail.
Reference Sequences of LRBA
The two reference transcripts for LRBA are NM_001199282.2 and NM_006726.4. No version of the reference sequence NM_001199282 was available at the time we did the sequencing. NM_001199282.2 produces a shorter protein product than does NM_006726.4 by omitting an exon of NM_006726.4 and one serine near the C terminus. We suggest that the primary protein product probably does not contain that exon.
The exon included in NM_006726.4 but omitted in NM_001199282.2 can be unambiguously identified by the Ensembl identifier ENSE00001249966. It comprises bases 6,291–6,323 of the LRBA reference RNA NM_006726.4 and leads to in-frame predicted translation of amino acids 2,016–2,026 (VCIFKLRENSK) in NP_006717.2. It is exon 39 in the intron-exon structure suggested by the Ensembl record for transcript ENST00000357115, which we used to guide the sequencing. We therefore refer to ENSE00001249966 as exon 39, but it is exon 40 in the GenBank record for NM_006726.4.
The sequence NM_006726.4 is a composite of at least four GenBank submissions that are not entirely consistent with one another. The evidence given by the National Center for Biotechnology Information (NCBI) Evidence Viewer in favor of inclusion of exon 39 is the GenBank sequences M83822.1 and AF216648.2. The sequence M83822.1 was published as part of the initial gene discovery. The sequence AF216648.2 replaced the much shorter sequence AF216648.1, which does not contain exon 39. The methods used for the identification of the sequence AF216648.1 were published, wherein the submitters reference the GenBank record M83822.1 but do not claim to have directly sequenced the cDNA. The GenBank record AF216648.2 explicitly states that it was not published in a journal. The lack of published evidence for prior sequencing of exon 39 in normal gene expression and the observation that exon 39 is skipped in all healthy donors sequenced in this study suggest that the primary transcript or transcripts of LRBA do not contain exon 39. Using Entrez for the alignment of NP_006717.2 to NCBI's Conserved Domain Database shows that the insertion of these 11 amino acids causes a gap in the alignment to the canonical sequence representing DUF1088, a domain of unknown function found in several proteins that also contain BEACH domains.
Determination of the Boundaries of the Large Deletion in P5
Standard PCR amplification and sequencing of LRBA were performed. In brief, 50 ng of genomic DNA was used in PCR employing specific primers with the following conditions: 95°C (2 min) for one cycle, 95°C (15 s), alignment (30 s), and 72°C (1 min) for 35 cycles each, and a final extension of 72°C (10 min). A long PCR kit (Roche, Basel, Switzerland) was used for long-range PCR amplification with the following conditions: 92°C for one cycle, 92°C (15 s), 56°C (30 s), and 68°C (6 min) for 10 cycles each, 92°C (15 s), 56°C (30 s), and 68°C (6 min) for 20 cycles each (20 s was added for each successive cycle), and a final extension of 68°C (10 min). The PCR products were gel purified (with a gel extraction kit [QIAGEN, Stockholm, Sweden]) and sequenced at the Macrogen Company (Seoul, South Korea). The sequences were analyzed with Lasergene software (DNAStar, Madison, WI, USA).
Generation of Epstein-Barr-virus Cell Line and LRBA Immunoblot
PBMCs from affected individuals and healthy donors were cultured in RPMI 1640 supplemented with 2 mM L-glutamine (Lonza Biologics, Basel, Switzerland), 50 U/ml penicillin, and 50 μg/ml streptomycin (GIBCO, Carlsbad, CA, USA) at a density of 1 × 106 cells/ml in the presence of 10 μg/ml of phytohemaglutinin (PHA [Sigma-Aldrich, St. Louis, MO, USA]) and a 1:2 dilution of Epstein-Barr virus (EBV) supernatant obtained from the marmoset cell line B95-8. Cells were maintained in culture for 1 month so that they would immortalize. EBV cell lines were used for immunoprecipitation and immunoblots.
LRBA levels were determined by immunoblot according to standard procedures. In brief, 25 μg of protein extracts from EBV cell lines was loaded onto a gradient gel (9%–18% polyacrylamide) and transferred onto polyvinylidene difluoride (PVDF) membranes for 4 hr at 130V in 5% isopropanol transfer buffer. Anti-LRBA (Sigma-Aldrich, St. Louis, MO, USA), which recognizes the amino acids 906–1,038, was used for the detection of a band of approximately 319 kDa.
B Cell Immunophenotyping
PBMCs were isolated by centrifugation through a Ficoll step gradient and stained for cell-surface-marker expression with the use of fluorochrome-conjugated antibodies against CD19 (APC-H7), CD10 (FITC), CD27 (PE-Cy5.5), CD38 (PE-Cy7), BAFF-R (PE [all from Becton Dickinson, Franklin Lakes, NJ, USA]), IgM (Cy5), IgA (Alexa 647), and IgG (FITC [all from Jackson Immunoresearch, West Grove, PA, USA]). A total of 5 × 105 cells were acquired with a FACS-Canto II flow cytometer with FACS-Diva software (BD Biosciences) and analyzed with FlowJo (Treestar, Ashland, OR).
B Cell Activation, Immunoglobulin Secretion, Plasmablast Generation, and Proliferation Detection
PBMCs of affected individuals and healthy controls were isolated by density gradient with LymphoprepTM (Axis-Shield). Naive B cells were separated from PBMCs by negative depletion with the use of Naive Isolation Kit II (Miltenyi Biotec, Bergisch-Gladbach, Germany). For stimulation, 50,000 naive B cells were cultured in U96-well plates in 250 μl of RPMI 1640 supplemented with fetal calf serum (FCS), L-glutamine, penicillin, and streptomycin. Cells were stimulated with 2.5 μg/ml of anti-human IgM (MP Biomedicals), 1 μg/ml of soluble recombinant CD40L (Enzo Life Sciences), 2.5 μg/ml of CpG oligodeoxynucleotides 2006 (InvivoGen, San Diego, CA, USA), and 75 ng/ml of recombinant BAFF (Peprotech, Rocky Hill, NJ, USA). Cells were maintained in culture for 13 days, and culture supernatants were collected for the determination of IgG production by ELISA (Bethyl Laboratories, Montgomery TX, USA).
In a second approach, 1 × 105 PBMCs were cultivated in U-bottom 96-well plates in Iscove's modified Dulbecco's medium (Biowest, East Sussex, UK) supplemented with 10% FCS (Lonza Biologics, Basel, Switzerland), nonessential amino acids (Invitrogen), fatty-acid supplement (Invitrogen), insulin (Sigma-Aldrich), and transferrin. The cells were activated by the addition of a soluble adiponectin-CD40L fusion protein combined with IL21-Fc or IL4 as described previously.17 After 4 days, cells were transferred to new wells in fresh medium and cultivated for an additional 3 days. Supernatants were tested for IgA, IgM, and IgG levels by sandwich ELISA with the use of goat anti-human Ig as capture and alkaline phosphatase-conjugated isotype-specific detection antibodies (Jackson Immunoresearch, West Grove, USA). The phenotype of activated B cells was analyzed by flow cytometry; cells were stained as described previously17 with antibodies specific to CD19, CD38, CD138, BAFF-R, CD43, and IgD (all Becton Dickinson, Franklin Lakes, NJ, USA), CD10 (Beckman Coulter, Brea, CA, USA), IgA, IgG, and IgM, (Jackson Immunoresearch), and CD27 (Biolegend, San Diego, CA, USA).
For proliferation assays, PBMCs were resuspended at 1 × 106 cells/ml in supplemented RPMI, and 1.5 × 105 cells were then added in triplicates into 96-well plates and stimulated with 10 μg/ml of Pokeweed mitogen (PWM [Sigma-Aldrich]). Cultures were incubated at 37°C for 72 hr in a humidified atmosphere containing 5% CO2. Proliferation was measured by the addition of 1 mCi/well of 3H-thymidine (Perkin-Elmer, Waltham, MA) for the last 18 hr of incubation. Cells were harvested onto glass-fiber filters (Wallac Oy, Turku, Finland) and resuspended in Betaplate Scint cocktail (Perkin Elmer Life Sciences, Cambridge, UK), and thymidine incorporation was determined with a 1205 Betaplate liquid scintillation counter (Perkin Elmer). Proliferative responses are expressed as stimulation index (SI) = net counts per minute (cpm) of PWM-stimulated cultures/cpm of unstimulated cultures.
Apoptosis Assay
Apoptosis levels were determined in EBV cells cultured under serum deprivation for 6 hr (1 × 106 cells/ml). After starvation, cells were harvested, and apoptosis was measured by Annexin V and propidium iodide (PI) staining (eBioscience, San Diego CA, USA). Immediately after staining, 10,000 events were acquired in an LSR II flow cytometer and were analyzed for the determination of the percentage of cells in early apoptosis (positive for Annexin V and negative for PI).
Transmission Electron Microscopy
Cells were fixed in 2.5% glutaraldehyde (Ladd, Burlington, Vermont, USA) in 0.1 M phosphate buffer (pH 7.4) at room temperature for 30 min. The cells were then transferred to a microcentrifuge tube and further fixed overnight in the refrigerator. After fixation, cells were rinsed in 0.1 M phosphate buffer and centrifuged. The pellets were then postfixed in 2% osmium tetroxide (TAAB, Berkshire, England) in 0.1 M phosphate buffer (pH 7.4) at 4°C for 2 hr, dehydrated in ethanol and then in acetone, and embedded in LX-112 (Ladd). Ultrathin sections (approximately 40–50 nm) were cut by a Leica ultracut UCT (Leica, Wien, Austria). Sections were contrasted with uranyl acetate and later with lead citrate and were examined in a Tecnai 12 Spirit Bio TWIN transmission electron microscope (Fei Company, Eindhhoven, the Netherlands) at 100kV. Digital images were taken with a Veleta camera (Olympus Soft Imaging Solutions, GmbH, Münster, Germany).28
Detection of Autophagic Flux
ImageStream (Amnis, Seattle, US), a multispectral flow cytometer combining standard microscopy with flow cytometry, was used for the analysis of autophagic flux. PBMCs or whole blood was stained with the violet LIVE/DEAD marker (L34955 Component A, Invitrogen, Carlsbad, CA, USA), a red lysosome-specific dye (LysoID, Enzo Life Sciences, Lausen, Switzerland), and a CD19 antibody. Cells were fixed and permeabilized (eBioscience kit) and stained with mouse-anti-LC3 (Nanotools, Teningen, Germany) and goat-anti-mouse IgG-AlexaFluor488 (Invitrogen). Single-stained controls were included for compensation. After image acquisition, the software package IDEAS 4.0.735 was used for analyzing live cells for colocalization of LC3 and lysosomes. We calculated autophagy levels by measuring percent colocalization (bright detailed similarity) of LC3 and LysoID gated on CD19+LC3+LysoID+ live cells. This analysis gives an estimation of the relative number of autophagolysosomes per cell in a sample of more than 20,000 cells and yields a powerful assessment of autophagic flux.
Results
Case Reports
In family A (Figures 1A and Figure S1), the index case (individual P1) was born in 1991 to first-cousin Arab parents. At 1 year of age, he presented with idiopathic thrombocytopenic purpura (ITP). At age 5, he was hospitalized with pleuropneumonia. A clinical workup (Table 1) revealed low immunoglobulin levels and led to the diagnosis of CVID and the initiation of intravenous immunoglobulin (IVIG) replacement. He progressively developed chronic lung disease; his computed tomography (CT) scans have shown bilateral bronchiectasis since was 6 years old. Lung biopsies were obtained and revealed a lymphoid interstitial pneumonia (LIP). Since the age of 2 years, his growth has been severely retarded (both weight and height are below the second percentile), and he has presented with significant clubbing. Recently, he developed strabismus as a result of abducens nerve palsy along with hemiplegia. During our study, a magnetic resonance image (MRI) of his head showed a cerebral mass, which is currently being evaluated for granuloma formation.
Figure 1.
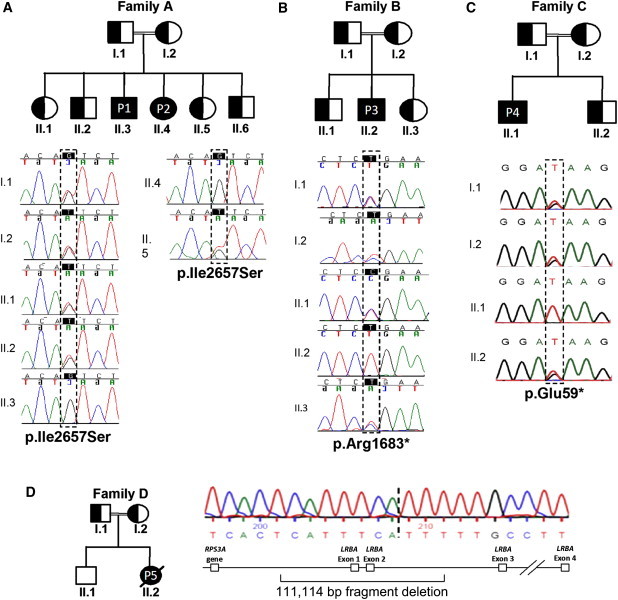
Mutations in LRBA
Shaded figures are affected individuals homozygous for a mutation in LRBA. Half-shaded figures are unaffected individuals heterozygous for a mutation in LRBA.
(A) Family A with two affected individuals showing a homozygous missense p.Ile2657Ser substitution.
(B) Family B with one affected individual who had homozygous nonsense substitution p.Arg1683∗.
(C) In family C, the amino acid substitution was nonsense: p.Glu59∗.
(D) The affected individual from family D has a large homozygous deletion including exons 1 and 2 and ∼60 kb upstream and ∼50 kb downstream. The parents in family D are heterozygous for the deletion, but the unaffected sibling does not carry the deletion.
Table 1.
Laboratory Values for Affected Individuals with Homozygous Mutations in LRBA
|
Affected Individual |
|||||
|---|---|---|---|---|---|
| P1 | P2 | P3 | P4 | P5 | |
| Substitution | p.Ile2657Ser | p.Ile2657Ser | p.Arg1683∗ | p.Glu59∗ | not applicable |
| Lymphocyte counts (first measurement) | |||||
| Age at measurement | 5 years | 14 years | 29 years | 19 years | 19 years |
| CD3 (cells/μl) | 900 (900–4,500) | 2,073 (800–3,500) | 2,350 (700–2,100) ↑ | 5,716 (700–2,100) ↑ | 355 (700–2,100) ↓ |
| CD4 (cells/μl) | 685 (500–2,400) | 767 (400–2,100) | 1,290 (300–1,400) | 1,061 (300–1,400) | 262 (300–1,400) ↓ |
| CD4 CD45RA | N.D | 179 (230–770)a | 201 (27–833)b | 72 (27–833)b | N.D |
| CD4 CD45RO | N.D | 553 (240–700)a | 887 (167–670)b ↑ | 588 (167–670)b | N.D |
| CD8 (cells/μl) | 380 (300–1,600) | 1,200 (200–1,200) | 1,032 (200–900) ↑ | 4,707 (200–900) ↑ | 165 (200–900) ↓ |
| CD8 CD45RA | N.D | 1,003 (240–710)a ↑ | 941 (19–508)b ↑ | 1,991 (19–508)b ↑ | N.D |
| CD8 CD45RO | N.D | 473 (10–142)b ↑ | 627 (15–275)b ↑ | 374 (15–275)b ↑ | N.D |
| Lymphocyte counts (second measurement) | |||||
| Age at measurement | 5 years | 10 years | 16 years | 16 years | 19 years |
| NK cells (cells/μl) | 260 (100–1,000) | 190 (70–1,200) | 35 (70–1,200) ↓ | N.D. | N.D. |
| CD19 (cells/μl) | 280 (200–2,100) | 300 (200–600) | 219 (200–600) | 69 (200–600) ↓ | 121 (100–500) |
| Switched memory B cells (CD19+ CD27+ IgM−, % of total B cells) | 0 (3.9–16.2) ↓ | 1 (3.85–16.5) ↓ | 0.8 (4–22.8) ↓ | 0.89 (4–22.8) ↓ | N.D. |
| Immunoglobulin levels | |||||
| Age at measurement | 5 years | 10 years | 16 years | 7 years | 19 years |
| IgG (g/l) | 1.6 (4.9–16.1) ↓ | 1.8 (5.4–16.1) ↓ | 3.23 (6.0–16.0) ↓ | 0.34 (5.4–16.1) ↓ | 3,4 (6.0–16.0) ↓ |
| IgM (g/l) | 0.22 (0.5–2) ↓ | 0.16 (0.5–1.8) ↓ | 0.72 (0.5–1.9) | 0.2 (0.5–1.8) ↓ | 0.2 (0.5–1.9) ↓ |
| IgA (g/l) | 0.1 (0.4–2) ↓ | 0 (0.7–2.5) ↓ | 0 (0.8–2.8) ↓ | 0.1 (0.5–2.4) ↓ | 0 (0.8–2.8) ↓ |
Age-adapted reference ranges are shown in parentheses. ↓ indicates a value below the reference ranges, and ↑ indicates a value above the reference ranges. Reference ranges that are not marked were obtained from Comans-Bitter et al.29 The following abbreviations are used: N.D., not done; and NK, natural killer.
Reference range obtained from Shearer et al.30
Reference range obtained from Mayo Medical Laboratories.
At 1 year of age, his sister (individual P2; Figure 1A and Figure S1), born in 1997, also had ITP, which was self-limiting. At 2 years of age, she developed asthma, which was satisfactorily treated with inhaled steroids. After developing serous otitis media, she underwent adenoidectomy and bilateral insertion of ventilation tubes at age 9. In the following year, a massive pneumonia along with loculated empyema required surgical intervention, including thoracotomy and drainage. Cultures from the pleural effusion revealed Streptococcus pneumoniae and Aspergillus. Low immunoglobulin levels were detected (Table 1), and she was treated with antibacterials, antifungals, and IVIG. Furthermore, she suffered from chronic lung disease with bilateral bronchiectasis. She was small for her age (both weight and height were below the fifth percentile since the age of 2). When she was 11 years old, reactive monoarthritis emerged in her right knee, and it responded well to local steroid injections.
Individual P3 (family B; Figure 1B), born in 1982, is of Sicilian origin. The parents were first cousins once removed. The first symptoms were recurrent warts and perineal molluscum contagiosum at age 12 and an increased frequency of mild respiratory infections. At age 16, he was hospitalized with ITP, lymphadenopathy, autoimmune haemolytic anemia (AIHA), atrophic gastritis with autoantibodies against intrinsic factor, and a submaxillar abscess (Staphylococcus aureus and Streptococcus viridians). An immunological workup revealed a moderate IgG hypogammaglobulinemia and complete IgA deficiency (Table 1) but a normal neutrophil count; IVIG replacement was started. He also had severe diarrhea in the absence of any detectable bacterial or parasitic infection. After undergoing a colonoscopy, he was diagnosed with a severe nonspecific colitis. During the following years, he developed severe recurrent pneumonias, including several interstitial pneumonias, sputum cultures of which did not grow any pathogen. At the time of the infections, IgM was intermittently elevated (5–8 g/l, data not shown) and swelling of hilar and mediastinal lymph nodes occurred. Biopsies of these lymph nodes were obtained, and the histological analysis showed a mixed lymphoid follicular hyperplasia with the absence of the follicular mantle zone. A pulmonary workup showed a lymphoid interstitial pneumonia and bronchiectasis. The interstitial lung disease was treated with methylprednisolone. Attempts to taper steroids were frequently associated with relapses, and long-term treatment with infliximab was initiated and allowed the discontinuation of methylprednisolone. Infliximab, however, led to little improvement of the chronic diarrhea. At age 26, the individual underwent an episode of seizures caused by a pseudotumoral lesion in the right temporal lobe. After surgical removal of the lesion, he recovered and the histopathological workup revealed a granulomatous infiltration with T cells, plasma cells, and macrophages but showed low B cell numbers. Recently, the seizures recurred, but no lesion was detectable in an MRI.
In Iranian family C with first-cousin parents (Figure 1C), individual P4, a male, was born in 1993 and presented with frequent upper-respiratory-tract infections, several episodes of pneumonia, AIHA, and ITP at the age of 2 years. Although he experienced recurrent respiratory and gastrointestinal infections, the diagnosis of CVID was not made until he was 7 years old, when he developed severe lower-respiratory-tract infections along with finger clubbing, hepatosplenomegaly, and failure to thrive. On the basis of these clinical findings and his low immunoglobulin levels (Table 1), regular IVIG replacement therapy was started. A pulmonary workup revealed an obstruction of the small airways and bronchiectasis. During 10 years of follow-up observation, he developed recurrent conjunctivitis and urticaria and a cor pulmonale with consecutive right-heart failure. However, the predominant clinical problems were ITP, AIHA, and an autoimmune enteropathy (which can be classified as Crohn disease), which was confirmed by gastrointestinal histology. Currently, he is being treated with IVIG replacement and antibiotic prophylaxis.
In family D, individual P5 (Figure 1D), born in 1983, was previously described.31 She was an Iranian woman who died at the age of 19 after respiratory failure. At 2 years of age, her medical problems began with allergic dermatitis followed by recurrent, chronic diarrhea and frequent upper-respiratory-tract infections, including sinusitis and otitis media. At age 15, after an episode of pneumonia, she was initially diagnosed with selective IgA and IgG2 deficiency. Within 2 years, her serum levels of IgG and IgM declined gradually, she developed bronchiectasis, and she was diagnosed with CVID. Additionally, she had autoimmune phenomena, hypothyroidism, and myasthenia gravis. However, there were no signs of ITP or AIHA. Because of chronic diarrhea and growth retardation, a gastrointestinal biopsy was performed and showed intestinal inflammation and subtotal villous atrophy.
Genotyping Families A and B
The analysis of family A SNP data identified two intervals (larger than 1 Mb) that segregated perfectly with disease status on chromosomes 4q and 5q. A perfectly segregating haplotype in family A had a multipoint LOD score of at least 2.08. There was also a near-perfect interval on 12q. The intervals on 4q, 5q, and 12q contain at most 81, 80, and 107 genes or predicted genes, respectively (Table S1).
Genotyping with microsatellites confirmed the perfect segregation on 4q and showed that the linked interval extended from 141.9 to 155.0 Mb at the very least and from 141.4 to 157.7 Mb at the most (Figure S1). The interval on 4q did not overlap with the 4q interval described previously in a dominant CVID family.24 Microsatellite genotyping also confirmed the intervals on 5q and 12q.
Before finding any mutation, we genotyped family B by using SNP chips. The largest perfectly segregating interval in family B was between 140.8–162.4 Mb on chromosome 4 and contained the largest perfect interval for family A. The multipoint LOD score was at least 1.36 in pedigree B, giving a total multipoint LOD score of at least 3.44 for this interval on chromosome 4q. Other than the shared large interval on chromosome 4q, there was no overlap between the large perfect intervals for family B (chr14: 65.7–82.7 Mb and chr21: 33.2–39.4 Mb) and other perfect or near perfect intervals for family A (chr5: 174.8–177.8 Mb and chr12: 103.4–111.0 Mb).
Homozygous Mutations in LRBA in Families A and B
Within the linkage interval on chromosome 4q, the eighth gene that we sequenced in family A was LRBA. We found a homozygous missense mutation, c.7970T>G, which introduces the amino acid substitution p.Ile2657Ser in exon 54 in individuals P1 and P2 (Figure 1A). All six unaffected members in family A were heterozygous for p.Ile2657Ser. We identified one more heterozygous healthy individual by sequencing DNA from 17 additional individuals, some of whom are relatives and all of whom reside in the same village as family A. DNA samples of 128 healthy donors of Arabic heritage were subsequently sequenced for exon 54 and gave only wild-type results, suggesting that p.Ile2657Ser is not a benign variant. In silico analysis of the p.Ile2657Ser substitution was performed with the bioinformatics tools SIFT32 and PolyPhen.33 SIFT predicted that the mutation is deleterious and has a most extreme possible score of 0.00. PolyPhen did not concur with this prediction, but the fact that PolyPhen used fewer than ten sequences homologous to LRBA limited its predictive power. Multiple alignment of homologs from several species shows that p.Ile2657 is a highly conserved residue (Figure S2).
To investigate whether p.Ile2657Ser affected LRBA levels, we performed immunoblot analysis in protein extracts from EBV cell lines from two affected homozygous individuals, three heterozygous individuals from family A, and two healthy controls (Figure 2B). No LRBA was detected in the affected individuals by immunoblot with the antibody HPA023597, which was raised against amino acids 906–1038 of LRBA. EBV cell lines from heterozygous individuals showed protein levels comparable to the variable amount of LRBA in healthy donor cells (Figure 2B).
Figure 2.
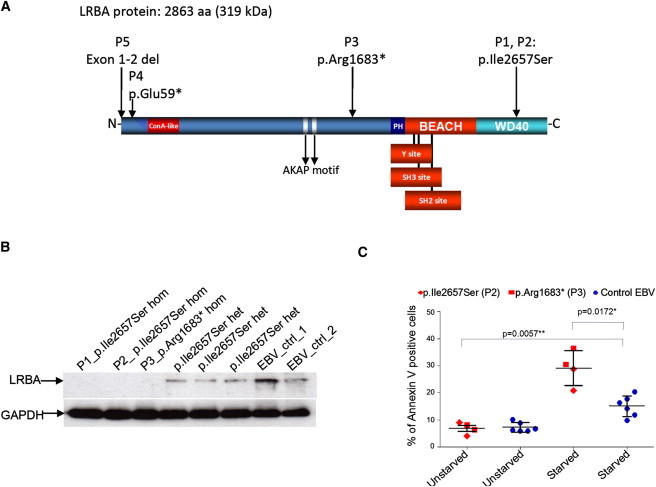
Homozygous LRBA Mutations Lead to Lack of Protein
(A) A schematic representation of LRBA shows domains and location of mutations at the protein level.
(B) Protein extracts of Epstein-Barr virus (EBV)-transformed B cell lines (EBV cells) from individuals P1, P2, and P3, from three heterozygous members of family A, and from two controls were loaded into a polyacrylamide gel. EBV cells from homozygous affected individuals show an absence of LRBA, whereas both heterozygous individuals and the healthy controls show protein expression.
(C) EBV cell lines from two affected individuals and three healthy donors were subjected to starvation. The proportion of early apoptotic cells was determined with Annexin V and propidium iodide staining. Both the percentage of early apoptotic EBV cells after starvation and the increase in apoptosis after starvation were significant (p values are 0.0172 and 0.0057, respectively, as determined by Welch t tests). We calculated the increase in apoptosis by subtracting the amount of unstarved EBV cells from the amount of starved EBV cells for both affected individuals and controls.
Because the data from family A strongly suggested LRBA as a candidate gene, we chose to sequence LRBA in family B. We found that the index case (P3) in family B has a homozygous c.5047C>T mutation, which introduced a stop codon (p.Arg1683∗) in exon 30 of LRBA, and that the four unaffected members of family B were heterozygous for this mutation (Figure 1B). As in family A, no LRBA was detected in EBV cell extracts of the affected individual (Figure 2B). Two hundred alleles from European healthy controls were sequenced for exon 30, and no mutation was found.
Additional Genetic Analysis and Sequencing
At this stage, we had evidence that homozygous mutations in LRBA might cause an early-onset form of hypogammaglobulinemia. We therefore analyzed genotype data and sequenced LRBA in a total of 14 additional individuals (12 Iranians and two Italians) for whom autosomal recessive (AR) CVID was suspected because of the early onset of symptoms and, in most cases, a consanguineous family structure. We sought other affected individuals with homozygous chromosome 4q intervals overlapping LRBA.
P4 in family C was homozygous for 112 consecutive markers in chr4: 140.8–172.4 Mb, whereas P5 in family D was homozygous for 104 consecutive markers in chr4: 136.7–164.2 Mb. For each of the two affected individuals, these regions were among the five longest homozygous intervals across all autosomes.
Ten additional Iranian simplex cases were each homozygous for 6–15 consecutive markers spanning LRBA in intervals of sizes ranging from 0.5 to 3.0 Mb. Each of these individuals had much longer homozygous intervals that did not contain LRBA. We concluded that P4 and P5 had strong evidence in favor of an LRBA mutation, whereas the other ten simplex cases had weak genotype evidence consistent with a mutation.
Two Italian families had perfectly segregating intervals of 60 markers spanning chr4: 151.4–152.5 Mb and 42 markers spanning chr4: 151.5–152.3 Mb, respectively. These intervals contain the majority of LRBA but not the entire gene. Because these two families were genotyped with the denser 250K mapping array, we can be confident of the homozygosity of the affected individuals, but the homozygous intervals are more than an order of magnitude shorter than the perfectly segregating intervals in families A and B. We concluded that there was enough evidence to justify sequencing LRBA in the affected individuals of these two families.
Sequencing and deletion assays (Figure S3) revealed homozygous mutations in the affected individuals from families C and D. Individual P4 from the Iranian family C (Figure 1C) had a homozygous LRBA mutation generating a stop codon in exon 1 (c.175G>T [p.Glu59∗]). This mutation was absent in 233 Iranian controls as determined by local sequencing. Individual P5 from the Iranian family D had a homozygous deletion of 111,114 bp (Figure 1D), including exons 1 and 2 and about 60 kb upstream and 50 kb downstream of these exons (the identification of the deletion boundaries is described in Figure S3 and Table S2). We identified the exact breakpoint of the deletion by directly sequencing a 2.5 kb product amplified after the use of primer pairs P11 and P18 (Table S2). Other members of families C and D were tested for the respective mutations, which confirmed AR inheritance (Figures 1C and 1D). Figure 2A depicts a schematic diagram of the mutations, and the mutations are listed in Table 2.
Table 2.
Summary of the LRBA Mutations
| Affected Individual | Substitution | Mutation | Exon | Accessiona |
|---|---|---|---|---|
| P1 | p.Ile2657Ser | c.7970T>G | 54 | ss479152582 |
| P2 | p.Ile2657Ser | c.7970T>G | 54 | ss479152582 |
| P3 | p.Arg1683∗ | c.5047C>T | 30 | ss479152581 |
| P4 | p.Glu59∗ | c.175G>T | 3 | ss479152583 |
| P5b | not applicable | g.152211739_152222852del111114 | 1–2del | nstd59 |
Accession numbers are for entries in dbSNP (P1–P4) or dbVar (P5).
For the deletion in P5, the positions are with respect to chromosome 4 (NCBI build 36).
No homozygous mutations in LRBA have been identified in the 12 additional families with suspected AR-CVID and weak linkage evidence. However, our sequencing efforts have been limited to identifying mutations in the coding regions of LRBA. Noncoding mutations might have been missed in this gene that has more than 50 exons and spans 752,397 bp of genomic DNA.
LRBA-Deficient B Cells Show Defective Activation and Survival
All individuals with homozygous LRBA mutations had low levels of IgA and IgG in serum, and all, with the exception of the individual who had the homozygous substitution p.Arg1683∗, had low levels of IgM (Table 1). In four of the five affected individuals, counts of CD19+ B cells were normal. Phenotypic analysis of B cell subsets revealed a strong reduction of IgM+ CD27+ marginal-zone-like B cells and IgG+/IgA+ CD27+ switched-memory B cells (Figure 3 and Table 1). Although IgM expression in CD27− cells was higher in B cells of P1 and P3 than in control cells analyzed in parallel, the proportion of transitional B cells in P1 (1.7% of CD19+ cells) was not larger than that in the control (0.5%–6% of CD19+ cells). Switched-memory B cells as defined by CD19+CD27+IgM− were low in all affected individuals tested (Table 1).
Figure 3.

Phenotypic Characterization of B Cells
PBMCs were gated for CD19+ CD20+ B cells (A) and were analyzed for IgM, CD27 (B), and BAFF-R expression (C), for CD10 and CD38 expression for the detection of transitional B cells (D), and for IgA and IgG expression for the detection of switched-memory B cells (E). P1 and P3 have fewer B cells than the healthy control analyzed in parallel (A). In addition, the IgMlow CD27−, the IgM+ CD27+ marginal-zone, and the IgM− CD27+ switched-memory B cell subsets are smaller, whereas the IgMhi CD27− B cell subset is larger, than in the control (B). BAFF-R expression is higher than in the control (BAFF-R mean fluorescence intensity of P1 = 2,476; P2 = 1,978; HD = 1,378). The proportion of CD10+ CD38+ transitional B cells in P1 is similar to that in the control (D), whereas IgA- or IgG-expressing switched-memory B cells are lacking (E).
To test whether the defect in B cell development correlates with defective B cell activation and/or immunoglobulin production, naive B cells from healthy controls and individuals with homozygous LRBA mutations were stimulated with anti-IgM, CD40L, BAFF, and CpG oligonucleotide (ODN). Stimulated naive B cells from healthy donors and individuals with heterozygous LRBA mutations showed robust IgG production in vitro, whereas naive B cells from three of the homozygous affected individuals produced extremely low levels of IgG (Figure S5). These results were confirmed when B cells of P1 were stimulated with CD40L combined with IL-21 or IL-4; under these conditions, IgG, IgA, and IgM secretion was reduced (Figure S5). In agreement with this, lymphocyte analysis after stimulation with CD40L and IL-4 or IL-21 showed poor cell survival and a dramatic reduction in the proportion of developing plasmablast cells (Figure S5). Lymphocyte proliferation in response to PWM (Figure S5) was also affected, which supports the defect of plasma cell development given that both processes are tightly linked.
LRBA-Deficient EBV Cells Show Increased Apoptosis
The defects observed in immunoglobulin production and B cell differentiation and proliferation might be due to increased cell death. It has been suggested previously that LRBA has a role in apoptosis.26 We explored the susceptibility to apoptosis in LRBA-deficient EBV cells and analyzed the amount of apoptotic cells after serum deprivation. In EBV cells derived from individuals P2 and P3, the proportion of apoptotic cells increased significantly after serum depletion; however, in cells from three healthy controls, the proportion of apoptotic cells showed only a slight increase (Figure 2C).
LRBA-Deficient B Cells Show Reduced Autophagy
Autophagy is the major process by which lysosomes degrade cellular material and is cytoprotective. In its absence, mammalian hematopoietic cells are prone to cell death.34 Because serum starvation of LRBA-deficient cells caused increased apoptosis, we hypothesized that LRBA deficiency would affect autophagy. Furthermore, there are homologs of LRBA with lysosomal functions, and mice with mutations in lysosomal proteins have similar phenotypes to mice with mutations in the autophagic pathway.35 To detect autophagosomes, we first compared LRBA-deficient B cells with healthy controls by electron microscopy. The controls showed a normal morphology and well-defined mitochondria, Golgi apparatus, and endoplasmic reticulum (ER) (Figure 4A, top left). This was in stark contrast with individual P4's B cells, which showed increased areas of Golgi apparatus (“G” in Figure 4A). Furthermore, we observed that many cells showed centrioles and accumulation of autophagosomes (Figure 4A, bottom).
Figure 4.
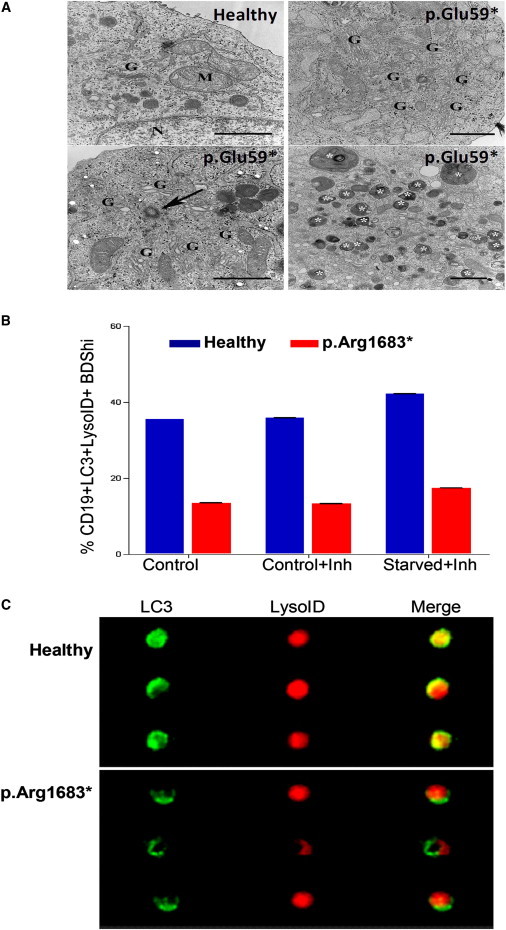
Evaluation of Autophagy in LRBA-Deficient and Control Cells
(A) In the top left panel, an image of a B cell from a control shows a normal ultrastructural morphology with well-defined mitochondria (M), Golgi apparatus (G), and ER (N, nucleus). An image from a B cell from P4 shows increased areas of Golgi apparatus (top right panel). Centrioles (arrow) were frequently found in the cytoplasm of B cells from P4 (bottom left panel), and accumulation of autophagosomes (indicated by white asterisks) in the cytoplasm was also frequently seen (bottom right panels). Scale bars represent 1 μm.
(B) Autophagic flux showing the colocalization of LC3 and LysoID in the presence or absence of the lysosomal inhibitors E64d/pepstatin on PBMCs (Inh). Cells from P3 (red) show low levels of colocalization of LC3 and LysoID in all conditions tested. Error bars are shown and show the variation in LysoID and LC3 colocalization displayed by the B cells acquired on the Image Stream (n = 20,000).
(C) Representative images of B cells from a healthy control and individual P3 show LC3 and lysoID colocalization.
Using a recently published technique to detect the autophagic flux in primary cells and cell lines by measuring the proportion of cells with autolysosomes, defined by the colocalization of both LC3 and a lysosomal marker,36 we found that LRBA-deficient B cells have a significantly reduced ability to induce autophagy in response to starvation (Figure 4B). Figure 4C shows representative images of B cells from one healthy donor and from individual P3. Additionally, autophagy in individual P2 was reduced (Figure S4). Data from individuals P2 and P3 cannot be pooled because their samples were obtained under different conditions. Interestingly, although lysosomal content and LC3-positive autophagosomes were increased, colocalization of these markers was reduced (data not shown). Together with the electron microscopy data, these results suggest that autophagic flux is impaired and does not reach completion possibly because either the fusion between autophagosomes and lysosomes is impaired or lysosomes are not functional. Therefore, cells are accumulating autophagosomes and LC3-containing vesicles, which might contribute to cell death.
Discussion
Homozygous mutations in LRBA cause a syndrome characterized by early-onset hypogammaglobulinemia with autoimmunity and inflammatory bowel disease. Affected individuals show reduced levels of at least two immunoglobulin isotypes (IgM, IgG, or IgA) and suffer from recurrent infections, autoimmunity, and chronic pulmonary and gastrointestinal disorders. We identified five individuals harboring four distinct mutations in LRBA (Table 2). Three of these mutations are probably functional null mutations because they represent either premature stop codons (before the principal domains of LRBA) or a large deletion. None of the mutations appear to disrupt MAB21L2 (MIM 604357), which is nested in LRBA.37 No previous study has reported LRBA germline mutations associated with any disease, but somatic mutations of LRBA have been reported in breast cancer.38
The following reasons led us to suspect LRBA as the affected gene in family A. (1) The first study of LRBA showed that LRBA is inducible by lipopolysaccharide (LPS), a component of bacteria that triggers an immune response.39 Although a portion of the sequence of LRBA cDNA was first discovered and deposited in GenBank in 1992,40 Wang et al., were the first to clone a full-length cDNA, and they proposed the current, descriptive name “LPS-responsive, beige-like anchor gene.”39 (2) LRBA shares some homology with a protein in the BCL family, and the fact that some proteins in that family are involved in B cell lymphomas indicates that LRBA is likely to be active in B cells.41 (3) LRBA shares substantial homology with LYST (MIM 606897), a lysosomal protein defective or absent in another primary immunodeficiency, Chediak-Higashi syndrome (CHS [MIM 214500]),42 a disease characterized in part by large lysosomal granules in leukocytes and giant melanosomes in melanocytes. Both mice and humans with CHS have normal levels of immunoglobulins and generally normal B cell function.43,44 (4) FAN (MIM 603043), another paralog of LRBA, is part of a pathway connecting CD40 (an important signaling molecule for B cells, monocytes, and other cells [MIM 109535]) to signaling for apoptosis via the sphingomyelin-ceramide pathway.45,46
LRBA is expressed in different cancer cell lines.26 Figure S6 shows that LRBA expression is widely distributed in different immune and nonimmune tissues. Although three different isoforms of murine Lrba have been described, there are no convincing data showing the existence of more than one isoform of LRBA. The NCBI does list two isoforms: RefSeq NP_001186211.2 (isoform 1; 2,851 amino acids) and NP_006717.2 (isoform 2; 2,863 amino acids). These differ in a dubious exon (exon 39), and we believe that isoform 1 is the principal LRBA isoform in B cells.
We show (Figure S6) that LRBA is expressed in many tissues. This is mostly consistent with an earlier study by Dynomin et al. and is consistent with the expression atlas BioGPS.41,47 This atlas shows that LRBA is much more highly expressed in immune cells, including T cells and B cells, than in other tissues. The original study on murine Lrba showed that at least one isoform is expressed in most tissues.39 It is interesting to contrast the wide expression pattern of Lrba with that of its paralog, Nbea, which has high levels of expression in brain and endocrine tissues and low or no expression in other tissues.48 Perhaps NBEA (MIM 604889) carries out some functions of LRBA in nonimmune cells of LRBA-deficient individuals. An overlap in LRBA and NBEA expression and function could reconcile the fact that autophagy happens in most cell types with our findings that LRBA-deficient lymphocytes have a defect in autophagy but that the phenotype of LRBA-deficient individuals is limited to the immune system.
Little is known about the exact function of LRBA in human biology. LRBA is a member of the BEACH-WD40 protein family.49 Members of this family include LYST, which is involved in the regulation of lysosome-related organelle size and movement;42 NBEAL2 (MIM 614169), which was recently identified as the gene mutated in gray platelet syndrome (MIM 139090);50–52 FAN (MIM 603043), a protein involved in TNFR signaling; and NBEA (MIM 604889), a protein with A-kinase anchoring and vesicular release at the neuromuscular junction.42,45,53,54 Interestingly, LRBA contains an A-kinase anchoring protein (AKAP) motif.55 AKAPs have an affinity for the regulatory subunits of protein kinase A (PKA RI or PKA RII) and other signaling enzymes such as protein kinase C (PKC). Their main function is to compartmentalize these signaling molecules to distinct membranes and organelles.55 The repeated WD40 domain is located at the C-terminal end and is preceded by the BEACH and Plekstrin Homology (PH) domains.49 The WD40 domain is highly conserved between species and is one of the most abundant domains in eukaryotic organisms. WD40 domains can bind multiple partners in different ways.56 They are important for protein-protein and protein-DNA interactions, and they participate in multiple cellular processes, such as signal transduction, vesicular trafficking, cytoskeleton assembly, the cell cycle, apoptosis, chromatin dynamics, and transcriptional regulation.56 The precise function of the BEACH domain is unknown; it might be important for protein-protein interaction.49 The PH domain of LRBA is weakly conserved. An intramolecular interaction between BEACH and PH domains has been shown, and both domains might therefore be important for protein folding and signaling.57
The p.Ile2657Ser substitution in P1 and P2 affects the WD40 domain of LRBA. However, the binding partners for the LRBA WD40 domain are unknown. Moreover, the p.Ile2657Ser substitution affects protein expression, suggesting that this amino acid substitution affects the stability of LRBA. Because p.Ile2657 is a conserved residue (Figure S2), it might be important for protein folding or might affect the binding with LRBA interaction partners. The p.Arg1683∗ change, if translated, would lead to a shorter protein lacking 1,180 residues at the C-terminal end of LRBA. This substitution would affect the expression of the PH, BEACH, and WD40 domains of LRBA. We did not detect any shorter form of LRBA in P3, and therefore, the shortened mRNA might be subject to nonsense-mediated decay. The mutation found in P4 introduces a stop codon at position 59 of the protein. If translated, it would give a very short product unlikely to have any function. In the case of P5, the protein should not be translated because the large deletion eliminates the translation start site (Figures 1D and 2A).
Overall, fewer than 20% of individuals with CVID have an affected relative.2 However, AR simplex cases, including three of the five affected individuals in our study, might not be recognized as familial cases. Among the family members summarized in Figure 1, the 15 individuals with heterozygous mutations are all asymptomatic. We infer that, unlike in the case of TNFRSF13B, LRBA is not likely to play a prominent role in dominantly-inherited antibody deficiencies.
All homozygous individuals with LRBA deficiency showed reduced counts of switched-memory B cells, as has been reported for individuals with CVID. P1 and P3 also lacked marginal-zone-like B cells. P4 had reduced counts of total B cells. P5 had low T cell levels. Natural killer (NK) cell counts were low in P3 and P4. Although we measured a reduced proportion of the CD16+CD56+ NK subpopulation in P3, no functional NK cell defect was detected for this individual as measured by normal CD107a translocation (data not shown).58
LRBA-deficient B cells cultured under conditions favoring class-switch recombination and plasmablast development failed to proliferate, differentiate into antibody-secreting cells, and induce expression of markers characteristic of plasmablasts. During antigen recognition, B cells proliferate for clonal expansion, a process important for differentiation to memory or plasma cells.59 Data provided here indicate that LRBA deficiency leads to defects in B cell differentiation. Analysis of tumor cells suggests that LRBA promotes proliferation and cell survival.26 Our results showing increased susceptibility to apoptosis in LRBA-deficient EBV cells support this interpretation.
LRBA deficiency resulted in reduced autophagy as shown by the abnormal accumulation of organelles in B cells. This observation was confirmed by the low colocalization of LC3 with lysosomes. These data suggest that LRBA plays a role in autophagy and that the increased susceptibility to apoptosis might be a consequence of defective autophagy. Supporting the effect of autophagy on B cell function, mice deficient in the autophagy-related Atg5 show defects in B cell development.60
Because autophagy and apoptosis have been reported to be defective in autoimmune conditions such as systemic lupus erythematosus,61 it is tempting to speculate that the autoimmune features seen in LRBA-deficient individuals are attributable to defective autophagy and increased apoptosis. Additionally, defects in autophagy genes have been associated with several conditions in humans, for example, X-linked myopathy with excessive autophagy (XMEA [MIM 310440]), Pompe disease (MIM 232300), and Parkinson disease (MIM 600116),62–64 suggesting that inherited defects in autophagy might lead to tissue-specific defects in cellular function. Additionally, autophagy has been linked to B cell germinal-center number and size in mice with a heterozygous deletion of the gene encoding the known autophagy protein beclin-1 (also known as Atg6). These mice have a reduced occurrence of autophagy and a reduced number and size of germinal centers.65 Lysosomal stability is known to be associated with germinal-center stability,66 but we could not mechanistically link LRBA to germinal-center formation. The description of beclin-1-heterozygous mice suggested the hypothesis that deficiency of LRBA would lead to reduced autophagy in lymphocytes. If cells are under stress and autophagy is reduced, then the more direct alternative, apoptosis, should be increased. Interestingly, the data regarding autophagy and apoptosis (Figure 4 and Figure S4) are consistent with the hypothesis that the lack of LRBA disrupts the autophagy pathway that includes beclin-1.67
We were enticed by the observation that several LRBA homologs, including LYST, are known to function in lysosomal vesicles.44,68,69 The movement of lysosomal vesicles in which these proteins participate might be part of an autophagy process. The principal subcellular-localization study was done by Wang et al.39 on the cell line RAW 264.7. They showed that LRBA colocalized with lysosomes, the trans-Golgi network, the ER, the perinuclear ER, and endocytic vacuoles. These localizations are consistent with a role for LRBA in autophagy because these organelles either play a role in the completion of the autophagosomal flux or are cleared by autophagy in the following ways. (1) Autophagosomal content is delivered to the lysosome; in this manuscript and our previous work, we measure autophagy levels by quantifying the extent of colocalization of lysosomal and autophagosomal markers.36 (2) Recent studies have shown show that autophagosomal membranes originate at the ER.70,71 Moreover, the existence of autophagy of ER components (ER-phagy) has been suggested.72 (3) The endocytic and autophagic pathways are linked, and components of functional endomembrane trafficking, the secretory pathway, and fusion machinery have been shown to have an impact on autophagy.73 (4) In work by Nishida et al., which describes an Atg5/7 independent pathway, autophagosome-like structures appear near the Golgi apparatus.74 Furthermore the Golgi-endosomal system is also thought to contribute to the Atg5/7-dependent conventional formation of autophagosomes.75 Finally, there is a possibility that clearance of the Golgi apparatus might be mediated by autophagy, possibly explaining the accumulation of Golgi in the absence of LRBA. The observed colocalizations do not identify the exact role of LRBA in the autophagic flux; further molecular studies are required.
Our study demonstrates the importance of LRBA for the human immune system and its role in host defense and autoimmune and autoinflammatory conditions. The finding that mutations in LRBA result in a severe phenotype of hypogammaglobulinemia and autoimmunity in four different families adds to the understanding of the participation of LRBA in apoptosis and suggests that individuals with the clinical phenotype described above should be screened for LRBA mutations. In our study, all affected individuals were clinically diagnosed with CVID, but the clinical data of LRBA deficiency show autoimmunity since early childhood. Our data support the participation of LRBA in apoptosis; however, future work is required for fully understanding the exact functions of LRBA.
Acknowledgments
The authors wish to thank Sary Workman, Astrid Petersen, Fariba Tahami, Eira Rawlings, Judy Levin, Charlotte Holden, and Francisco Espinosa Rosales for their assistance. This study was supported by the German Federal Ministry of Education and Research (BMBF 01 EO 0803). The research was funded in part by the European Community's 6th and 7th Framework Programmes FP7/2007-2013 under grant agreements HEALTH-F2-2008-201549 (EURO-PADnet) and Health-F5-2008-223292 (Euro Gene Scan) and by a Marie Curie Excellence Grant to B.G. (MEXT-CT-2006-042316, MC-PIAID); European Research Council starting grant (24511-ImmunoSwitch); grants from the Swedish Research Council and Swedish Cancer Society; a Glaxo-Smith-Kline grant managed by the Primary Immunodeficiency Association UK; and a grant from the Fondazione C. Golgi, Brescia, and Associazione Italiana Immunodeficienze Primitive. This research is also supported in part by the Intramural Research Program of the National Institutes of Health, National Library of Medicine, USA. Gabriela Lopez-Herrera received partial scholarship 93910 from CONACyT, Mexico. Giacomo Tampella is a PhD student of a joint PhD program of the Universities of Brescia and Trieste, Italy. Hans Stauss was supported by grants from Leukaemia and Lymphoma Research UK and from the Medical Research Council. Kanchan Phadwal and Anna Katharina Simon were supported by the National Institute for Health Research Biomedical Research Centre, Oxford, UK.
Supplemental Data
Web Resources
The URLs for the data presented herein are as follows:
Mayo Medical Laboratories, http://www.mayomedicallaboratories.com/test-catalog/print/89319
OMA Browser, http://omabrowser.org/cgi-bin/gateway.pl
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen, http://genetics.bwh.harvard.edu/pph
SIFT, http://sift.jcvi.org
Accession Numbers
The LRBA sequences reported in this paper have been deposited in dbSNP under accession numbers ss479152582, ss479152582, ss479152581, and ss479152583 (for patients 1–4) and in dbVar under accession number nstd59 (for patient 5).
References
- 1.Plebani A., Soresina A., Rondelli R., Amato G.M., Azzari C., Cardinale F., Cazzola G., Consolini R., De Mattia D., Dell'Erba G., Italian Pediatric Group for XLA-AIEOP Clinical, immunological, and molecular analysis in a large cohort of patients with X-linked agammaglobulinemia: An Italian multicenter study. Clin. Immunol. 2002;104:221–230. doi: 10.1006/clim.2002.5241. [DOI] [PubMed] [Google Scholar]
- 2.Vetrie D., Vorechovský I., Sideras P., Holland J., Davies A., Flinter F., Hammarström L., Kinnon C., Levinsky R., Bobrow M. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361:226–233. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 3.Conley M.E., Broides A., Hernandez-Trujillo V., Howard V., Kanegane H., Miyawaki T., Shurtleff S.A. Genetic analysis of patients with defects in early B-cell development. Immunol. Rev. 2005;203:216–234. doi: 10.1111/j.0105-2896.2005.00233.x. [DOI] [PubMed] [Google Scholar]
- 4.Conley M.E., Dobbs A.K., Quintana A.M., Bosompem A., Wang Y.-D., Coustan-Smith E., Smith A.M., Perez E.E., Murray P.J. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. J. Exp. Med. 2012;209:463–470. doi: 10.1084/jem.20112533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Burg M., Gennery A.R. Educational paper. The expanding clinical and immunological spectrum of severe combined immunodeficiency. Eur. J. Pediatr. 2011;170:561–571. doi: 10.1007/s00431-011-1452-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ehrlich M., Sanchez C., Shao C., Nishiyama R., Kehrl J., Kuick R., Kubota T., Hanash S.M. ICF, an immunodeficiency syndrome: DNA methyltransferase 3B involvement, chromosome anomalies, and gene dysregulation. Autoimmunity. 2008;41:253–271. doi: 10.1080/08916930802024202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Durandy A., Peron S., Fischer A. Hyper-IgM syndromes. Curr. Opin. Rheumatol. 2006;18:369–376. doi: 10.1097/01.bor.0000231905.12172.b5. [DOI] [PubMed] [Google Scholar]
- 8.Yong P.F., Thaventhiran J.E., Grimbacher B. “A rose is a rose is a rose,” but CVID is Not CVID common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:47–107. doi: 10.1016/B978-0-12-385991-4.00002-7. [DOI] [PubMed] [Google Scholar]
- 9.Cunningham-Rundles C., Bodian C. Common variable immunodeficiency: Clinical and immunological features of 248 patients. Clin. Immunol. 1999;92:34–48. doi: 10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- 10.Quinti I., Soresina A., Spadaro G., Martino S., Donnanno S., Agostini C., Claudio P., Franco D., Maria Pesce A., Borghese F., Italian Primary Immunodeficiency Network Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J. Clin. Immunol. 2007;27:308–316. doi: 10.1007/s10875-007-9075-1. [DOI] [PubMed] [Google Scholar]
- 11.Wehr C., Kivioja T., Schmitt C., Ferry B., Witte T., Eren E., Vlkova M., Hernandez M., Detkova D., Bos P.R. The EUROclass trial: Defining subgroups in common variable immunodeficiency. Blood. 2008;111:77–85. doi: 10.1182/blood-2007-06-091744. [DOI] [PubMed] [Google Scholar]
- 12.Vorechovský I., Zetterquist H., Paganelli R., Koskinen S., Webster A.D.B., Björkander J., Smith C.I.E., Hammarström L. Family and linkage study of selective IgA deficiency and common variable immunodeficiency. Clin. Immunol. Immunopathol. 1995;77:185–192. doi: 10.1006/clin.1995.1142. [DOI] [PubMed] [Google Scholar]
- 13.van Zelm M.C., Reisli I., van der Burg M., Castaño D., van Noesel C.J.M., van Tol M.J.D., Woellner C., Grimbacher B., Patiño P.J., van Dongen J.J.M., Franco J.L. An antibody-deficiency syndrome due to mutations in the CD19 gene. N. Engl. J. Med. 2006;354:1901–1912. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- 14.Kuijpers T.W., Bende R.J., Baars P.A., Grummels A., Derks I.A.M., Dolman K.M., Beaumont T., Tedder T.F., van Noesel C.J.M., Eldering E., van Lier R.A. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J. Clin. Invest. 2010;120:214–222. doi: 10.1172/JCI40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Zelm M.C., Smet J., Adams B., Mascart F., Schandené L., Janssen F., Ferster A., Kuo C.-C., Levy S., van Dongen J.J.M., van der Burg M. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J. Clin. Invest. 2010;120:1265–1274. doi: 10.1172/JCI39748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiel J., Kimmig L., Salzer U., Grudzien M., Lebrecht D., Hagena T., Draeger R., Völxen N., Bergbreiter A., Jennings S. Genetic CD21 deficiency is associated with hypogammaglobulinemia. J. Allergy Clin. Immunol. 2012;129 doi: 10.1016/j.jaci.2011.09.027. 801–810.e6. [DOI] [PubMed] [Google Scholar]
- 17.Warnatz K., Salzer U., Rizzi M., Fischer B., Gutenberger S., Böhm J., Kienzler A.K., Pan-Hammarström Q., Hammarström L., Rakhmanov M. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc. Natl. Acad. Sci. USA. 2009;106:13945–13950. doi: 10.1073/pnas.0903543106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grimbacher B., Hutloff A., Schlesier M., Glocker E., Warnatz K., Dräger R., Eibel H., Fischer B., Schäffer A.A., Mages H.W. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat. Immunol. 2003;4:261–268. doi: 10.1038/ni902. [DOI] [PubMed] [Google Scholar]
- 19.Salzer U., Chapel H.M., Webster A.D.B., Pan-Hammarström Q., Schmitt-Graeff A., Schlesier M., Peter H.H., Rockstroh J.K., Schneider P., Schäffer A.A. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat. Genet. 2005;37:820–828. doi: 10.1038/ng1600. [DOI] [PubMed] [Google Scholar]
- 20.Castigli E., Wilson S.A., Garibyan L., Rachid R., Bonilla F., Schneider L., Geha R.S. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat. Genet. 2005;37:829–834. doi: 10.1038/ng1601. [DOI] [PubMed] [Google Scholar]
- 21.Pan-Hammarström Q., Salzer U., Du L., Björkander J., Cunningham-Rundles C., Nelson D.L., Bacchelli C., Gaspar H.B., Offer S., Behrens T.W. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat. Genet. 2007;39:429–430. doi: 10.1038/ng0407-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sekine H., Ferreira R.C., Pan-Hammarström Q., Graham R.R., Ziemba B., de Vries S.S., Liu J., Hippen K., Koeuth T., Ortmann W. Role for Msh5 in the regulation of Ig class switch recombination. Proc. Natl. Acad. Sci. USA. 2007;104:7193–7198. doi: 10.1073/pnas.0700815104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salzer U., Bacchelli C., Buckridge S., Pan-Hammarström Q., Jennings S., Lougaris V., Bergbreiter A., Hagena T., Birmelin J., Plebani A. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009;113:1967–1976. doi: 10.1182/blood-2008-02-141937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finck A., Van der Meer J.W.M., Schäffer A.A., Pfannstiel J., Fieschi C., Plebani A., Webster A.D.B., Hammarström L., Grimbacher B. Linkage of autosomal-dominant common variable immunodeficiency to chromosome 4q. Eur. J. Hum. Genet. 2006;14:867–875. doi: 10.1038/sj.ejhg.5201634. [DOI] [PubMed] [Google Scholar]
- 25.Schäffer A.A., Pfannstiel J., Webster A.D.B., Plebani A., Hammarström L., Grimbacher B. Analysis of families with common variable immunodeficiency (CVID) and IgA deficiency suggests linkage of CVID to chromosome 16q. Hum. Genet. 2006;118:725–729. doi: 10.1007/s00439-005-0101-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J.-W., Gamsby J.J., Highfill S.L., Mora L.B., Bloom G.C., Yeatman T.J., Pan T.C., Ramne A.L., Chodosh L.A., Cress W.D. Deregulated expression of LRBA facilitates cancer cell growth. Oncogene. 2004;23:4089–4097. doi: 10.1038/sj.onc.1207567. [DOI] [PubMed] [Google Scholar]
- 27.Glocker E.-O., Hennigs A., Nabavi M., Schäffer A.A., Woellner C., Salzer U., Pfeifer D., Veelken H., Warnatz K., Tahami F. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 2009;361:1727–1735. doi: 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cámara Y., Asin-Cayuela J., Park C.B., Metodiev M.D., Shi Y., Ruzzenente B., Kukat C., Habermann B., Wibom R., Hultenby K. MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 2011;13:527–539. doi: 10.1016/j.cmet.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 29.Comans-Bitter W.M., de Groot R., van den Beemd R., Neijens H.J., Hop W.C.J., Groeneveld K., Hooijkaas H., van Dongen J.J.M. Immunophenotyping of blood lymphocytes in childhood. Reference values for lymphocyte subpopulations. J. Pediatr. 1997;130:388–393. doi: 10.1016/s0022-3476(97)70200-2. [DOI] [PubMed] [Google Scholar]
- 30.Shearer W.T., Rosenblatt H.M., Gelman R.S., Oyomopito R., Plaeger S., Stiehm E.R., Wara D.W., Douglas S.D., Luzuriaga K., McFarland E.J., Pediatric AIDS Clinical Trials Group Lymphocyte subsets in healthy children from birth through 18 years of age: The Pediatric AIDS Clinical Trials Group P1009 study. J. Allergy Clin. Immunol. 2003;112:973–980. doi: 10.1016/j.jaci.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 31.Aghamohammadi A., Mohammadi J., Parvaneh N., Rezaei N., Moin M., Español T., Hammarström L. Progression of selective IgA deficiency to common variable immunodeficiency. Int. Arch. Allergy Immunol. 2008;147:87–92. doi: 10.1159/000135694. [DOI] [PubMed] [Google Scholar]
- 32.Ng P.C., Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mortensen M., Ferguson D.J., Edelmann M., Kessler B., Morten K.J., Komatsu M., Simon A.K. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc. Natl. Acad. Sci. USA. 2010;107:832–837. doi: 10.1073/pnas.0913170107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ballabio A. Disease pathogenesis explained by basic science: Lysosomal storage diseases as autophagocytic disorders. Int. J. Clin. Pharmacol. Ther. 2009;47(Suppl 1):S34–S38. doi: 10.5414/cpp47034. [DOI] [PubMed] [Google Scholar]
- 36.Phadwal K., Alegre-Abarrategui J., Watson A.S., Pike L., Anbalagan S., Hammond E.M., Wade-Martins R., McMichae A., Kleneman P., Simon A.K. A novel method for autophagy detection in primary cells: Impaired levels of macroautophagy in immunosenescent T cells. Autophagy. 2012;8 doi: 10.4161/auto.18935. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsang W.H., Shek K.F., Lee T.Y., Chow K.L. An evolutionarily conserved nested gene pair - Mab21 and Lrba/Nbea in metazoan. Genomics. 2009;94:177–187. doi: 10.1016/j.ygeno.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 38.Sjöblom T., Jones S., Wood L.D., Parsons D.W., Lin J., Barber T.D., Mandelker D., Leary R.J., Ptak J., Silliman N. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 39.Wang J.-W., Howson J., Haller E., Kerr W.G. Identification of a novel lipopolysaccharide-inducible gene with key features of both A kinase anchor proteins and chs1/beige proteins. J. Immunol. 2001;166:4586–4595. doi: 10.4049/jimmunol.166.7.4586. [DOI] [PubMed] [Google Scholar]
- 40.Feuchter A.E., Freeman J.D., Mager D.L. Strategy for detecting cellular transcripts promoted by human endogenous long terminal repeats: Identification of a novel gene (CDC4L) with homology to yeast CDC4. Genomics. 1992;13:1237–1246. doi: 10.1016/0888-7543(92)90041-p. [DOI] [PubMed] [Google Scholar]
- 41.Dyomin V.G., Chaganti S.R., Dyomina K., Palanisamy N., Murty V.V.V.S., Dalla-Favera R., Chaganti R.S.K. BCL8 is a novel, evolutionarily conserved human gene family encoding proteins with presumptive protein kinase A anchoring function. Genomics. 2002;80:158–165. doi: 10.1006/geno.2002.6822. [DOI] [PubMed] [Google Scholar]
- 42.Kaplan J., De Domenico I., Ward D.M. Chediak-Higashi syndrome. Curr. Opin. Hematol. 2008;15:22–29. doi: 10.1097/MOH.0b013e3282f2bcce. [DOI] [PubMed] [Google Scholar]
- 43.Elin R.J., Edelin J.B., Wolff S.M. Infection and immunoglobulin concentrations in Chediak-Higashi mice. Infect. Immun. 1974;10:88–91. doi: 10.1128/iai.10.1.88-91.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farhoudi A., Chavoshzadeh Z., Pourpak Z., Izadyar M., Gharagozlou M., Movahedi M., Aghamohamadi A., Mirsaeid Ghazi B., Moin M., Rezaei N. Report of six cases of Chediak-Higashi syndrome with regard to clinical and laboratory findings. Iran. J. Allergy Asthma Immunol. 2003;2:189–192. [PubMed] [Google Scholar]
- 45.Adam-Klages S., Adam D., Wiegmann K., Struve S., Kolanus W., Schneider-Mergener J., Krönke M. FAN, a novel WD-repeat protein, couples the p55 TNF-receptor to neutral sphingomyelinase. Cell. 1996;86:937–947. doi: 10.1016/s0092-8674(00)80169-5. [DOI] [PubMed] [Google Scholar]
- 46.Ségui B., Andrieu-Abadie N., Adam-Klages S., Meilhac O., Kreder D., Garcia V., Bruno A.P., Jaffrézou J.-P., Salvayre R., Krönke M., Levade T. CD40 signals apoptosis through FAN-regulated activation of the sphingomyelin-ceramide pathway. J. Biol. Chem. 1999;274:37251–37258. doi: 10.1074/jbc.274.52.37251. [DOI] [PubMed] [Google Scholar]
- 47.Wu C., Orozco C., Boyer J., Leglise M., Goodale J., Batalov S., Hodge C.L., Haase J., Janes J., Huss J.W., 3rd, Su A.I. BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su Y., Balice-Gordon R.J., Hess D.M., Landsman D.S., Minarcik J., Golden J., Hurwitz I., Liebhaber S.A., Cooke N.E. Neurobeachin is essential for neuromuscular synaptic transmission. J. Neurosci. 2004;24:3627–3636. doi: 10.1523/JNEUROSCI.4644-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Lozanne A. The role of BEACH proteins in Dictyostelium. Traffic. 2003;4:6–12. doi: 10.1034/j.1600-0854.2003.40102.x. [DOI] [PubMed] [Google Scholar]
- 50.Gunay-Aygun M., Falik-Zaccai T.C., Vilboux T., Zivony-Elboum Y., Gumruk F., Cetin M., Khayat M., Boerkoel C.F., Kfir N., Huang Y. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet α-granules. Nat. Genet. 2011;43:732–734. doi: 10.1038/ng.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Albers C.A., Cvejic A., Favier R., Bouwmans E.E., Alessi M.-C., Bertone P., Jordan G., Kettleborough R.N.W., Kiddle G., Kostadima M. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat. Genet. 2011;43:735–737. doi: 10.1038/ng.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kahr W.H.A., Hinckley J., Li L., Schwertz H., Christensen H., Rowley J.W., Pluthero F.G., Urban D., Fabbro S., Nixon B. Mutations in NBEAL2 encoding a BEACH protein, cause gray platelet syndrome. Nat. Genet. 2011;43:738–740. doi: 10.1038/ng.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huynh C., Roth D., Ward D.M., Kaplan J., Andrews N.W. Defective lysosomal exocytosis and plasma membrane repair in Chediak-Higashi/beige cells. Proc. Natl. Acad. Sci. USA. 2004;101:16795–16800. doi: 10.1073/pnas.0405905101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang X., Herberg F.W., Laue M.M., Wüllner C., Hu B., Petrasch-Parwez E., Kilimann M.W. Neurobeachin: A protein kinase A-anchoring, beige/Chediak-Higashi protein homolog implicated in neuronal membrane traffic. J. Neurosci. 2000;20:8551–8565. doi: 10.1523/JNEUROSCI.20-23-08551.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pidoux G., Taskén K. Specificity and spatial dynamics of protein kinase A signaling organized by A-kinase-anchoring proteins. J. Mol. Endocrinol. 2010;44:271–284. doi: 10.1677/JME-10-0010. [DOI] [PubMed] [Google Scholar]
- 56.Xu C., Min J. Structure and function of WD40 domain proteins. Protein Cell. 2011;2:202–214. doi: 10.1007/s13238-011-1018-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jogl G., Shen Y., Gebauer D., Li J., Wiegmann K., Kashkar H., Krönke M., Tong L. Crystal structure of the BEACH domain reveals an unusual fold and extensive association with a novel PH domain. EMBO J. 2002;21:4785–4795. doi: 10.1093/emboj/cdf502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Warren H.S. Target-induced natural killer cell loss as a measure of NK cell responses. J. Immunol. Methods. 2011;370:86–92. doi: 10.1016/j.jim.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 59.LeBien T.W., Tedder T.F. B lymphocytes: How they develop and function. Blood. 2008;112:1570–1580. doi: 10.1182/blood-2008-02-078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller B.C., Zhao Z., Stephenson L.M., Cadwell K., Pua H.H., Lee H.K., Mizushima N.N., Iwasaki A., He Y.W., Swat W., Virgin H.W., 4th The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4:309–314. doi: 10.4161/auto.5474. [DOI] [PubMed] [Google Scholar]
- 61.Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Malicdan M.C., Noguchi S., Nonaka I., Saftig P., Nishino I. Lysosomal myopathies: An excessive build-up in autophagosomes is too much to handle. Neuromuscul. Disord. 2008;18:521–529. doi: 10.1016/j.nmd.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 63.Shea L., Raben N. Autophagy in skeletal muscle: Implications for Pompe disease. Int. J. Clin. Pharmacol. Ther. 2009;47(Suppl 1):S42–S47. doi: 10.5414/cpp47042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schapira A.H., Jenner P. Etiology and pathogenesis of Parkinson's disease. Mov. Disord. 2011;26:1049–1055. doi: 10.1002/mds.23732. [DOI] [PubMed] [Google Scholar]
- 65.Qu X., Yu J., Bhagat G., Furuya N., Hibshoosh H., Troxel A., Rosen J., Eskelinen E.-L., Mizushima N., Ohsumi Y. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Nierop K., Muller F.J.M., Stap J., Van Noorden C.J.F., van Eijk M., de Groot C. Lysosomal destabilization contributes to apoptosis of germinal center B-lymphocytes. J. Histochem. Cytochem. 2006;54:1425–1435. doi: 10.1369/jhc.6A6967.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maiuri M.C., Criollo A., Tasdemir E., Vicencio J.M., Tajeddine N., Hickman J.A., Geneste O., Kroemer G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007;3:374–376. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 68.de Souza N., Vallier L.G., Fares H., Greenwald I. SEL-2, the C. elegans neurobeachin/LRBA homolog, is a negative regulator of lin-12/Notch activity and affects endosomal traffic in polarized epithelial cells. Development. 2007;134:691–702. doi: 10.1242/dev.02767. [DOI] [PubMed] [Google Scholar]
- 69.Lim A., Kraut R. The Drosophila BEACH family protein, blue cheese, links lysosomal axon transport with motor neuron degeneration. J. Neurosci. 2009;29:951–963. doi: 10.1523/JNEUROSCI.2582-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hayashi-Nishino M., Fujita N., Noda T., Yamaguchi A., Yoshimori T., Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 71.Cuervo A.M. The plasma membrane brings autophagosomes to life. Nat. Cell Biol. 2010;12:735–737. doi: 10.1038/ncb0810-735. [DOI] [PubMed] [Google Scholar]
- 72.Bernales S., Schuck S., Walter P. ER-phagy: Selective autophagy of the endoplasmic reticulum. Autophagy. 2007;3:285–287. doi: 10.4161/auto.3930. [DOI] [PubMed] [Google Scholar]
- 73.Popovic D., Akutsu M., Novak I., Harper W., Behrends C., Dikic I. Rab GAPase-Activating Proteins in Autophagy: Regulation of Endocytic and Autophagy Pathways by Direct Binding to Human ATG8 modifiers. Mol. Cell. Biol. 2012;32:1733–1744. doi: 10.1128/MCB.06717-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nishida Y., Arakawa S., Fujitani K., Yamaguchi H., Mizuta T., Kanaseki T., Komatsu M., Otsu K., Tsujimoto Y., Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 75.Ohashi Y., Munro S. Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol Biol Cell. 2010;21:3998–4008. doi: 10.1091/mbc.E10-05-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.