

Background
Torquere is Latin for ‘to twist’ and gave rise to ‘torque’ in English and, in French, to ‘torsade’ a ‘twisted fringe, cord or ribbon’. Torsade de pointes (TdP) is a ballet sequence in which the dancer twists around her ‘pointes’ (dancing en pointe means ‘on the tip’ and is a classical ballet technique, usually performed using special shoes called pointes). Dessertenne used this phrase to describe a distinctive form of ventricular tachycardia in an elderly woman, because the electrical axis of the ventricular complexes varied cyclically giving the electrocardiogram (ECG) a twisted appearance (Figure 1) [1]. Similar polymorphic ventricular tachycardia had previously been identified as the cause of loss of consciousness during quinidine therapy (‘quinidine syncope’) [2]. TdP can be fatal and occurs in several clinical settings associated with prolongation of the ventricular action potential, notably drug treatment and inherited abnormalities of cardiac ion channels [3]. Prolongation of the ventricular action potential is recognized non-invasively by QT segment prolongation on the ECG (a useful but tricky surrogate end-point for risk of clinically important TdP – see below). We have commented previously on drug-induced QT prolongation and its importance in drug development [4–7].
Figure 1.
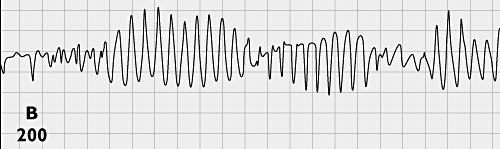
Classical appearance of torsades de pointes in an 80-year-old woman with complete intermittent atrioventricular block [1]. This figure was published in : Dessertenne F. La tachycardie ventriculaire à deux foyers opposés variables. Archives des maladies du cœur et des vaisseaux, 1966; 59 : 263–272. Copyright © 1966 Elsevier Masson SAS. All rights reserved
Several antidysrhythmic drugs, for example disopyramide and procainamide, as well as quinidine, cause TdP including some (flecainide, encainide and d-sotalol) that have increased mortality in randomized controlled trials [8, 9]. Several non-cardiac drugs can also, albeit less frequently cause TdP. These include domperidone (in high dose intravenously – see below), methadone, arsenic trioxide, and several antihistamines, antipsychotic drugs and anti-infective drugs [3]. Not only do drugs differ in their potential to cause TdP, but several patient factors also strongly influence the risk [3]. One of the strongest of these is female sex [10] perhaps because testosterone, which shortens QT interval [11], is lower in women than in men. Several other susceptibility factors can be understood in terms of their influence on cardiac repolarization. These include poor left ventricular function, hypokalaemia, severe hypomagnesaemia and genetic polymorphisms in cardiac ion channels associated with inherited long QT syndromes (LQTS). Drug–drug interactions are clinically important, notably the interaction between ketoconazole (a potent inhibitor of CYP3A) and terfenadine [12], which contributed to the withdrawal of terfenadine from the market after several years of use, including over the counter sales. Diet (especially fruit juice consumption) also influences drug disposition in complex ways [13] underscoring the difficulties for regulators concerned about the potential for low frequency but severe drug harms.
Mechanisms of drug-induced QT prolongation
Most drugs that cause TdP prolong the QT interval by blocking cardiac potassium ion channels, specifically the rapid delayed rectifier channel Kv11.1 [14], which is responsible for the rapid repolarizing current IKr. The mouth of this channel on the inner side of the cell membrane is unusually wide for a potassium ion-selective channel, permitting access to many drugs [15]. This part of the channel is made of α-subunits with many aromatic amino acid residues, favouring binding of lipid-soluble drugs (it is no coincidence that several drugs that prolong QT act in the central nervous system and have been designed to pass lipid barriers readily). The α-subunits of Kv11.1 are coded by the human ether-a-go-go-related gene (HERG) located on the long arm of chromosome 7 [15]. Many (but not all) biologicals (e.g. monoclonal antibodies) do not penetrate cell membranes and are inactive in HERG assays in vitro[16] and hence unlikely to prolong QT by this mechanism. However, a few drugs (including arsenic trioxide and the antiprotozoal drug pentamidine isethionate) prolong QT by causing abnormal trafficking of Kv11.1 and hence reduced channel density rather than channel blockade [17]. Altered channel trafficking can be a consequence of actions on a scorpion toxin binding site on the outside of Kv11.1 [18], and regulatory authorities have understandably taken a conservative position in not automatically exempting biologicals from specific in vivo QT studies, even if they are inactive in the HERG assay in vitro[17].
Mechanism of TdP
Two mechanisms are implicated in drug-induced TdP: after-depolarization and (especially) re-entry [3]. Prolongation of the cardiac action potential increases Ca2+ entry during the plateau phase of the action potential and hence increases [Ca2+]i. This triggers a transient inward current via Na+/Ca2+ exchange (which has a stoichiometry of one Ca2+ ion out for three Na+ ions in) and by opening non-selective cation channels in the cell membranes. After-depolarization can initiate a sequence of ventricular extra beats. Digoxin treatment also increases [Ca2+]i but shortens QT (via vagal activation). It predisposes to TdP [3, 4], presumably through this mechanism, as well as by causing bradycardia. Inhomogeneity between repolarization in different cardiac cells (epicardial, mid-myocardial and endocardial myocytes and Purkinje fibres) facilitates re-entry, which is believed to be the main mechanism of TdP in long QT syndrome [19]. This may help to explain why some drugs, notably amiodarone, bind to the HERG channel, block Kv11.1 and markedly prolong QT yet have a rather low propensity to cause TdP. It is hypothesized that such drugs influence different cardiac cells similarly and hence do not increase heterogeneity or facilitate re-entry [3]. Why amiodarone should prolong QT so uniformly while other drugs do not is an unsolved riddle. An alternative explanation is that therapeutic doses of amiodarone have such a large effect in prolonging the ventricular action potential that this prevents re-entry by prolonging the absolute refractory period.
Regulatory aspects and current concerns
From this discussion, it will be appreciated that if a drug interacts with Kv11.1 in vitro and/or prolongs QT in vivo, this will represent a signal of considerable concern. Indeed, QT prolongation with TdP has been a common cause of withdrawal or restriction of marketed drugs [20], and with increased regulatory awareness [21–23] QT prolongation is now a common cause of failure during development of new drugs with diverse primary pharmacologies and intended therapeutic applications. However, it will also be appreciated that the relation between these surrogates and the risk of TdP is complex and somewhat unpredictable. Thus, while detailed guidance for preclinical and early human studies as to heart rate correction and other technical, statistical and trial-design issues has been published, slavish and uncritical extension of such advice to other early phase studies (distinct from those specifically focused on detecting a QT signal) risks throwing the baby out with the bathwater.
The benefit to harm balance of drugs that prolong QT and increase the risk of TdP, yet have a distinct therapeutic niche, such as arsenic trioxide for relapsed promyelocytic leukaemia, may be finely balanced, making for difficult specific individual clinical judgements [3]. From a general perspective, are we abandoning the development of safe and potentially very valuable agents because of false positive QT signals (i.e. ones that do not translate into appreciable increased risks of TdP)? Concerns as to the possible demise of neuropsychopharmacology [24] are a case in point: one factor may be the (enhanced) potential for lipid soluble drugs that penetrate the blood–brain barrier to prolong QT, as mentioned above.
Three papers in the current issue of the Journal attest to the continued importance of drug effects on the QT interval [25–27]. van Gorp and colleagues describe the pharmacokinetics and pharmacodynamics of escitalopram in overdose (78 patients, median dose 140 mg). The effect of escitolapram on QT interval lags behind the increase in drug plasma concentration. They also found that a single dose of activated charcoal reduced the fraction of the escitolapram dose that was absorbed and the risk of QT prolongation [25]. Since 2010 the ICH E14 guidelines [23] have been adopted in Japan, and Sugiyama and colleagues addressed the question of whether specific studies are needed in this ethnic group using levofloxacin (an anti-infective drug known to prolong QT modestly without serious risk of TdP, and sometimes used as an alternative to moxifloxacin as a positive control in rigorous studies on QT interval) [26]. No significant difference between Japanese and Caucasian subjects was detected, and a trend suggested that, if anything, Caucasians may be more sensitive than Japanese in this regard. Domperidone is a competitive antagonist of dopamine at D2 receptors in the chemoreceptor trigger zone and is a well established and clinically valuable anti-emetic drug. It has a good safety record, but early studies of high doses administered intravenously revealed QT prolongation and TdP. Like terfenadine, it is metabolized by CYP3A, and Boyce and his colleagues have investigated the pharmacokinetic interaction and QT effects at steady-state in healthy volunteers of domperidone and ketoconazole [27]. They used an elegant, randomized, placebo-controlled, double-blind, crossover design. Therapeutic doses of domperidone and ketoconazole were administered by mouth and in men each drug at steady-state modestly prolonged QTcF (the QT interval corrected for heart rate by the Fredericia method). In combination, ketoconazole tripled the plasma concentrations of domperidone and increased QTcF to a clinically important extent (overall adjusted mean difference from placebo 15.9 ms, 95% CI 12.47, 19.33 ms). The authors argue that domperidone (a uniquely useful anti-emetic, because its selective distribution in the central nervous system means that it does not block D2 receptors in the basal ganglia and so can usefully be co-administered with dopamine receptor agonists) should not be co-administered with ketoconazole. In women, QTcF did not differ significantly from placebo during treatment with either of the drugs singly or in combination. This requires confirmation but is fascinating and completely unexpected in view of the strong predilection of drug-induced TdP for females [10]. Doubtless there will be further surprises around the corner!
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.Dessertenne F. La tachycardie ventriculaire à deux foyers opposés variables. Arch Mal Coeur Vaiss. 1966;59:263–72. [PubMed] [Google Scholar]
- 2.Allison FG. Syncopal attacks following quinidine administration. Can Med Assoc J. 1958;78:949–50. [PMC free article] [PubMed] [Google Scholar]
- 3.Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–22. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- 4.Ritter JM. Editors' view: Drug-induced long QT syndrome and drug development. Br J Clin Pharmacol. 2008;66:341–4. doi: 10.1111/j.1365-2125.2008.03275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Molokhia M, Pathak A, Lapeyre-Mestre M, Pharm LC, Montastruc JL. L'Association française des centres régionaux de pharmacovigilance (CRPV), McKeigue P. Case ascertainment and estimated incidence of drug-induced long-QT syndrome: study in Southwest France. Br J Clin Pharmacol. 2008;66:386–95. doi: 10.1111/j.1365-2125.2008.03229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dixon R, Job S, Oliver R, Tompson D, Wright JG, Maltby K, Lorch U, Taubel J. Lamotrigine does not prolong QTc in a thorough QT/QTc study in healthy subjects. Br J Clin Pharmacol. 2008;66:396–404. doi: 10.1111/j.1365-2125.2008.03250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hennessy S, Leonard CE, Newcomb C, Kimmel SE, Bilker WB. Cisapride and ventricular arrhythmia. Br J Clin Pharmacol. 2008;66:375–85. doi: 10.1111/j.1365-2125.2008.03249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effects of encainide and flecainide on mortality in a randomised trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:406–12. doi: 10.1056/NEJM198908103210629. [DOI] [PubMed] [Google Scholar]
- 9.Waldo AL, Camm AJ, de Ruyder H, Friedman PL, MacNeil DJ, Pauls JF, Pitt B, Pratt CM, Schwartz PJ, Veltri EP. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. Lancet. 1996;348:7–12. doi: 10.1016/s0140-6736(96)02149-6. [DOI] [PubMed] [Google Scholar]
- 10.Makkar RR, Fromm BS, Steinman RT, Meissner MD, Lehman MH. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA. 1993;270:2590–7. doi: 10.1001/jama.270.21.2590. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Ouyang P, Post WS, Dalal D, Vaidya D, Blasco-Colmenares E, Soliman EZ, Tomaselli GF, Guallar E. Sex steroid hormones and electrocardiographic QT-interval duration: findings from the third National Health and Nutrition Examination Survey and the Multi-Ethnic Study of Atherosclerosis. Am J Epidemiol. 2011;174:403–11. doi: 10.1093/aje/kwr172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monahan BP, Ferguson CL, Killeavy ES, Lloyd BK, Troy J, Cantilena LR., Jr Torsades de pointes occurring in association with terfenadine use. JAMA. 1990;264:2788–90. [PubMed] [Google Scholar]
- 13.Somogyi A, Loke KY, Ferro A, Lewis LD, Cohen AF, Ritter JM. Editors' pick 2011. Br J Clin Pharmacol. 2012;73:4–8. [Google Scholar]
- 14.Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roden DM. Cellular basis of drug-induced torsades de pointes. Br J Pharmacol. 2008;154:1502–7. doi: 10.1038/bjp.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vargas HM, Bass AS, Breidenbach A, Feldman HS, Gintant GA, Harmer AR. Scientific review and recommendations on preclinical cardiovascular safety evaluation of biologics. J Pharmacol Toxicol Methods. 2008;58:72–6. doi: 10.1016/j.vascn.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Salvi V, Karnad DR, Panicker GK, Kothari S. Update on the evaluation of new drug for effects on cardiac repolarization in humans: issues in early drug development. Br J Pharmacol. 2010;159:34–48. doi: 10.1111/j.1476-5381.2009.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piccini JP, Whellan DJ, Berridge BR, Finkle J, Pettit SD, Stockbridge N, Valentin JP, Vargas HM, Krucoff MW. Current challenges in the evaluation of cardiac safety during drug development: translational medicine meets the Critical Path Initiative. Am Heart J. 2009;158:317–26. doi: 10.1016/j.ahj.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 19.Akar FG, Yan GX, Antzlevitch C, Rosenbaum DS. Unique topographical distribution of M cells underlies re-entrant mechanism of torsade de pointes in the long-QT syndrome. Circulation. 2002;105:1247–53. doi: 10.1161/hc1002.105231. [DOI] [PubMed] [Google Scholar]
- 20.Lasser KE, Allen PD, Woolhandle SJ, Himmelstein DU, Wolfe SM, Bor DH. Timing of new black box warnings and withdrawals for prescription medications. JAMA. 2002;287:2215–20. doi: 10.1001/jama.287.17.2215. [DOI] [PubMed] [Google Scholar]
- 21.Haverkamp W, Breithardt G, Camm AJ, et al. The potential for QT prolongation and proarrhythmia by non-ant-arrhythmic drugs: clinical and regulatory implications. Eur Heart J. 2000;21:1216–31. doi: 10.1053/euhj.2000.2249. [DOI] [PubMed] [Google Scholar]
- 22.Shah RR. The significance of QT interval in drug development. Br J Clin Pharmacol. 2002;54:188–202. doi: 10.1046/j.1365-2125.2002.01627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guidance for Industry. E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. Available at http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM129357.pdf (last accessed 4 February 2012)
- 24.van Gerven J, Cohen A. Vanishing clinical psychopharmacology. Br J Clin Pharmacol. 2011;72:1–5. doi: 10.1111/j.1365-2125.2011.04021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Gorp F, Duffull S, Hackett LP, Isbister GK. Population pharmacokinetics and pharmacodynamics of escitolapram in overdose and the effect of activated charcoal. Br J Clin Pharmacol. 2012;73:402–10. doi: 10.1111/j.1365-2125.2011.04091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugiyama A, Nakamura Y, Nishimura S, Adachi-Akahane S, Kumagai Y, Gayed J, Naseem A, Ferber G, Taubel J, Camm J. Comparison of the effects of levofloxacin on QT/QTc interval assessed in both healthy Japanese and Caucasian subjects. Br J Clin Pharmacol. 2012;73:455–9. doi: 10.1111/j.1365-2125.2011.04110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyce MJ, Baisley KJ, Warrington SJ. Pharmacokinetic interaction between domperidone and ketokinazole leads to QT prolongation in healthy volunteers: a randomized, placebo-controlled, double-blind, crossover study. Br J Clin Pharmacol. 2012;73:411–21. doi: 10.1111/j.1365-2125.2011.04093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]