

Abstract
Cantú syndrome is a rare disorder characterized by congenital hypertrichosis, neonatal macrosomia, a distinct osteochondrodysplasia, and cardiomegaly. Using an exome-sequencing approach applied to one proband-parent trio and three unrelated single cases, we identified heterozygous mutations in ABCC9 in all probands. With the inclusion of the remaining cohort of ten individuals with Cantú syndrome, a total of eleven mutations in ABCC9 were found. The de novo occurrence in all six simplex cases in our cohort substantiates the presence of a dominant disease mechanism. All mutations were missense, and several mutations affect Arg1154. This mutation hot spot lies within the second type 1 transmembrane region of this ATP-binding cassette transporter protein, which may suggest an activating mutation. ABCC9 encodes the sulfonylurea receptor (SUR) that forms ATP-sensitive potassium channels (KATP channels) originally shown in cardiac, skeletal, and smooth muscle. Previously, loss-of-function mutations in this gene have been associated with idiopathic dilated cardiomyopathy type 10 (CMD10). These findings identify the genetic basis of Cantú syndrome and suggest that this is a new member of the potassium channelopathies.
Main Text
Cantú syndrome (MIM 239850) is a rare but recognizable disorder characterized by congenital hypertrichosis, neonatal macrosomia, a distinct osteochondrodysplasia, and cardiomegaly. The hypertrichosis leads to thick scalp hair, which extends onto the forehead, and a general increase in body hair. In addition, macrocephaly and coarse facial features, including a broad nasal bridge, epicanthal folds, a wide mouth, and full lips, can be suggestive of a storage disorder.1–3 About half of individuals with Cantú syndrome are macrosomic and oedematous at birth, whereas in childhood they usually have a muscular appearance with little subcutaneous fat.3 Although the skeletal changes reported in a more recent review of ten individuals were mild,3 a thickened calvarium, narrow thorax, wide ribs, flattened or ovoid vertebral bodies, coxa valga, osteopenia, enlarged medullary canals, and metaphyseal widening of long bones have been reported previously.1,4 Motor development is usually delayed due to hypotonia, most individuals have a mild speech delay, and a small percentage has learning difficulties or intellectual disability. Notably, cardiac manifestations such as patent ductus arteriosus, ventricular hypertrophy, pulmonary hypertension, and pericardial effusions are present in ∼80% of cases.1,3
Despite the identification of Cantú syndrome as a distinct clinical entity ∼30 years ago, its underlying genetic cause has not been identified to date.5 Initially, autosomal-recessive inheritance was suggested on the basis of the two affected siblings in the original report and consanguinity in another family.5,6 However, using segregation analysis, Robertson et al. showed that Cantú syndrome is most likely a dominant disorder.4 In addition, of the 37 cases of Cantú syndrome in the literature, the majority are simplex cases; also, two known parent-child transmissions suggest an autosomal-dominant inheritance pattern.1–12 Except for a distal 1p36 deletion and a 4q26q27 duplication in two individuals with only mild phenotypic similarities to Cantú syndrome, no recurrent chromosomal aberrations in individuals clinically diagnosed with Cantú syndrome have been reported that would allow the identification of the genetic cause.3,7,8
To identify the underlying genetic cause of Cantú syndrome, we studied a cohort of 14 individuals, of which seven were simplex cases and seven were familial cases. The familial cases consisted of two sisters and their mother (individuals 2a, 2b, and 2c), a father-daughter pair (individuals 8a and 8b) and a sibling pair (individuals 9a and 9b). In four families, the parents were consanguineous (individuals 1, 5, 8b, 9a, and 9b). The clinical details of these individuals are presented in Table 1, Figure 1A, and Figures S1A and S1B (available online). The four exome-sequenced individuals were selected from the cohort as those most resembling the phenotype initially outlined by Cantú et al.5
Table 1.
Phenotype of Individuals with Cantú Syndrome
| Clinical Features |
Affected Individuals |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2a | 2b | 2c | 3 | 4 | 5 | 6 | 7 | 8a | 8b | 9a | 9b | 10 | |
| Gender | M | F | F | F | M | F | M | F | M | F | M | M | F | M |
| Mutation (cDNA) | 3460C>T | 3461G>A | 3461G>A | 3461G>A | 3461G>A | 3460C>T | 3460C>T | 3128G>A | 3461G>T | 1433C>T | 1433C>T | - | - | - |
| Alteration (protein) | Arg1154Trp | Arg1154Gln | Arg1154Gln | Arg1154Gln | Arg1154Gln | Arg1154Trp | Arg1154Trp | Cys1043Tyr | Arg1154Gln | Ala478Val | Ala478Val | - | - | - |
| Inherited | de novo | inherited | inherited | de novo | de novo | de novo | de novo | de novo | inherited | |||||
| Consanguinity | + | - | - | - | - | - | + | - | - | - | + | + | + | - |
| Age at evaluation | 4 m | 16 yrs | 10 yrs | 39 yrs | 8 yrs | 21 yrs | 3.5 m | 4.5 yrs | 9.8 yrs | 4 m | 32 yrs | 6 yrs | 4 yrs | 3 m |
| Alive | - | + | + | + | + | + | + | + | + | + | + | + | - | + |
| Congenital hypertrichosis | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Macrosomia at birth | + | - | + | + | - | + | - | + | - | + | + | - | - | + |
| Macrocephaly | + | + | + | + | + | - | + | + | + | + | + | + | + | + |
| ID∗ and/or developmental delay | - | - | - | - | + | - | + | - | - | + | - | + | + | + |
| Facial Features | ||||||||||||||
| Coarse face | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Epicanthal folds | + | + | + | + | + | + | + | + | + | + | + | + | + | - |
| Abundant and/or curly eyelashes | + | + | + | - | + | - | + | + | + | + | - | + | + | + |
| Broad and/or flat nasal bridge | + | + | + | + | + | - | + | + | + | + | + | + | + | + |
| Small nose and/or anteverted nostrils | + | + | + | + | + | + | + | + | + | + | - | - | - | + |
| Prominent mouth and/or thick lips | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Long philtrum | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| High and/or narrow palate | + | + | + | + | - | + | + | - | - | - | + | + | + | - |
| Macroglossy | + | + | + | + | - | + | + | - | - | - | + | - | - | - |
| Anterior open bite | - | + | + | - | - | + | - | - | - | - | - | - | - | |
| Gingival hyperplasia | + | + | + | + | - | + | + | - | - | + | - | - | - | - |
| Short neck | - | - | - | + | + | - | + | + | + | - | - | + | + | + |
| Cardiac Features | ||||||||||||||
| Structural cardiac anomalies | + | - | - | - | - | + | + | - | + | + | - | - | + | |
| Pulmory hypertension | + | - | - | - | - | - | - | - | + | - | - | - | - | |
| Pericardial effusion | - | + | + | - | + | - | - | - | - | - | - | - | ||
| Cardiomegaly | - | + | + | + | + | + | - | - | + | - | + | + | + | + |
| Hypertrophic and/or dilated cardiomyopathy | + | + | + | + | - | - | - | - | - | - | + | + | + | |
| Radiological Findings | ||||||||||||||
| Generalized osteopenia | - | - | - | - | - | + | - | + | + | + | + | |||
| Thick calvarium | + | + | + | + | - | - | + | - | - | + | + | + | + | |
| Delayed bone age | + | - | - | + | - | + | + | + | + | |||||
| Enlarged sella turcica, vertical skull base | - | - | - | - | - | - | + | - | - | - | + | + | + | |
| Narrow shoulders | - | - | - | - | - | - | - | - | - | - | - | + | + | + |
| Narrow thorax | - | - | + | - | + | - | - | - | + | - | + | + | + | + |
| Broad ribs | - | + | + | + | - | - | + | + | + | + | - | - | - | + |
| Vertebral endplate irregularities | - | - | - | - | + | - | - | + | - | - | - | + | + | - |
| Platyspondyly | + | + | - | + | - | + | - | - | - | - | - | + | + | - |
| Ovoid vertebral bodies | + | - | - | - | - | - | + | + | - | - | - | + | + | + |
| Hypoplastic ischium and pubic bones | - | - | - | - | - | - | - | - | - | - | - | + | + | - |
| Narrow obturator foramen | - | + | - | + | - | - | - | - | ||||||
| Erlenmeyer-flask-like long bones | + | + | + | + | - | - | + | - | + | + | + | - | ||
| Bilateral coxa valga | - | - | + | - | + | - | - | - | - | + | - | - | + | |
| Metaphyseal flare with enl. medul. ca.∗ | + | + | + | + | + | - | + | - | + | + | + | + | - | |
| Transverse metaphyseal bands | - | - | - | - | - | - | - | - | - | - | + | - | - | - |
| Other Features | ||||||||||||||
| Preaxial distal phalangeal hypoplasia | - | - | - | - | - | + | - | - | + | - | - | - | ||
| Short, broad first toe | + | - | - | - | - | - | + | + | + | + | + | + | + | |
| Umbilical hernia | + | - | - | - | - | - | + | + | - | + | + | - | - | + |
| Pyloric stenosis | + | - | - | - | - | - | - | - | - | - | - | - | - | + |
| Immune deficiency | - | - | - | - | + | - | - | + | - | + | + | - | ||
| Wrinkled and/or loose skin | + | - | - | - | - | - | + | - | - | + | + | + | + | - |
| Deep palmar creases | + | - | - | - | - | - | + | - | - | + | + | + | + | - |
| Finger pads | + | - | - | - | - | + | + | - | - | - | - | - | - | |
| Hyperextensibility interphalangeal joints | + | - | + | - | - | + | + | - | + | - | + | - | - | - |
| Pectus carinatum | + | - | - | - | - | + | - | - | - | - | - | - | - | - |
| Genital anomalies | + | - | - | - | - | - | + | - | - | + | - | - | - | |
| Scoliosis | + | + | + | + | - | - | - | - | - | - | - | - | - | - |
| Lymphedema | - | + | + | + | - | + | - | - | - | - | + | + | + | - |
| Increased tendency for upper GI∗ bleeding | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Renal anomalies | - | - | - | - | - | - | - | + | - | - | - | |||
| Previously published | yes1 | yes1 | yes 1 | yes12 | ||||||||||
RefSeq accession number NM_020297.2 was used in naming mutations. Features that were not available were left blank. Adapted from Engels et al. 2002.12
Abbreviations: enl. medul. ca., enlarged medullary canal; F, female; GI = gastrointestinal; ID, intellectual disability; m, months; M, male; yrs, years.
Figure 1.
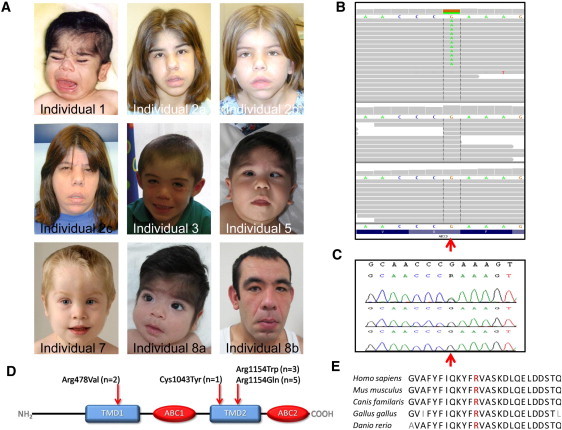
Mutations in ABCC9 Cause Cantú Syndorme
(A) Portrait photographs of Cantú syndrome individuals with ABCC9 mutations, identified by exome sequencing or Sanger sequencing. Note the coarse facial appearance, including a broad nasal bridge, a short nose, a long philtrum, a wide mouth, and full lips.
(B) De novo ABCC9 mutation g.21995261G>A (c.3460C>T; p. Arg1154Trp) identified by exome sequencing of an affected individual and his parents. The upper panel shows the next generation sequencing reads of the child (individual 1) followed by the reads of the father (middle) and mother (bottom).
(C) Sanger validation in the same trio; Sanger traces of the child (individual 1, top), father (middle), and mother (bottom). The point mutation (g.21995261G>A; c.3460C>T) is marked with a red arrow.
(D) Schematic overview of SUR2, with Cantú syndrome-causing mutations depicted by the arrow. Abbreviations: ABC, ATP-binding cassette transporter domain (red); TMD, ABC transmembrane domain type-1 (blue).
(E) Amino-acid conservation of the mutation hot spot p.Arg1154 for multiple species (human, mouse, dog, chicken, zebrafish); the highly conserved arginine is depicted in red.
Initially, we applied an exome-sequencing approach to data from three individuals using a SOLiD v4 sequencing instrument and Agilent's SureSelect v1 (38 Mb) exome-enrichment kit. Detected variants were prioritized as described previously (Table S1).13,14 Only one gene (RP1L1 [MIM 608581]) harbored variants in all three individuals, but these were not validated by Sanger sequencing. Therefore, we performed additional exome sequencing on a simplex case (individual 1) and his parents (Table S2). De novo analysis (as described in Vissers et al.15) resulted in 15 potential de novo mutations in 15 candidate genes (Figures 1B and 1C and Table S2). Systematic validation through Sanger sequencing of all 15 candidates showed that only a mutation in ABCC9 (c.3460C>T) could be validated in the affected individual.15 Furthermore, of these 15 candidate genes, private, nonsynonymous variants could only be identified in ABCC9 (MIM 601439), in two of the three initially sequenced individuals (individuals 3 and 4; c.3461G>A). Visual inspection of the exome-sequencing data of the third individual (individual 2a, c.3460C>T) identified an additional potential mutation in ABCC9 at very low sequence coverage. Notably, all mutations resided within exon 27 of ABCC9.
In the remaining cohort of ten individuals with Cantú syndrome, five additional missense mutations were found in ABCC9 through Sanger sequencing (NM_020297.2; Table S3). Thus, in total, 11/14 cases had an ABCC9 mutation. Interestingly, eight mutations occured in a mutation hot spot, affecting the Arg1154 residue in exon 27, which affected the second type 1 transmembrane region (TMD2: transmembrane domain 2) of the protein encoded by ABCC9, sulfonylurea receptor (SUR2) (Figure 1D). All other mutations (c.3128G>A [p.Cys1043Tyr] and c.1433C>T [p.Ala478Val]) also affected either TMD1 or TMD2. All mutations but one (that found in individuals 8a and b) have been predicted to be possibly or probably damaging by multiple in silico prediction programs. The base-pair conservation (measured by phyloP) was relatively high, whereas the Arg1154 residue was conserved up to zebrafish (Figure 1E). Importantly, in all six simplex cases, the mutations occurred de novo (Figure S1). Furthermore, none of the mutations have been identified in any of the over 5,000 publicly available exomes (Exome Variant Server, NHLBI Exome Sequencing Project [ESP], Seattle). All procedures were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national), and proper informed consent was obtained.
ABCC9 is a member of the ATP-binding cassette subfamily C, also known as the CFTR/MDR (cystic fibrosis transmembrane conductance regulator and multidrug resistance protein) family, and encodes the channel regulator SUR2, which contains TMD1 and TMD2, an N-terminal domain (TMD0), and two nucleotide-binding folds (NBF1 and NBF2), comprising Walker A and Walker B nucleotide-binding motifs and other conserved sequences.16,17 Together with a member of the Kir channel family, SUR2 forms ATP-sensitive potassium channels (KATP channels) consisting of four SUR2 subunits and four pore-forming Kir subunits.17 Alternative RNA splicing of the terminal exon of ABCC9 produces two SUR2 isoforms: SUR2A, predominantly expressed in cardiac and skeletal muscle cells, and SUR2B in smooth muscle.18 Interestingly, the genes for Kir6.1 (KCNJ8 [MIM 600935]) and SUR2 are located in a cluster of genes on chromosome 12p12.1, suggesting coregulation at the gene level.19 A similar situation is observed for Kir6.2 (KCNJ11 [MIM 600937]) and SUR1 (ABCC8 [MIM 600509]), which are also located in a cluster of genes but on chromosome 11p15.1. This provides evidence that these regions were produced in an ancient duplication event, which is in line with its preservation from zebrafish to human (Figure 1E).20 Whereas the chromosome 12 cluster-encoded KATP channel functions primarily in heart, skeletal, and smooth muscle, the chromosome 11 cluster-encoded KATP channel shows a predominant role in the neuroendocrine system, and mutations in these genes may lead to hyperinsulinemic hypoglycemia and neonatal diabetes.18
KATP channels open and close in response to intracellular changes in the ADP/ATP ratio, thereby linking the metabolic state of the cell to its membrane potential.17,18,21 Inhibition of KATP channel activity causes membrane depolarization and thereby activation of voltage-dependent Ca2+ channels, leading to Ca2+ influx and a rise in intracellular [Ca2+].19
Correct maintenance of calcium handling is essential for the normal functioning of the heart, and dysfunctional myocellular calcium homeostasis contributes to the pathogenesis of dilated cardiomyopathy (CMD10 [MIM 608569]).22–24 In Sur2−/− mice, KATP channel activity is essentially absent, and these animals show hypertension, coronary artery vasospasm, and sudden cardiac death.
Previously, mutations in exon 38 of ABCC9, which encodes the C terminus of SUR2A, have been reported in two individuals with idiopathic CMD10.25 One of these mutations was later reported as a variant of unknown frequency in dbSNP (rs72559751). Interestingly, the mutations described for CMD10 affect an exon that is only transcribed in the isoform SUR2A, which shows high cardiac muscle expression, whereas this mutation does not affect the SUR2B isoform, which is expressed predominantly in vascular smooth muscle. This may explain why the phenotype remains restricted to the heart, even though the frameshift mutation affects a gene which might have a more general function, as implicated by the various clinical problems in individuals with Cantú syndrome.
The individuals with Cantú syndrome that have a causative mutation in ABCC9 are clinically as severely affected as those for whom no mutation could be detected. Further studies are required to determine whether this reflects genetic heterogeneity.
Remarkably, in 2006, Grange et al. noted the overlapping phenotype in individuals with Cantú syndrome and individuals treated with Minoxidil, a KATP channel opener.1,26 This drug is used for treatment of hypertension as well as baldness, and treated individuals showed a similarly increased body-hair pattern and pericardial effusions.27 Other reported effects of Minoxidil include increased elastin expression in smooth muscle cells and edema.28 Minoxidil acts as a KATP channel agonist to increase potassium permeability, resulting in a decrease in cytoplasmic [Ca2+] and subsequent smooth muscle relaxation.29,30 This reduced vascular resistance and higher cardiac output may result in increased cardiac muscle mass. Because of the clinical overlap between the effects of Minoxidil treatment and Cantú syndrome, one could speculate that they have the same underlying mechanism for opening the KATP channel.
None of our individuals had deletions or protein-truncating mutations, and the phenotypes of four individuals, 139, 262714, 263616, and 255953 as reported in DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources), with overlapping deletions and a duplication, respectively, do not resemble Cantú syndrome. These results support the role of activating mutations as an underlying mechanism in Cantú syndrome. An indirect proof that the Cantú syndrome-causing mutations are activating mutations comes from homologous mutations identified in ABCC8 (which encodes SUR1) that are a common cause of neonatal diabetes.31,32 The authors of Babenko et al. showed an activating effect of ABCC8 mutations, one of which affected the amino acid homologous to our identified hot spot (Arg1154).31 They concluded that mutations overactivate KATP channels, and that the enhanced stimulatory action of the mutant receptor is sufficient to keep KATP channels open, even at an elevated ratio of ATP to ADP.31 Interestingly, the majority of these individuals are treated by sulphonylurea tablets,31,33 a drug that binds to the SUR1 subunit to cause channel closure independently of ATP.33 Depending on the exact functional consequence of the mutations, the determination of which will require further studies, one could consider therapeutic options for individuals with Cantú syndrome using a similar strategy.
In summary, we identified the underlying genetic cause of Cantú syndrome, mutations in ABCC9, and propose that this syndrome be added to the list of potassium channelopathies with potential therapeutic options.
Acknowledgments
We thank the patients and their parents for their participation and personnel from the Sequencing Facility Nijmegen for technical assistance. This study was financially supported by (1) the Netherlands Organization for Health Research and Development (ZonMW grants 917-66-36 and 911-08-025 to J.A.V., 917-86-319 to B.B.A.d.V., and 916-12-95 to A.H.); (2) the European Union-funded TECHGENE project (Health-F5-2009-223143 to J.A.V.); and (3) the AnEUploidy project (LSHG-CT-2006-37627 to A.H., B.W.M.v.B., H.G.B., B.B.A.d.V., and J.A.V.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://projects.tcag.ca/variation/
DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources), http://decipher.sanger.ac.uk/
Exome Variant Server, NHLBI Exome Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Grange D.K., Lorch S.M., Cole P.L., Singh G.K. Cantu syndrome in a woman and her two daughters: Further confirmation of autosomal dominant inheritance and review of the cardiac manifestations. Am. J. Med. Genet. A. 2006;140:1673–1680. doi: 10.1002/ajmg.a.31348. [DOI] [PubMed] [Google Scholar]
- 2.Lazalde B., Sánchez-Urbina R., Nuño-Arana I., Bitar W.E., de Lourdes Ramírez-Dueñas M. Autosomal dominant inheritance in Cantú syndrome (congenital hypertrichosis, osteochondrodysplasia, and cardiomegaly) Am. J. Med. Genet. 2000;94:421–427. doi: 10.1002/1096-8628(20001023)94:5<421::aid-ajmg15>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 3.Scurr I., Wilson L., Lees M., Robertson S., Kirk E., Turner A., Morton J., Kidd A., Shashi V., Stanley C. Cantú syndrome: report of nine new cases and expansion of the clinical phenotype. Am. J. Med. Genet. A. 2011;155A:508–518. doi: 10.1002/ajmg.a.33885. [DOI] [PubMed] [Google Scholar]
- 4.Robertson S.P., Kirk E., Bernier F., Brereton J., Turner A., Bankier A. Congenital hypertrichosis, osteochondrodysplasia, and cardiomegaly: Cantú syndrome. Am. J. Med. Genet. 1999;85:395–402. [PubMed] [Google Scholar]
- 5.Cantú J.M., García-Cruz D., Sánchez-Corona J., Hernández A., Nazará Z. A distinct osteochondrodysplasia with hypertrichosis- Individualization of a probable autosomal recessive entity. Hum. Genet. 1982;60:36–41. doi: 10.1007/BF00281261. [DOI] [PubMed] [Google Scholar]
- 6.Rosser E.M., Kaariainen H., Hurst J.A., Baraitser M., Hall C.M., Clayton P., Leonard J.V. Three patients with the osteochondrodysplasia and hypertrichosis syndrome—Cantu syndrome. Clin. Dysmorphol. 1998;7:79–85. doi: 10.1097/00019605-199804000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Kurban M., Kim C.A., Kiuru M., Fantauzzo K., Cabral R., Abbas O., Levy B., Christiano A.M. Copy number variations on chromosome 4q26-27 are associated with Cantu syndrome. Dermatology (Basel) 2011;223:316–320. doi: 10.1159/000333800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan T.Y., Bankier A., Slater H.R., Northrop E.L., Zacharin M., Savarirayan R. A patient with monosomy 1p36, atypical features and phenotypic similarities with Cantu syndrome. Am. J. Med. Genet. A. 2005;139:216–220. doi: 10.1002/ajmg.a.31013. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Cruz D., Sánchez-Corona J., Nazará Z., Garcia-Crúz M.O., Figuera L.E., Castañeda V., Cantú J.M. Congenital hypertrichosis, osteochondrodysplasia, and cardiomegaly: further delineation of a new genetic syndrome. Am. J. Med. Genet. 1997;69:138–151. doi: 10.1002/(sici)1096-8628(19970317)69:2<138::aid-ajmg5>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 10.García-Cruz D., Mampel A., Echeverria M.I., Vargas A.L., Castañeda-Cisneros G., Davalos-Rodriguez N., Patiño-Garcia B., Garcia-Cruz M.O., Castañeda V., Cardona E.G. Cantu syndrome and lymphoedema. Clin. Dysmorphol. 2011;20:32–37. doi: 10.1097/MCD.0b013e32833d015c. [DOI] [PubMed] [Google Scholar]
- 11.O'Brien J.J., Ririe D.G. Anesthetic experience in a patient with Cantú syndrome. Paediatr. Anaesth. 2008;18:1255–1257. doi: 10.1111/j.1460-9592.2008.02760.x. [DOI] [PubMed] [Google Scholar]
- 12.Engels H., Bosse K., Ehrbrecht A., Zahn S., Hoischen A., Propping P., Bindl L., Reutter H. Further case of Cantú syndrome: exclusion of cryptic subtelomeric chromosome aberrations. Am. J. Med. Genet. 2002;111:205–209. doi: 10.1002/ajmg.10560. [DOI] [PubMed] [Google Scholar]
- 13.Hoischen A., van Bon B.W., Gilissen C., Arts P., van Lier B., Steehouwer M., de Vries P., de Reuver R., Wieskamp N., Mortier G. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 2010;42:483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 14.Hoischen A., van Bon B.W., Rodríguez-Santiago B., Gilissen C., Vissers L.E., de Vries P., Janssen I., van Lier B., Hastings R., Smithson S.F. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat. Genet. 2011;43:729–731. doi: 10.1038/ng.868. [DOI] [PubMed] [Google Scholar]
- 15.Vissers L.E., de Ligt J., Gilissen C., Janssen I., Steehouwer M., de Vries P., van Lier B., Arts P., Wieskamp N., del Rosario M. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 16.Higgins C.F. ABC transporters: physiology, structure and mechanism—an overview. Res. Microbiol. 2001;152:205–210. doi: 10.1016/s0923-2508(01)01193-7. [DOI] [PubMed] [Google Scholar]
- 17.Nichols C.G. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 18.Bryan J., Muñoz A., Zhang X., Düfer M., Drews G., Krippeit-Drews P., Aguilar-Bryan L. ABCC8 and ABCC9: ABC transporters that regulate K+ channels. Pflugers Arch. 2007;453:703–718. doi: 10.1007/s00424-006-0116-z. [DOI] [PubMed] [Google Scholar]
- 19.Flagg T.P., Enkvetchakul D., Koster J.C., Nichols C.G. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol. Rev. 2010;90:799–829. doi: 10.1152/physrev.00027.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Catchen J.M., Conery J.S., Postlethwait J.H. Automated identification of conserved synteny after whole-genome duplication. Genome Res. 2009;19:1497–1505. doi: 10.1101/gr.090480.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Wet H., Fotinou C., Amad N., Dreger M., Ashcroft F.M. The ATPase activities of sulfonylurea receptor 2A and sulfonylurea receptor 2B are influenced by the C-terminal 42 amino acids. FEBS J. 2010;277:2654–2662. doi: 10.1111/j.1742-464X.2010.07675.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmitt J.P., Kamisago M., Asahi M., Li G.H., Ahmad F., Mende U., Kranias E.G., MacLennan D.H., Seidman J.G., Seidman C.E. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299:1410–1413. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- 23.Chien K.R., Ross J., Jr., Hoshijima M. Calcium and heart failure: the cycle game. Nat. Med. 2003;9:508–509. doi: 10.1038/nm0503-508. [DOI] [PubMed] [Google Scholar]
- 24.Cartwright E.J., Mohamed T., Oceandy D., Neyses L. Calcium signaling dysfunction in heart disease. Biofactors. 2011;37:175–181. doi: 10.1002/biof.149. [DOI] [PubMed] [Google Scholar]
- 25.Bienengraeber M., Olson T.M., Selivanov V.A., Kathmann E.C., O'Cochlain F., Gao F., Karger A.B., Ballew J.D., Hodgson D.M., Zingman L.V. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat. Genet. 2004;36:382–387. doi: 10.1038/ng1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Russ U., Lange U., Löffler-Walz C., Hambrock A., Quast U. Binding and effect of K ATP channel openers in the absence of Mg2+ Br. J. Pharmacol. 2003;139:368–380. doi: 10.1038/sj.bjp.0705238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Messenger A.G., Rundegren J. Minoxidil: mechanisms of action on hair growth. Br. J. Dermatol. 2004;150:186–194. doi: 10.1111/j.1365-2133.2004.05785.x. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi A., Suzuki T., Wachi H., Tajima S., Nishikawa T., Murad S., Pinnell S.R. Minoxidil stimulates elastin expression in aortic smooth muscle cells. Arch. Biochem. Biophys. 1994;315:137–141. doi: 10.1006/abbi.1994.1482. [DOI] [PubMed] [Google Scholar]
- 29.Meisheri K.D., Cipkus L.A., Taylor C.J. Mechanism of action of minoxidil sulfate-induced vasodilation: a role for increased K+ permeability. J. Pharmacol. Exp. Ther. 1988;245:751–760. [PubMed] [Google Scholar]
- 30.Winquist R.J., Heaney L.A., Wallace A.A., Baskin E.P., Stein R.B., Garcia M.L., Kaczorowski G.J. Glyburide blocks the relaxation response to BRL 34915 (cromakalim), minoxidil sulfate and diazoxide in vascular smooth muscle. J. Pharmacol. Exp. Ther. 1989;248:149–156. [PubMed] [Google Scholar]
- 31.Babenko A.P., Polak M., Cavé H., Busiah K., Czernichow P., Scharfmann R., Bryan J., Aguilar-Bryan L., Vaxillaire M., Froguel P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med. 2006;355:456–466. doi: 10.1056/NEJMoa055068. [DOI] [PubMed] [Google Scholar]
- 32.de Wet H., Rees M.G., Shimomura K., Aittoniemi J., Patch A.M., Flanagan S.E., Ellard S., Hattersley A.T., Sansom M.S., Ashcroft F.M. Increased ATPase activity produced by mutations at arginine-1380 in nucleotide-binding domain 2 of ABCC8 causes neonatal diabetes. Proc. Natl. Acad. Sci. USA. 2007;104:18988–18992. doi: 10.1073/pnas.0707428104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edghill E.L., Flanagan S.E., Ellard S. Permanent neonatal diabetes due to activating mutations in ABCC8 and KCNJ11. Rev. Endocr. Metab. Disord. 2010;11:193–198. doi: 10.1007/s11154-010-9149-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.