

Abstract
Congenital lipomatous overgrowth with vascular, epidermal, and skeletal anomalies (CLOVES) is a sporadically occurring, nonhereditary disorder characterized by asymmetric somatic hypertrophy and anomalies in multiple organs. We hypothesized that CLOVES syndrome would be caused by a somatic mutation arising during early embryonic development. Therefore, we employed massively parallel sequencing to search for somatic mosaic mutations in fresh, frozen, or fixed archival tissue from six affected individuals. We identified mutations in PIK3CA in all six individuals, and mutant allele frequencies ranged from 3% to 30% in affected tissue from multiple embryonic lineages. Interestingly, these same mutations have been identified in cancer cells, in which they increase phosphoinositide-3-kinase activity. We conclude that CLOVES is caused by postzygotic activating mutations in PIK3CA. The application of similar sequencing strategies will probably identify additional genetic causes for sporadically occurring, nonheritable malformations.
Main Text
Syndromes can be genetic in origin but not necessarily heritable. Happle postulated that such disorders arise as the result of somatic rather than germline mutations, i.e., because complete heterozygosity for a causative mutation would either be lethal to the affected individual or be incapable of transmission through egg or sperm.1 Happle's hypothesis was confirmed by the discovery of somatic mosaic mutations in several diseases, including McCune-Albright syndrome (MIM 174800) and Proteus syndrome (MIM 176920).2,3
Isolated malformations and nonhereditary syndromes with malformation as a component feature could also be caused by a somatic mutation. One such candidate is the recently described disorder CLOVES (congenital lipomatous asymmetric overgrowth of the trunk with lymphatic, capillary, venous, and combined-type vascular malformations, epidermal nevi, and skeletal anomalies [MIM 612918]) (Figure 1).4–8 Therefore, we sought to identify postzygotic mutations in individuals with CLOVES by employing massively parallel sequencing of DNA and RNA from affected tissue to look for mutations that are present at low frequencies. The study was approved by the institutional review board at Boston Children's Hospital. Study participants provided written informed consent to participate in the study and to authorize the publication of clinical images.
Figure 1.
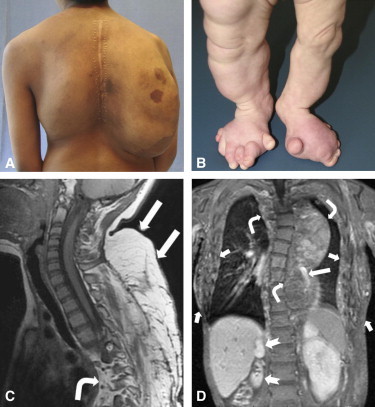
Clinical Features of CLOVES Syndrome
(A) Participant CL5 at age 15 years. Note large, bilateral, posterior thoracic fatty masses with overlying capillary malformation on the right side.
(B) Participant CL2 at age 18 months. She has overgrowth of her lower extremities, polydactyly, and wide feet with an expanded first interdigital space.
(C) A sagittal T1-MRI (magnetic resonance image) of participant CL5 demonstrates cervicothoracic lipomatous overgrowth (straight arrows) extending into the posterior mediastinum and paraspinal region (bent arrow) and scoliosis.
(D) A coronal postcontrast T1-MRI of participant CL3 shows bilateral truncal (short arrows) and mediastinal (bent arrows) fatty overgrowth, phlebectasia (long arrow), scoliosis, and asymmetrical kidneys due to right renal hypoplasia (notched arrows).
We used fresh or frozen affected tissue from four CLOVES-affected individuals, each of whom had undergone resection of a lipomatous overgrowth or a vascular malformation with lipomatous overgrowth (Tables 1 and 2), and we produced barcoded DNA sequencing libraries as described previously.9 We assumed that lipomatous tissue would contain mutant cells given that incompletely resected lesions often regrow.5 We also recovered mRNA and generated barcoded cDNA sequencing libraries10 from these same specimens. For two of these individuals, we prepared DNA sequencing libraries from white blood cell (blood or saliva) DNA. We assumed that this unaffected tissue would not contain a CLOVES causative mutation. Formalin-fixed paraffin-embedded tissue blocks were available from two other individuals (Table 2) from whom we recovered DNA by using thin sections to produce additional barcoded sequencing libraries (see Figure S1, available online, for the experimental design).9 Saliva DNA was available from one of these individuals and was used for the preparation of a library.
Table 1.
Summary of Participants with CLOVES Syndrome
| CL1 | CL2 | CL3 | CL4 | CL5 | CL6 | |
|---|---|---|---|---|---|---|
| Age (years)a | 2 | 1 | 14 | 1 | 15 | 18 |
| Sex | male | female | female | male | male | female |
| PIK3CA mutation | c.3140A>G (p.His1047Arg) | c.3140A>G (p.His1047Arg) | c.1624G>A (p.Glu542Lys) | c.1624G>A (p.Glu542Lys) | c.1258T>C (p.Cys420Arg) | c.1258T>C (p.Cys420Arg) |
| Lipomatous overgrowth | ||||||
| Trunk | + | + | + | + | + | + |
| Limb(s) | – | + | + | + | – | + |
| Vascular anomalies | ||||||
| Lymphatic malformation | + | – | + | + | NA | + |
| Capillary malformation | NA | NA | + | + | + | + |
| Venous malformation | + | + | + | + | + | + |
| Fast-flow malformation | – | NA | NA | – | + | – |
| Musculoskeletal | ||||||
| Wide hands or feet | – | + | + | + | + | + |
| Foot “sandal-gap” deformity | + | + | – | + | – | + |
| Macrodactyly | – | + | + | + | + | + |
| Limb asymmetry | NA | + | + | + | + | + |
| Paraspinal mass | NA | – | NA | NA | + | NA |
| Scoliosis | + | – | + | – | + | + |
| Renal | NA | hypoplastic left kidney and Wilms tumor | hypoplastic right kidney | – | – | NA |
| Other findings | rib anomalies and pterygium coli | NA | NA | NA | NA | abnormal marginal venous system and splenomegaly |
The following abbreviation is used: NA, not available.
At the time the tissue sample was obtained.
Table 2.
Summary of PIK3CA Mutations in Participants with CLOVES Syndrome
| Subject | Identified Mutation | Lesional Tissue |
Pathologic Components |
DNA Source |
Mutant Allele Frequency |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lipomatous Overgrowth | Vascular Component | Massively Parallel DNA Sequence | Genomic DNA PCR Subclones | RT-PCR Subclonesa | |||||||
| CL1 | c.3140A>G (p.His1047Arg) | resected truncal mass | yes | – | FFPE | 5/24 | (21%) | 8/49 | (16%) | NA | NA |
| CL2 | c.3140A>G (p.His1047Arg) | debulked tissue from both feet | yes | – | FFPE | 13/64 | (20%) | 16/70 | (23%) | NA | NA |
| saliva | 0/19 | (0%) | 0/69 | (0%) | |||||||
| CL3 | c.1624G>A (p.Glu542Lys) | resected truncal mass | yes | LM | frozen | 1/13 | (8%) | 4/48 | (8%) | 0/80 | (0%) |
| CL4 | c.1624G>A (p.Glu542Lys) | resected buttock and thigh mass | yes | combined LM and VM | frozen | 2/16 | (13%) | 3/48 | (6%) | 5/48 | (10%) |
| blood | 0/18 | (0%) | 0/40 | (0%) | |||||||
| CL5 | c.1258T>C (p.Cys420Arg) | resected truncal mass | yes | combined AVM | fresh | 8/44 | (18%) | 2/70 | (3%) | 16/62 | (26%) |
| blood | NA | NA | 0/68 | (0%) | |||||||
| CL6 | c.1258T>C (p.Cys420Arg) | left-leg amputation below the knee | yes | combined LM and VM | fresh | 12/40 | (30%) | 8/70 | (11%) | 23/65 | (35%) |
| saliva | 0/55 | (0%) | 0/70 | (0%) | |||||||
The following abbreviations are used: FFPE, formalin-fixed paraffin-embedded; NA, not available; LM, lymphatic malformation; VM, venous malformation; and AVM, arteriovenous malformation.
cDNA prepared from the same RNA stock that was used for massively parallel RNA sequencing.
We enriched each DNA library generated from the fresh or frozen samples for exonic sequences by using the SureSelect human exome kit (Agilent Technologies, Santa Clara, CA, USA). For DNA libraries generated from the paraffin tissue blocks, we employed a custom-designed enrichment array that contained exonic sequences from 77 genes involved in signaling pathways for several growth factors. We had previously employed this targeted array to screen individuals with metachondromatosis.9 Several genes included in this array, e.g., PTEN (MIM 601728), AKT1 (MIM 164730), AKT2 (MIM 164731), and AKT3 (MIM 611223), have been implicated in other overgrowth syndromes.3,11–14 We performed RNA sequencing in case the gene responsible for CLOVES was abundantly transcribed in affected tissue. We reasoned that detecting low-level mosaicism would be easier in an abundantly expressed transcript for which read depth can be greater than 200× as compared to only 20× for a whole-exome sequence.
After we employed massively parallel sequencing and filtering to remove PCR duplicates, we found that the whole-exome capture data provided >20× coverage for 85% of the exome for three samples and for 50% of the exome for one sample. The 77 gene capture array sequence yielded >20× coverage for 95% of the array for both samples. RNA-seq provided >20× coverage for the ∼2,500 most abundantly expressed transcripts. We next filtered the data to remove SNPs that were present in dbSNP build 132, the 1000 Genomes Project, or the National Heart, Lung, and Blood Institute (NHBLI) whole-exome database (Table S1). We then filtered for variants present in greater than 5% of reads in affected tissue and ranked these variants with respect to the fold coverage for that nucleotide. For example, we ranked a variant that was present in 3 of 50 reads (6%) higher than a variant present in one of five reads (20%) by assuming that the former was more likely to be a true positive and that the latter was more likely to be a false-positive sequencing error. Finally, we focused on highly ranked variants that either were solely observed in the affected tissue or were more abundant in affected tissue than in unaffected tissue (blood or saliva).
Each CLOVES-affected individual for whom DNA from fresh or frozen affected tissue was sequenced had a missense PIK3CA (MIM 171834) mutation that was not present in the blood or saliva DNA sequence (when available). Participants CL3 and CL4 had a c.1624G>A (p.Glu542Lys) mutation, and participants CL5 and CL6 had a c.1258T>C (p.Cys420Arg) mutation based on RefSeq NM_006218.2 (Figure 2 and Table 2). PIK3CA was among the 77 genes included in the targeted-capture array. Individuals CL1 and CL2, whose paraffin DNA samples were used in that array, both had a c.3140A>G (p.His1047Arg) missense mutation in PIK3CA (Table 2). The PIK3CA sequence was poorly represented in the RNA sequence data (<2× coverage), and missense mutations were not found (Table S2). Nevertheless, we detected in three participants the same mutations observed in the whole-exome sequence data when we performed gene-specific RT-PCR of PIK3CA by using total RNA from frozen affected tissue as the template. We confirmed that all mutations detected by massively parallel sequencing were present in the participants by reanalyzing the original tissue samples and by PCR amplifying, subcloning, and sequencing individual amplimers (Figure 2, Table 2, and Table S3).
Figure 2.
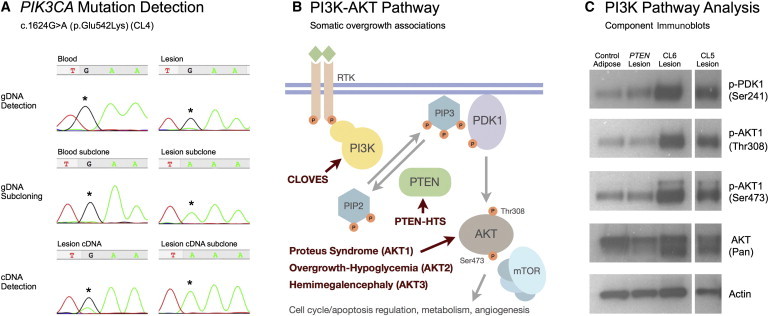
Somatic Activating PIK3CA Mutations in CLOVES Syndrome
(A) Demonstration of the PIK3CA c.1624G>A somatic mosaic mutation in participant CL4. Massively parallel sequencing of this individual's lesional tissue identified a c.1624G>A mutation in 2 of 16 reads (Table 2). With blood DNA and lesional-tissue DNA as templates, PCR amplimers encompassing the candidate mutation were generated and Sanger sequenced. On the top left is an electropherogram of blood-DNA amplimers showing only a wild-type sequence. On the top right is an electropherogram of lesional-tissue amplimers showing wild-type and mutant sequences. Blood and lesional-tissue amplimers were subcloned, and 48 individual colonies were sequenced. In the middle on the left, all subclones from blood-DNA amplimers contain a wild-type sequence; a representative electropherogram of a wild-type sequence from a single clone is shown. In the middle on the right, 3 of 48 subclones from lesional-DNA amplimers contain a mutant sequence; a representative electropherogram of a mutant sequence from a single clone is shown. With mRNA recovered from lesional tissue as a template, RT-PCR was performed with primers in exons flanking the exon containing the candidate mutation. On the bottom left, a Sanger-sequence electropherogram of the RT-PCR amplimers shows wild-type and mutant sequences. Five of the 48 subclones from the RT-PCR amplimers contain a mutant sequence, one of which is shown at the bottom right.
(B) A schematic of the PI3K-AKT signaling pathway indicates the overgrowth syndromes currently associated with mutations in this pathway. Binding of a growth factor to a receptor-tyrosine kinase activates a PI3K family member, including PIK3CA, which converts phosphatidylinositol-3,4-bisphosphate (PIP2) to the −3,4,5-triphosphate (PIP3) in a reaction that is antagonized by PTEN. Membrane-associated PIP3 facilitates the localization and phosphorylation of PDK1, which then activates AKT by phosphorylation at Thr308. AKT is further activated by Ser742 phosphorylation by the PDK2 complex including mTOR (FRAP1 [MIM 601231]). The following abbreviation is used: PTEN-HTS, PTEN Hamartoma Tumor Syndrome.
(C) PIK3CA mutations constitutively activate the PI3K-AKT pathway. In the first column are immunoblot luminescence images of protein lysates prepared from normal subcutaneous adipose tissue. In the second column is hamartomatous tissue from an individual with a known PTEN mutation. In the right columns is lipomatous tissue from CLOVES-affected participants CL5 and CL6 (the blank lane between samples was removed). Lysates were separated by 4%–8% SDS-PAGE, transferred to immobilon P, and immunodetected with antibodies (Cell Signaling, Cambridge, MA, USA) that recognize total AKT, phosphorylated forms of PDK1 and AKT1, and beta-actin (as a loading control). Compared to lysates from adipose tissue from an unaffected individual or lysates from lesional tissue from an individual with a heterozygous PTEN mutation, lysates from the CLOVES lipomatous tissue show marked increases in the activated forms of PDK1 and AKT1.
One individual with CLOVES syndrome required lower-extremity amputation. Thus, we collected fresh lipomatous tissue from which we separated adipocytes from fibroblasts and vascular endothelial cells. We also recovered DNA from several affected tissues, including a cutaneous lymphatic malformation and a marginal vein, in the amputated limb. We found that the purified adipocytes from the lipomatous tissue and each of the affected tissue samples were mosaic for the same mutant allele (Figure 3).
Figure 3.
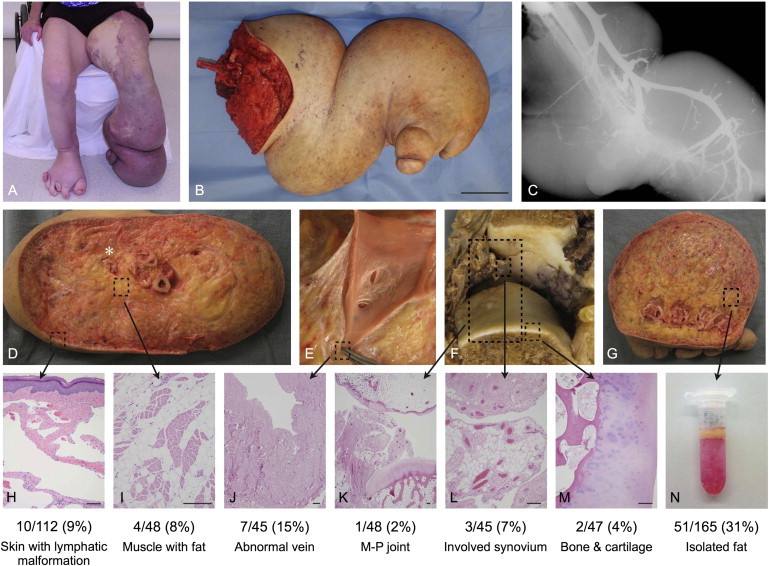
Detection of PIK3CA Somatic Mosaicism in Multiple Tissue Types
(A) At 18 years of age, participant CL6 shows overgrowth of the lower limbs, right-foot polydactyly, and lymphatic and venous anomalies of the lower-left extremity.
(B) The participant's resected lower limb. The scale bar represents 10 cm.
(C) The participant's radiograph (after venous contrast injection) demonstrates a dilated and aberrant venous system.
(D–M) Resection specimen and photomicrographs of hematoxylin- and eosin-stained sections from sampled areas used for DNA isolation. The locations of tissue sampling are indicated by boxes. The scale bar indicates 200 μm.
(D) A transverse section at the base of the calf shows massive lipomatous overgrowth involving subcutaneous tissue and skeletal muscle (I) and obliteration of tissue planes. The stranded appearance of subcutaneous tissue is due to extensive lymphatic malformation (H). The asterisk shows an abnormally dilated vein that was dissected (E) and sampled (J).
(F) A metatarsal-phalangeal joint with degenerative articular changes (top) and synovial expansion by fat and venous malformation. Also shown are sampled sections of the total joint (K), synovium with fat and venous malformation (L), and articular cartilage and bone (M).
(G) A transmetatarsal section shows extensive involvement of lipomatous overgrowth and lymphatic malformation.
(N) A tube containing adipocytes (yellow layer) freshly isolated after collagenase treatment by centrifugation.
DNA was amplified from each of the aforementioned tissues. Amplimers were subcloned, and the frequency of mutant alleles was determined. The numbers and percentages of mutant alleles are listed below the corresponding tissue samples.
The PIK3CA mutations we discovered have been previously identified as somatic alterations in several types of cancer.15 Cell biologic studies of these mutations indicate that they activate phosphoinositide-3-kinase (PI3K) activity in the absence of growth-factor signaling.16 Because PI3K activity normally leads to phosphorylation of AKT family members, we extracted protein from two participants' lipomatous overgrowths to compare its level of AKT phosphorylation to that of protein extracted from normal adipose tissue and from an adipose containing PTEN hamartoma of soft tissue (PHOST)12 from an individual with a PTEN mutation. Affected tissue from the participants with CLOVES syndrome had higher levels of AKT phosphorylation than did the other samples (Figure 2).
Massively-parallel-sequencing technologies have facilitated the identification of causes of heritable diseases. Challenges in applying these technologies to sporadically occurring somatic malformations include having sufficient numbers of individuals with the same phenotype and procuring and distinguishing affected from unaffected tissue from these individuals. We successfully identified somatic mutations (for which mutant allele frequencies ranged from 8% to 30% in lesional tissue) in individuals who have CLOVES syndrome by performing exome sequencing of fresh or frozen lesional tissue with paired blood and saliva samples and by performing targeted genomic sequencing of archival paraffin tissue samples. We did not detect mutations in blood or saliva DNA from participants who had mutations in their anomalous tissue. This finding is consistent with earlier studies that demonstrated that constitutive activation of the PI3K-AKT pathway is detrimental to hematopoiesis.17 Selection against somatic mutations in hematopoietic stem cells or their derivatives has been described for other disorders.18 Therefore, discovering the cause of other somatic disorders might require sequencing DNA recovered from the lesional tissue, not deep sequencing of blood-derived DNA.
PIK3CA encodes the 110-kD catalytic alpha subunit of PI3K, which in response to tyrosine kinase receptor ligand binding is activated and converts phosphatidylinositol (3,4)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-triphosphate (PIP3) (Figure 2). This leads to the translocation and phosphorylation of PDK1 (PDPK1 [MIM 605213]) at the cell membrane. PDK1 then phosphorylates AKT to initiate downstream cellular effects. Regulation of this pathway is partly achieved by PTEN, which catalyzes the conversion of PIP3 to PIP2 (Figure 2). Each of the missense mutations we observed in participants with CLOVES syndrome has previously been identified in several types of adult-onset cancer, including cancers of the gastrointestinal tract, brain, breast, bladder, lung, and thyroid.15 Rather than directly causing transformation in these cancers, the PIK3CA missense mutations are thought to enhance the tumor's growth or aggressiveness. The fact that conditional activation of PIK3CA missense mutant alleles in two different mouse cancer models increased the incidence and severity of cancer is consistent with this theory.19,20 At the cellular level, the missense mutations we observed in participants with CLOVES syndrome have been shown to increase the activity of PI3K and lead to an abundance of phosphorylated AKT.16,21 The fact that we observed increased AKT phosphorylation in affected tissues from participants who have CLOVES syndrome is consistent with these findings (Figure 2C). Interestingly, we did not observe increased AKT phosphorylation in the affected tissue from the individual with the PTEN mutation; explanations for the lack of increased AKT phosphorylation in the PTEN lesion compared to the PIK3CA lesions might include a stoichiometric difference between heterozygous PTEN inactivation and PIK3CA activation, a biologic difference between a vascular malformation with adiposity and a lipomatous overgrowth, or a difference in how the different samples had been processed and stored prior to protein extraction.
When overexpressed, PIK3CA missense mutations identified in participants with CLOVES syndrome have the ability to transform cells.21,22 We have detected Wilms tumor (MIM 194070) in two CLOVES-affected individuals, including participant CL2 in this study. Consequently, endogenous expression of missense PIK3CA mutants could be transformative in some human cell types. We hypothesize that the low rate of malignant transformation in individuals with CLOVES syndrome is due to the low level of endogenous PIK3CA expression in most cells. For example, among the four frozen samples in which we performed RNA sequencing, PIK3CA was the 19,000th most abundant RNA transcript. This explains why we did not find PIK3CA mutations in our RNA sequence data and also probably explains the difference in the rate of cell transformation that occurs when mutant PIK3CA levels are overexpressed in vitro versus endogenously expressed in vivo.19,20
An individual in whom we extracted DNA from several different lesions was mosaic for the same PIK3CA mutation at each site. Different cell types were used as the source of DNA from the various locations. For example, we sequenced DNA from adipocytes that had been recovered after dissociation and density centrifugation from a lipomatous lesion and from endothelial, smooth-muscle, and fibrocytic cells from the malformed embryonic marginal venous system (Figure 3). The fact that the same mutant allele was detected at each location suggests that the mutation arose early enough in development so as to affect several cell lineages. Interestingly, the mutant allele frequency was 51/165 (31%) in adipocyte DNA purified from a lipomatous overgrowth. This frequency is significantly lower than the 50% (p < 0.005) expected if all adipocytes within the lesion contained the mutation, suggesting that lipomatous overgrowth can result from paracrine signaling from mutant to wild-type cells. The low mutant allelic frequency we observed in other tissues is also consistent with the hypothesis that several CLOVES malformations are the result of paracrine signaling.
Our findings add CLOVES to a growing list of overgrowth syndromes that result from somatic activation of the PI3K-AKT pathway. Individuals with PTEN loss-of-function mutations (somatic and germline) have asymmetric soft-tissue overgrowth in association with vascular anomalies.11,12 Missense mutations in each of the AKT family members have been reported in individuals with Proteus syndrome (AKT1), individuals with asymmetric overgrowth and hypoglycemia (AKT2), and an individual with hemimegalencephaly (AKT3).3,13,14 The nonoverlapping phenotypes associated with the different mutations might reflect the stage of development at which the genetic alterations arose and the restricted ability of a cell type or tissue to be affected by the consequences. In a PIK3CA activating mutation, it is reasonable to speculate that differing phenotypes in the CLOVES spectrum might occur depending upon when the mutation arises during development; such is the case for the broad range of phenotypes that occur in individuals with somatic mutations in GNAS and PTEN.11,23 Similar to CLOVES syndrome, Klippel-Trenaunay syndrome (KTS [MIM 149000]) exhibits features such as capillary, lymphatic, and venous anomalies with overgrowth.24 Therefore, KTS might also result from mutations in components of the PI3K-AKT pathway. We have begun testing this hypothesis by screening DNA extracted from lesional tissue in individuals with KTS for the c.1258T>C and the c.3140A>G PIK3CA missense mutations, and we have found mosaicism for c.3140A>G in 3 of 15 individuals examined (Figure S2).
Massively parallel sequencing of DNA or RNA recovered from anomalous tissue can facilitate the identification of a somatically arising mutation that is responsible for the disorder. We employed this strategy to identify activating mutations in PIK3CA as the cause of CLOVES syndrome. These same activating mutations have been detected in several types of cancer, and pharmacologic inhibitors of PIK3CA are being developed for the treatment of these tumors.25 These same inhibitors might have therapeutic applications for individuals with CLOVES syndrome or other overgrowth anomalies that are also the result of somatic activating mutations in PIK3CA.
Acknowledgments
We are grateful for the active support of the CLOVES syndrome community (www.clovessyndrome.org and www.clovesfoundation.org) and to the individuals and families that participated in this study. We also acknowledge Cameron Trenor III, Arin Greene, Joseph Upton III, Rebecca Fevurly, Denise Adams, and all of the members of the Vascular Anomalies Center, Boston Children's Hospital for their dedication to the care of individuals with CLOVES and other vascular problems. We thank Mr. Ryan Neff for providing computation assistance, and we thank Joseph Gleeson and colleagues, who shared information about similar findings in individuals with hemimegalencephaly. This work was funded in part by the Manton Center for Orphan Disease Research (pilot grant 94824-01 to K.C.K.) and the Stuart and Jane Weitzman Family Vascular Anomalies Fund at Boston Children's Hospital, the National Institutes of Health-National Institute of Arthritis and Musculoskeletal and Skin Diseases grant AR053237, and the Howard Hughes Medical Institute.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org/index.html
biomaRt, http://www.bioconductor.org/packages/2.2/bioc/html/biomaRt.html
dBSNP Build 132, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi?build_id=132
GATK, http://www.broadinstitute.org/gsa/wiki/index.php/Home_Page
GenomicFeatures, http://www.bioconductor.org/packages/2.9/bioc/html/GenomicFeatures.html
Integrative Genomics Viewer, http://www.broadinstitute.org/igv
NHLBI Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
RNA-Seq Unified Mapper (RUM), http://www.cbil.upenn.edu/RUM/
University of California-Santa Cruz Genome Bioinformatics, http://www.genome.ucsc.edu
References
- 1.Happle R. Lethal genes surviving by mosaicism: A possible explanation for sporadic birth defects involving the skin. J. Am. Acad. Dermatol. 1987;16:899–906. doi: 10.1016/s0190-9622(87)80249-9. [DOI] [PubMed] [Google Scholar]
- 2.Weinstein L.S., Shenker A., Gejman P.V., Merino M.J., Friedman E., Spiegel A.M. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N. Engl. J. Med. 1991;325:1688–1695. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 3.Lindhurst M.J., Sapp J.C., Teer J.K., Johnston J.J., Finn E.M., Peters K., Turner J., Cannons J.L., Bick D., Blakemore L. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N. Engl. J. Med. 2011;365:611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sapp J.C., Turner J.T., van de Kamp J.M., van Dijk F.S., Lowry R.B., Biesecker L.G. Newly delineated syndrome of congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE syndrome) in seven patients. Am. J. Med. Genet. A. 2007;143A:2944–2958. doi: 10.1002/ajmg.a.32023. [DOI] [PubMed] [Google Scholar]
- 5.Alomari A.I. Characterization of a distinct syndrome that associates complex truncal overgrowth, vascular, and acral anomalies: A descriptive study of 18 cases of CLOVES syndrome. Clin. Dysmorphol. 2009;18:1–7. doi: 10.1097/MCD.0b013e328317a716. [DOI] [PubMed] [Google Scholar]
- 6.Alomari A.I. CLOVE(S) syndrome: Expanding the acronym. Am. J. Med. Genet. A. 2009;149A:294–295. doi: 10.1002/ajmg.a.32632. [author reply] [DOI] [PubMed] [Google Scholar]
- 7.Alomari A.I., Burrows P.E., Lee E.Y., Hedequist D.J., Mulliken J.B., Fishman S.J. CLOVES syndrome with thoracic and central phlebectasia: Increased risk of pulmonary embolism. J. Thorac. Cardiovasc. Surg. 2010;140:459–463. doi: 10.1016/j.jtcvs.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 8.Alomari A.I., Chaudry G., Rodesch G., Burrows P.E., Mulliken J.B., Smith E.R., Fishman S.J., Orbach D.B. Complex spinal-paraspinal fast-flow lesions in CLOVES syndrome: Analysis of clinical and imaging findings in 6 patients. AJNR Am. J. Neuroradiol. 2011;32:1812–1817. doi: 10.3174/ajnr.A2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowen M.E., Boyden E.D., Holm I.A., Campos-Xavier B., Bonafé L., Superti-Furga A., Ikegawa S., Cormier-Daire V., Bovée J.V., Pansuriya T.C. Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome. PLoS Genet. 2011;7:e1002050. doi: 10.1371/journal.pgen.1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christodoulou D.C., Gorham J.M., Herman D.S., Seidman J.G. Construction of normalized RNA-seq libraries for next-generation sequencing using the crab duplex-specific nuclease. Curr. Protoc. Mol. Biol. 2011;94:4.12.1–4.12.11. doi: 10.1002/0471142727.mb0412s94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan M.-H., Mester J., Peterson C., Yang Y., Chen J.-L., Rybicki L.A., Milas K., Pederson H., Remzi B., Orloff M.S., Eng C. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet. 2011;88:42–56. doi: 10.1016/j.ajhg.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurek K.C., Howard E., Tennant L.B., Upton J., Alomari A.I., Burrows P.E., Chalache K., Harris D.J., Trenor C.C., 3rd, Eng C. PTEN hamartoma of soft tissue: A distinctive lesion in PTEN syndromes. Am. J. Surg. Pathol. 2012;36:671–687. doi: 10.1097/PAS.0b013e31824dd86c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hussain K., Challis B., Rocha N., Payne F., Minic M., Thompson A., Daly A., Scott C., Harris J., Smillie B.J.L. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334:474. doi: 10.1126/science.1210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poduri A., Evrony G.D., Cai X., Elhosary P.C., Beroukhim R., Lehtinen M.K., Hills L.B., Heinzen E.L., Hill A., Hill R.S. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74:41–48. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samuels Y., Wang Z., Bardelli A., Silliman N., Ptak J., Szabo S., Yan H., Gazdar A., Powell S.M., Riggins G.J. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 16.Kang S., Bader A.G., Vogt P.K. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc. Natl. Acad. Sci. USA. 2005;102:802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kharas M.G., Okabe R., Ganis J.J., Gozo M., Khandan T., Paktinat M., Gilliland D.G., Gritsman K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood. 2010;115:1406–1415. doi: 10.1182/blood-2009-06-229443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu H.H., Ji K., Alderson N., He Z., Li S., Liu W., Zhang D.-E., Li L., Feng G.-S. Kit-Shp2-Kit signaling acts to maintain a functional hematopoietic stem and progenitor cell pool. Blood. 2011;117:5350–5361. doi: 10.1182/blood-2011-01-333476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan W., Stawiski E., Janakiraman V., Chan E., Durinck S., Edgar K.A., Kljavin N.M., Rivers C.S., Gnad F., Roose-Girma M. Conditional activation of Pik3ca(H1047R) in a knock-in mouse model promotes mammary tumorigenesis and emergence of mutations. Oncogene. 2012 doi: 10.1038/onc.2012.53. Published online February 27, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinross K.M., Montgomery K.G., Kleinschmidt M., Waring P., Ivetac I., Tikoo A., Saad M., Hare L., Roh V., Mantamadiotis T. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J. Clin. Invest. 2012;122:553–557. doi: 10.1172/JCI59309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bader A.G.A., Kang S.S., Vogt P.K.P. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc. Natl. Acad. Sci. USA. 2006;103:1475–1479. doi: 10.1073/pnas.0510857103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams J.R., Xu K., Liu J.C., Agamez N.M., Loch A.J., Wong R.G., Wang W., Wright K.L., Lane T.F., Zacksenhaus E. Cooperation between Pik3ca and p53 mutations in mouse mammary tumor formation. Cancer Res. 2011;71:2706–2717. doi: 10.1158/0008-5472.CAN-10-0738. [DOI] [PubMed] [Google Scholar]
- 23.Diaz A., Danon M., Crawford J. McCune-Albright syndrome and disorders due to activating mutations of GNAS1. J. Pediatr. Endocrinol. Metab. 2007;20:853–880. doi: 10.1515/jpem.2007.20.8.853. [DOI] [PubMed] [Google Scholar]
- 24.Cohen M.M., Jr. Klippel-Trenaunay syndrome. Am. J. Med. Genet. 2000;93:171–175. doi: 10.1002/1096-8628(20000731)93:3<171::aid-ajmg1>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y., Wang B.-C., Xiao Y. PI3K: A potential therapeutic target for cancer. J. Cell. Physiol. 2012;227:2818–2821. doi: 10.1002/jcp.23038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.