

Abstract
AIM
To determine if reported lower plasma concentrations of artemisinin derivatives for malaria in pregnancy result from reduced oral bioavailability, expanded volume of distribution or increased clearance.
METHODS
In a sequentially assigned crossover treatment study, pregnant women with uncomplicated falciparum malaria received i.v. artesunate (i.v. ARS) (4 mg kg−1) on the first day and oral ARS (4 mg kg−1) on the second, or, oral on the first and i.v. on the second, in both groups followed by oral ARS (4 mg kg−1 day−1) for 5 days. Plasma concentrations of ARS and dihyroartemisinin (DHA) were measured by liquid chromatography-mass-spectrometry on days 0, 1, 2 and 6. Controls were the same women restudied when healthy (3 months post partum).
RESULTS
I.v. ARS administration resulted in similar ARS and DHA pharmacokinetics in pregnant women with malaria (n = 20) and in controls (n = 14). Oral administration resulted in higher total drug exposure in pregnancy [AUC (95% CI) in (ng ml−1 h)/(mg kg−1)] of 55.1 (30.1, 100.0) vs. 26.5 (12.2, 54.3) for ARS, P = 0.002 and 673 (386, 1130) vs. 523 (351, 724) for DHA, P = 0.007. The corresponding median absolute oral bioavailability (F%) was 21.7 (12.6, 75.1) vs. 9.9 (6.0, 36.81) for ARS (P = 0.046) and 77.0 (42.2, 129) vs. 72.7 (42.0, 87.7) for DHA, P = 0.033. Total DHA exposure was lower at day 6 in pregnant women with malaria (P < 0.001) compared with day 0 or 1, but not in the controls (P = 0.084).
CONCLUSIONS
This study demonstrates the effects of malaria on oral ARS drug disposition are greater than those of pregnancy. This probably results from a disease related reduction in first pass metabolism. The data are reassuring regarding current dosing recommendations.
Keywords: artesunate, dihyroartemisinin, malaria, pharmacokinetics, post partum, pregnancy
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Lowered plasma concentrations of artesunate have been reported in uncomplicated malaria in pregnancy which could risk the life of the mother and fetus.
The reason for lowered plasma concentrations in pregnancy is unexplained.
Oral artesunate is hydrolyzed rapidly in the stomach to the biologically active metabolite dihyroartemisinin.
WHAT THIS STUDY ADDS
Following i.v. artesunate administration for malaria in pregnancy the disposition of artesunate and dihydroartemisinin were similar to controls (the same women studied post partum without malaria).
Higher concentrations of artesunate and dihydroartemisinin were observed after oral administration i.e the disease reduced the pre-systemic metabolism of artesunate.
This study did not confirm previous reports and is reassuring regarding current dosing for artesunate in pregnancy.
Introduction
The pharmacokinetic properties of many antimalarials are altered during pregnancy [1]. There is often an expanded apparent volume of distribution and increased clearance resulting in lower drug exposure which contributes to higher treatment failure rates. In low transmission settings treatment responses to antimalarial drug regimens are worse in pregnant women than in age-matched women from the same location who are not pregnant [2]. The artemisinin derivatives are now central to the treatment of falciparum malaria and artemisinin combination treatments are recommended in the second and third trimesters of pregnancy. Previous studies have suggested that plasma concentrations of the artemisinin derivatives and their common active metabolite dihydroartemisinin (DHA) are low in pregnancy [3, 4]. Finding the correct dose of artesunate (ARS) in pregnancy is necessary not only to obtain optimal cure rates for the individual patient but also to limit the emergence and spread of resistance [5].
The recommendations for dosing of ARS and artemisinin based combination therapies were based initially on Chinese studies with different dosing durations and were modified by later pharmacodynamic assessments in non-immune patients with uncomplicated falciparum malaria [6]. ARS is rapidly hydrolyzed in vivo to DHA which also has antimalarial activity [7]. There are three pharmacokinetic studies of artemisinin derivatives in pregnant women [4, 8, 9]. One reports ARS and DHA pharmacokinetics using liquid chromatography-mass-spectrometry (LC-MS) in 24 pregnant women with acute uncomplicated P. falciparum malaria treated with oral ARS 4 mg kg−1 day−1 (and atovaquone-proguanil) [4]. DHA maximum concentration and drug exposure were 4.2- and 1.8-fold lower in pregnant women compared with non-pregnant Thai adults (17 males, three females) with acute uncomplicated malaria given approximately half the dose (1.79 mg kg−1) [10]. The corresponding estimates of the apparent volumes of distribution and oral clearance were 2.3- and 2.7-fold higher in pregnant women compared with Thai adults [10]. Similar findings were reported for artemether given as artemether-lumefantrine (Coartem®) for uncomplicated P. falciparum[3, 4]. The maximum concentration and exposure for artemether were lower by approximately 1.9- and 3.2-fold and for DHA, 1.2 and 1.6 fold, in 13 pregnant women [3] compared with 25 adult male Thai patients with acute uncomplicated falciparum malaria [11]. The relative contributions of decreased absorption or increased clearance or volume of distribution to the reductions in plasma concentrations were not quantified. Furthermore the earlier studies were carried out before some major sources of preventable errors in measuring ARS and DHA were discovered [12]. Thermolability of ARS and DHA, haemolytic products related to sample collection and malaria infection in combination with organic solvents during sample processing have been shown to cause ex vivo degradation of ARS and DHA resulting in apparently lowered plasma concentrations [12]. Recently, two publications originating from a single study reported on the disposition of ARS and DHA comparing pregnant women with malaria, the same women post partum without malaria and a control group of non-pregnant women with malaria [8, 9]. The results were at odds depending on which group was used as a control. The non-compartmental pharmacokinetic analysis, showed DHA exposure to be less when comparing pregnant women with malaria and the same women studied post partum after oral ARS, but the difference was not statistically different [9]. However, when compared with non-pregnant controls with malaria, DHA exposure was significantly lower in pregnant women with malaria. The report using a population modelling approach excluded data from post partum women because of erratic ARS absorption, which made it difficult to identify a predictive structural model to describe the data. The analysis indicated a 42.3% increase in DHA clearance in pregnant women compared with non-pregnant controls. The authors concluded that dosing in pregnant women with malaria needed to be revised [8].
The aim of this project was to characterize ARS and DHA pharmacokinetics definitively following intravenous administration of ARS and thereby determine whether reduced oral absorption or increased distribution and elimination affected plasma concentrations of ARS and DHA in pregnant women with uncomplicated P. falciparum infections.
Methods
Subjects
Twenty pregnant women (second and third trimester) with acute uncomplicated falciparum malaria, participated in the study after having given written informed consent or thumb print if the patient was unable to read or write. This study took place in the clinics of the Shoklo Malaria Research Unit on the North Western border of Thailand. Enrolment commenced in April 2008 and the last post partum sampling was in March 2009.
Since 1986, when malaria was the main cause of maternal death in this area, the Shoklo Malaria Research Unit has run a weekly antenatal clinic programme to detect and treat all parasitaemic episodes during pregnancy, to prevent malaria mortality and reduce morbidity [13]. Blood smears are taken for the detection of malaria parasites at each visit. At the time of protocol submission falciparum malaria in the second and third trimesters of pregnancy was routinely treated with a 7 day course of ARS. Anaemic women (haematocrit <25%) were not eligible for inclusion.
Ethical approval
Approval for the study was granted by the Faculty of Tropical Medicine, Mahidol University Ethics Committee (TM-IR 029/2005) and Oxford Tropical Research Ethics Committee (OXTREC 007-05).
Study design
Consecutively enrolled pregnant women were assigned alternatively to the two groups. Group 1 treatment commenced with i.v. ARS while group 2 started with oral ARS and received the i.v. ARS dose the next day. Each group received the same total dose of ARS as follows:
Group 1: i.v. ARS (Guilin Pharmaceutical Factory, Guangxi, China repackaged in Thailand by Atlantic Pharmaceuticals) 4 mg kg−1 on day 0 followed by oral ARS 4 mg kg−1 orally for 6 days or,
Group 2: oral artesunate (Guilin Pharma, Guilin, Peoples Republic of China repackaged in Thailand by Atlantic Pharmaceuticals) 4 mg kg−1 on day 0 followed by i.v. ARS 4 mg kg−1 on day 1 followed by oral artesunate 4 mg kg−1 daily for 5 days. Oral treatment was directly observed and given with water.
The same women were restudied in a healthy state 3 months post partum and were prescribed the same treatment regimen.
In the post partum period women were followed monthly. At 3 months post partum, each woman was examined by a physician and screened for malaria. If she was unwell, e.g. malaria, or any other illness, this was treated and the sampling was postponed to a later date.
Sampling regimen
In group 1, 2 ml of blood was drawn at baseline on day 0 and then at 5, 15, 30, 60, 120, 180, 240 and 360 min after the i.v. dose and on day 1 at baseline and after the oral dose at 15, 30, 60, 90, 120, 180, 240 and 360 min.
In group 2, 2 ml of blood was drawn at baseline on day 0 and then at 15, 30, 60, 90, 120, 180, 240 and 360 min after the oral dose and at 5, 15, 30, 60, 120, 180, 240 and 360 min after the i.v. dose on day 1.
In both groups on day 6, 2 ml samples were obtained before (baseline) and after the oral dose at 60, 120 and 240 min.
Blood samples were collected into 2 ml tubes containing sodium fluoride/potassium oxalate as an anticoagulant. The tubes were pre-chilled on wet ice prior to use. After inverting the tube five to six times to ensure mixing the tube was placed directly back onto the wet ice and processed as soon as possible after collection. Whole blood was centrifuged at 4°C, 2000 g for 7 min to obtain plasma. Immediately after centrifugation, the plasma was transferred into a screw cap cryovial (Nalgene No. 5000 0012) and frozen in liquid nitrogen. The sampling and storage time were recorded and a note made in the case of visible haemolysis.
Samples were transferred to the main laboratory daily where they were stored at −80°C. After all samples were collected they were transferred on dry ice for ARS and DHA drug analysis at the Clinical Pharmacology Laboratory, MORU, Bangkok, Thailand [14].. ARS and DHA were quantified with liquid chromatography-tandem mass-spectroscopy [14]. This analytical method has been validated according to USA FDA guidelines. As stipulated by USA FDA guidelines, quality control samples at low, middle and high concentration were analyzed within each analytical batch to ensure accuracy and precision during analysis. The total assay coefficients of variation were less than 8% at every level for this study. The lower limit of quantification (LLOQ) was 1.2 ng ml−1 for ARS and 2.0 ng ml−1 for DHA.
Pharmacokinetic analysis
Individual concentration–time data were evaluated using a non-compartmental analysis approach in WinNonlin version 5.0 (Pharsight Corporation, California, USA). Total drug exposure up to the last measured concentration (AUC(0,last)) was calculated using the linear trapezoidal method for ascending concentrations and the logarithmic trapezoidal method for descending concentrations. Drug exposure was extrapolated from the last observed concentration to time infinity by Clast/λZ for each individual subject to compute total drug exposure (AUC(0,∞)). The terminal elimination half-life (t1/2) was estimated by log-linear regression of three to six concentrations in the terminal elimination phase. Maximum concentration (Cmax) and time to maximum concentration (tmax) were taken directly from the observed data. Apparent volume of distribution (VD) and clearance (CL) were computed individually according to standard procedures. Complete in vivo conversion of ARS into DHA was assumed and the administered dose of DHA was calculated by adjusting for the difference in molecular weight. Individual values of dose-adjusted total drug exposure after oral administration divided by dose-adjusted total drug exposure after i.v. administration were used to calculate absolute oral bioavailability (F).
Individual pharmacokinetic parameter estimates and individual ARS and DHA concentrations at day 0, day 1 and day 6 were investigated in both pregnant and post partum women using data collected at the same time points (i.e. 1, 2, 4 and 6 h after dose) to evaluate the magnitude of metabolic auto-induction and/or a possible disease effect.
Statistical analysis
Pharmacokinetic parameter estimates for the pregnant women with malaria were compared with estimates for the same women when studied post partum and without malaria using the Wilcoxon matched-pairs signed-ranks test in STATA v.10. Individual ratios (pregnant : post partum) of parameter estimates for each woman were calculated and summarized to illustrate trends and the direction of significant differences. Comparisons were made using Mann-Whitney U-test for non-parametric data. All women were categorized into two groups ‘mildly unwell’ and ‘moderately unwell’ on the basis of admission laboratory and clinical findings. Women with raised (≥25th centile) plasma creatinine or blood urea nitrogen or raised (≥25th centile) liver function tests, including bilirubin (total or direct), aspartate aminotransferase or alanine aminotransferase, or fever (≥37.5°C) and tachycardia (>100 beats min−1) were categorized as moderately unwell, and if none of these conditions was satisfied, mildly unwell.
Efficacy and safety assessment
A full medical history and examination (including obstetric evaluation) was carried out by a physician and a midwife. Gestational age of the pregnancy was obtained from a routine ultrasound obtained at the woman's first antenatal consultation and fetal viability confirmed by ultrasound. Complete blood count, biochemistry and parasite count were measured on admission. Blood smears (thin and thick films) were prepared using Giemsa staining and were read for 200 fields before being declared negative. Parasite densities were counted per 500 white blood cells or per 1000 red blood cells (RBC) and expressed as parasites per microlitre. A total of 200 high powered fields on the thick film were read before declaring a smear negative. All stages of the parasites were recorded (asexual and gametocytes). Three spots of blood on filter paper were obtained for PCR genotyping using the method described by Brockman and colleagues [15] using MSP1, MSP2 and GLURP to differentiate recrudescent and novel infections.
Daily malaria smears and temperature measurements were made for assessment of parasite clearance. Patients had a daily evaluation including: clinical and obstetric examination, drug administration and recording of subjective and objective side effects on a case record form. Thereafter women were seen weekly for 63 days or until delivery, whichever occurred later. Anaemia (haematocrit <30%) was screened for each week for 63 days and thereafter every second week. Anaemic women received ferrous sulphate and folic acid supplements.
In the case of reappearance of falciparum parasites during the follow-up period a PCR blood spot sample was obtained and the patient was treated with ARS (2 mg kg−1 day−1; 50 mg tablets Guilin Pharma, Guilin, Peoples Republic of China repackaged in Thailand by Atlantic Pharmaceuticals) and clindamycin (300 mg three times daily; 300 mg capsule from Siam Pharmaceutical Company Ltd, Bangkok, Thailand) both for 7 days and followed up weekly for 6 weeks or until delivery.
All women were asked to deliver in the Shoklo Malaria Research Unit obstetric facility and data on pregnancy outcomes were collected including gender, birth weight (Electronic SECA medical: scales, precision 10g) and labour complications. Each baby was examined by a trained physician for the presence of congenital abnormalities. Mothers and their babies were invited to monthly follow-up visits and their infants were assessed for motor and neurological development, according to the methods developed and validated at this site and described previously [16].
Results
Twenty-one pregnant women were enrolled. One patient withdrew consent before any sampling or drug administration took place and she has not been included in the analysis. Women who received the oral drug first (group 2) were a mean of 6 years older and, as a consequence, had higher gravidity and parity (Table 1). The remaining admission characteristics were not significantly different (Table 1). As expected women who returned for post partum sampling (malaria smear negative) had significantly lower temperature, pulse rate, respiratory rate, body mass index and higher blood pressure and haematocrit on individual paired testing (P < 0.05 for all, data not shown).
Table 1.
Baseline characteristics of women in groups 1 (first dose i.v.) and 2 (first dose oral), and according to disease severity
| Characteristic | Group 1-i.v. first n = 10 | Group 2-Oral first n = 10 | P value | Moderately unwell n = 13 | Mildly unwell n = 7 | P value |
|---|---|---|---|---|---|---|
| Age (years) | 24 (17–40) | 32 (22–40) | 0.052 | 25 (17–40) | 27 (25–40) | 0.275 |
| Gravidity | 1 (1–6) | 4 (1–8) | 0.043 | 1 (1–8) | 4 (1–6) | 0.438 |
| Parity | 0 (0–5) | 2 (0–6) | 0.052 | 0 (0–6) | 2 (0–5) | 0.351 |
| Primigravida (%) (n) | 60 (6/10) | 20 (2/10) | 0.170 | 53.8 (7/13) | 14.3 (1/7) | 0.158 |
| Smoker % (n) | 20.0 (2/10) | 20.0 (2/10) | 1.000 | 7.7 (1/13) | 42.9 (3/7) | 0.101 |
| Body weight (kg) | 48 (40–59) | 50 (40–64) | 0.315 | 48 (40–64) | 50 (42–59) | 0.643 |
| Body mass index (kg m−2) | 19.8 (17.3–24.2) | 20.9 (18.8–25.3) | 0.190 | 20.4 (17.3–25.3) | 19.8 (19.2–24.2) | 0.588 |
| Temperature (°C) | 37.8 (36.0–40.0) | 37.1 (36.1–39.0) | 0.853 | 38 (36.2–40.0) | 36.2 (36.0–37.8) | 0.005 |
| Febrile (%) (n) | 60.0 (6/10) | 40.0 (4/10) | 0.656 | 69.2 (9/13) | 14.3 (1/7) | 0.057 |
| History fever (%) (n) | 100.0 (10/10) | 90.0 (9/10) | 1.000 | 100.0 (13/13) | 85.7 (6/7) | 0.350 |
| Fever (days) | 2 (1–7) | 3 (1–4) | 0.604 | 2 (1–7) | 1.5 (1–3) | 0.210 |
| Gestational age (weeks) | 24.8 (17.1–33.5) | 25.5 (14.0–37.4) | 0.912 | 24 (14–37) | 25 (14–34) | 0.699 |
| Third trimester (%) (n) | 40.0 (4/10) | 50.0 (5/10) | 1.000 | 46.2 (6/13) | 42.9 (3/7) | 1.00 |
| Haematocrit (%) | 30 (25–46) | 30 (27–34) | 0.625 | 30 (25–34) | 32 (27–46) | 0.938 |
| Anaemic (%) (n) | 33.0 (4/9) | 40.0 (4/10) | 1.000 | 50.0 (6/12) | 28.6 (2/7) | 0.633 |
| GM parasitaemia (/µl) | 7 993 (575–63 096) | 2 864 (129–51 286) | 0.317 | 6 281 (288–51 286) | 1 792 (129–63 096) | 0.277 |
| Moderately unwell (%) (n) | 70% (7/10) | 60% (6/10) | 1.000 | n.a | n.a | n.a |
Data are median (range) or proportion % (n).
Based on the criteria described above 13 women were in the moderately unwell group and seven were in the mildly unwell group. Apart from fever, which was one of the differentiating criteria, there were no other significant demographic differences between the two groups (Table 1).
Pharmacokinetics
ARS and DHA pharmacokinetics were well characterized in pregnant women with malaria and in the same women post partum in a healthy state, with the designed sampling schedule (Figures 1 and 2, Table 2A,B). There were no significant differences in ARS or DHA pharmacokinetics between pregnant women with malaria compared with the same women restudied post partum in a healthy state after i.v. administration of ARS. By contrast after oral administration, pregnant women with malaria had significantly higher total drug exposure (AUC) than the same women post partum in a healthy state (PARS = 0.002, PDHA = 0.007) (Table 2B). The median absolute oral bioavailability (F%) assessed by comparison of AUCs following oral and i.v. administration was 21.7% for ARS and 77.0% for DHA in pregnant women with uncomplicated falciparum malaria (Table 2B) compared with 9.9% for ARS (P = 0.046) and 72.7% for DHA (P = 0.033) in the the same women post partum in a healthy state (Table 2B). This increased bioavailability resulted in significantly lower estimates for oral clearance (CL/F, PARS = 0.002, PDHA = 0.013) and apparent volume of distribution (V/F, PDHA = 0.0330) after oral administration (Table 2B). The low bioavailability of ARS confirms a major pre-systemic metabolism and/or breakdown of ARS into DHA after oral administration.
Figure 1.
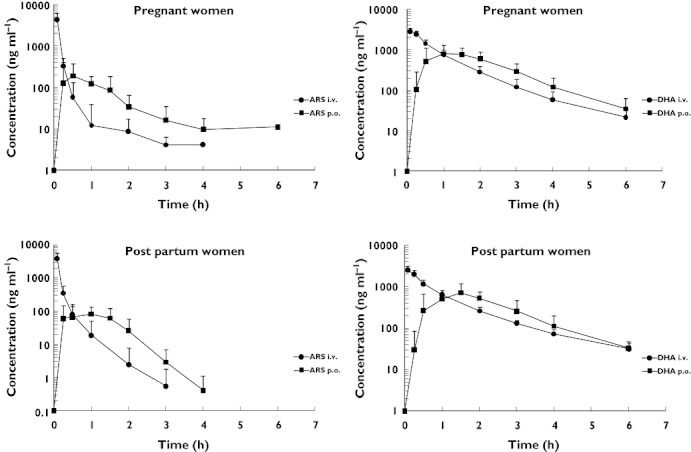
Artesunate and dihydroartemisinin plasma concentration–time profiles in pregnant women with uncomplicated falciparum malaria and in the same women post partum in a healthy state
Figure 2.
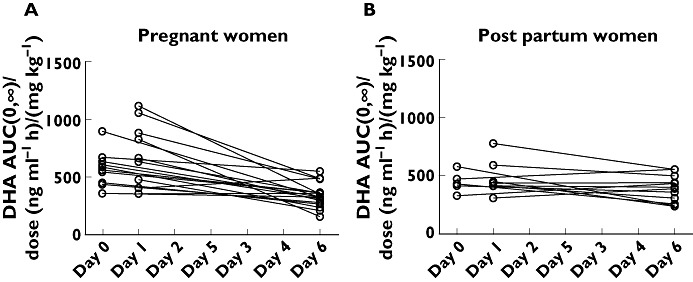
Dose-normalized DHA exposure (AUC(0,∞)/dose) at day 0 and day 1 vs. DHA exposure at day 6 after repeated oral administration of artesunate (4 mg kg−1 day−1) in pregnant women with malaria (A) and in the same women post partum in a healthy state (B)
Table 2.
Non-compartmental analysis of ARS and DHA pharmacokinetics in pregnant women with malaria and in the same women post partum in a healthy state
| (A) i.v. administration | ||||||
|---|---|---|---|---|---|---|
| ARS | DHA | |||||
| Pregnant (n = 20) | Post partum (n = 14) | P value | Pregnant (n = 20) | Post partum (n = 14) | P value | |
| Body weight (kg) | 48.0 (40.0–64.0) | 46.5 (37.0–52.0) | 0.011 | 48.0 (40.0–64.0) | 46.5 (37.0–52.0) | 0.011 |
| Dose (mg kg−1) | 4.00 (3.33–4.05) | 4.00 (3.96–4.05) | 0.232 | 2.96 (2.47–3.00) | 2.96 (2.93–3.00) | 0.232 |
| Number of points lambda | 3.00 (3.00–5.00) | 3.00 (3.00–5.00) | 0.359 | 4.00 (4.00–6.00) | 5.00 (4.00–5.00) | 0.014 |
| C0 (ng ml−1) | 15 700 (3 860–28 700) | 12 200 (5 490–23 900) | 0.975 | 3210 (1570–4360) | 2930 (856–3980) | 0.272 |
| C0/dose (ng ml−1/mg kg−1) | 3 910 (976–7 110) | 3 070 (1 390–5 900) | 0.925 | 1110 (535–1450) | 984 (292–1340) | 0.246 |
| CL (l h−1) | 196.0 (101.0–410.0) | 213.0 (81.5–467.0) | 0.730 | 60.6 (32.8–107.0) | 61.9 (35.3–99.7) | 0.925 |
| CL (l h−1 kg−1) | 4.19 (2.29–10.20) | 5.05 (2.20–9.93) | 0.551 | 1.20 (0.683–2.14) | 1.35 (0.905–1.99) | 0.470 |
| V (l) | 35.6 (17.0–208.0) | 53.3 (22.3–94.6) | 0.594 | 87.5 (40.5–196.0) | 108 (44.9–225.0) | 0.300 |
| V (l kg−1) | 0.76 (0.39–4.15) | 1.18 (0.46–1.89) | 0.594 | 1.76 (0.84–3.92) | 2.37 (1.15–4.51) | 0.124 |
| t1/2 (h) | 0.12 (0.11–0.61) | 0.13 (0.11–0.42) | 0.730 | 1.03 (0.65–1.46) | 1.15 (0.88–1.57) | 0.074 |
| AUC(0,∞) (ng ml−1 h) | 955 (395–1 735) | 792 (398–1 840) | 0.975 | 2450 (1370–4340) | 2220 (1470–3270) | 0.363 |
| AUC(0,∞)/dose (ng ml−1 h/mg kg−1) | 239 (98–437) | 198 (101–454) | 0.875 | 831 (467–1460) | 741 (502–1100) | 0.246 |
| Back ext. AUC (%) | 68.5 (32.3–76.6) | 71.2 (51.2–76.2) | 0.826 | 10.1 (5.3–15.7) | 9.7 (4.8–13.1) | 0.925 |
| Ext. AUC (%) | 0.05 (0.02–0.28) | 0.06 (0.03–0.20) | 0.551 | 0.92 (0.15–2.80) | 1.80 (0.69–5.79) | 0.022 |
| (B) oral administration | ||||||
|---|---|---|---|---|---|---|
| ARS | DHA | |||||
| Pregnant (n = 20) | Post partum (n = 14) | P value | Pregnant (n = 19) | Post partum (n = 14) | P value | |
| Body weight (kg) | 48.0 (40.0–64.0) | 47.0 (37.0–52.0) | 0.015 | 48.0 (40.0–64.0) | 46.5 (37.0–52.0) | 0.015 |
| Dose (mg kg−1) | 3.99 (3.87–4.09) | 3.99 (3.85–4.33) | 0.972 | 2.96 (2.87–3.03) | 2.96 (2.85–3.21) | 0.972 |
| Number of points lambda | 4 (3–5) | 3 (3–5) | 0.157 | 4 (3–5) | 4 (3–5) | 0.272 |
| Cmax (ng ml−1) | 212 (30–1 240) | 119 (35–326) | 0.055 | 1040 (635–2110) | 915 (384–2090) | 0.133 |
| Cmax/dose (ng ml−1/mg kg−1) | 52.9 (7.8–310.0) | 30.9 (8.9–80.4) | 0.033 | 351 (219–712) | 307 (133–652) | 0.152 |
| tmax (h) | 1.00 (0.22–1.55) | 1.00 (0.25–2.00) | 0.140 | 1.50 (0.50–3.05) | 1.50 (0.50–3.00) | 0.527 |
| CL/F (l h−1) | 877 (545–1 540) | 1 640 (792–3 940) | 0.002 | 75.8 (35.3–128) | 89.8 (55.1–128) | 0.039 |
| CL/F (l h−1 kg−1) | 17.8 (10.3–34.2) | 37.8 (18.4–82.1) | 0.002 | 1.53 (0.88–2.57) | 1.91 (1.38–2.85) | 0.013 |
| V/F (l) | 503 (102–5 420) | 643 (173–1 810) | 0.087 | 94 (50–295) | 137 (57–196) | 0.033 |
| V/F (l kg−1) | 10.2 (2.03–113.0) | 13.1 (4.67–40.1) | 0.087 | 1.91 (1.13–5.91) | 2.87 (1.45–4.17) | 0.033 |
| t1/2 (h) | 0.35 (0.10–4.32) | 0.24 (0.13-0.62) | 0.916 | 0.86 (0.67–1.59) | 1.02 (0.71–1.30) | 0.279 |
| AUC(0,∞) (ng ml−1 h) | 217 (117–392) | 106 (48–221) | 0.003 | 1940 (1140–3400) | 1550 (1010–2180) | 0.007 |
| AUC(0,∞)/dose (ng ml−1 h/mg kg−1) | 55.1 (30.1–100) | 26.5 (12.2–54.3) | 0.002 | 673 (386–1130) | 523 (351–724) | 0.007 |
| Ext. AUC (%) | 1.08 (0.21–40.60) | 0.87 (0.16–3.10) | 0.701 | 1.78 (0.52–3.94) | 2.70 (1.56–5.71) | 0.133 |
| F (%) | 21.7 (12.6–75.1) | 9.86 (6.0–36.8) | 0.046 | 77.0 (42.2–129.0) | 72.7 (42.0–87.7) | 0.033 |
All estimates are given as median (range). P values are given using the Wilcoxon matched-pairs signed-ranks test. Number of points lambda number of observations used in the log-linear regression in the terminal elimination phase, C0 initial predicted plasma concentration after i.v. administration, Cmax maximum observed plasma concentration after oral administration, tmax observed time to reach Cmax, CL clearance, V volume of distribution, t1/2 terminal elimination half-life, AUC(0,∞) predicted area under the plasma concentration–time curve after the last dose from zero time to infinity, Back ext. AUC percentage of AUC(0,∞) back-extrapolated from the first observation to time zero, Ext. AUC percentage of AUC(0,∞) extrapolated from the last observation to infinity and F (%) oral bioavailability.
There were no significant differences in derived ARS or DHA pharmacokinetic variables between day 0 and day 1 after i.v. or oral administration in pregnant women with malaria as illustrated for DHA (Table 3). There was a significantly lower total DHA exposure at day 6 compared with day 0 and day 1 after repeated oral administration of ARS in pregnant women with malaria (Figure 2, Table 3) which was not seen in the same women post partum in a healthy state. There were insufficient data points above the LOQ to perform a non-compartmental analysis for ARS but individual drug concentrations at day 0 and at day 1 compared with drug concentrations at day 6 show the same effect for ARS as for DHA in pregnant women with malaria. There were no significant differences in individual ARS concentrations at day 0 and at day 1 compared with day 6 for the same women post partum in a healthy state. These results suggest that a disease effect is a more likely explanation than metabolic auto-induction for the findings in the pregnant group with acute malaria.
Table 3.
Statistical comparison of DHA pharmacokinetics between day 0 and day 1 vs. day 6 after oral administration of artesunate in pregnant women with uncomplicated malaria and in the same women post partum in a healthy state*
| DHA Day 0 and Day 1 | DHA Day 6 | Ratio Day0 and 1 : Day6 | P value | |
|---|---|---|---|---|
| Pregnant women | n = 19 | n = 19 | n = 19 | |
| Cmax/dose (ng ml−1/mg kg−1) | 329.0 (92.5–664.0) | 191.0 (69.8–399.0) | 1.97 (0.34–5.27) | 0.007 |
| tmax (h) | 1.00 (0.97–2.03) | 1.05 (0.983–2.10) | 1.00 (0.50–2.00) | 0.565 |
| CL/F (l h−1 kg−1) | 1.65 (0.90–3.57) | 3.06 (1.82–6.38) | 0.55 (0.19–1.21) | <0.001 |
| V/F (l kg−1) | 2.23 (1.11–5.95) | 4.00 (2.00–8.29) | 0.54 (0.15–2.34) | 0.004 |
| t1/2 (h) | 0.84 (0.66–1.47) | 0.84 (0.63–1.34) | 0.98 (0.81–2.32) | 0.968 |
| AUC(0,∞)/dose ( ng ml−1 h/mg kg−1) | 605 (356–1120) | 327 (157–550) | 1.83 (0.83–5.28) | <0.001 |
| Post partum women | n = 12 | n = 12 | n = 12 | |
| Cmax/dose (ng ml−1/(mg kg−1)] | 231 (133–404) | 152 (74–324) | 1.39 (0.59–2.82) | 0.182 |
| tmax (h) | 1.00 (1.00–2.00) | 1.58 (1.00–4.00) | 0.93 (0.25–2.00) | 0.523 |
| CL/F (l h−1 kg−1) | 2.30 (1.28–3.22) | 2.52 (1.79–4.17) | 0.86 (0.42–1.32) | 0.136 |
| V/F (l kg−1) | 3.32 (1.85–4.33) | 3.46 (2.60–8.33) | 0.92 (0.40–1.23) | 0.117 |
| t1/2 (h) | 1.00 (0.76–1.26) | 1.04 (0.78–1.50) | 0.95 (0.76–1.41) | 0.347 |
| AUC(0,∞)/dose (ng ml−1 h/mg kg−1) | 435 (311–779) | 397 (240–557) | 1.17 (0.76–2.38) | 0.084 |
Non-compartmental analysis was performed using drug concentrations measured at the same time points (i.e. 1, 2, 4 and 6 h after dose) at day 0, day 1 and day 6 for all groups. Data are median (range). Cmax, maximum observed plasma concentration after oral administration, tmax, observed time to reach Cmax, CL clearance, V, volume of distribution, t1/2, terminal elimination half-life, AUC(0,∞), predicted area under the plasma concentration–time curve after the last dose from zero time to infinity and F, oral bioavailability.
The antimalarial effect derives both from ARS and its metabolite DHA. There was a significantly higher combined drug exposure of ARS and DHA after oral (P < 0.001) and i.v. (P = 0.001) treatment in women who were moderately unwell compared with women who were mildly unwell (Table 4, Figure 3).
Table 4.
Statistical comparison of ARS and DHA pharmacokinetics in mildly and moderately unwell pregnant women with uncomplicated malaria
| (a) i.v. administration | ||||||
|---|---|---|---|---|---|---|
| ARS | DHA | |||||
| Mildly unwell (n = 7) | Moderately unwell (n = 13) | P value | Mildly unwell (n = 7) | Moderately unwell (n = 13) | P value | |
| Body weight (kg) | 50.0 (42.0–59.0) | 48.0 (40.0–64.0) | 0.605 | 50.0 (42.0–59.0) | 48.0 (40.0–64.0) | 0.605 |
| Dose (mg kg−1) | 3.97 (3.96–4.00) | 4.03 (3.33–4.05) | 0.039 | 2.94 (2.93–2.96) | 2.99 (2.47–3.00) | 0.039 |
| Number of points lambda | 3 (3–5) | 3 (3–4) | 0.202 | 4 (4–5) | 5 (4–6) | 0.259 |
| C0 (ng ml−1) | 10 900 (3 860–21 500) | 16 900 (6 140–28 700) | 0.191 | 3030 (1570–3780) | 3240 (2510–4360) | 0.362 |
| C0/dose (ng ml−1/mg kg−1) | 2 730 (976–5 430) | 4 160 (1 520–7 110) | 0.166 | 1020 (535–1290) | 1140 (850–1450) | 0.251 |
| CL (l h−1) | 246 (159–273) | 182 (101–410) | 0.104 | 71 (55–107) | 48 (33–66) | 0.005 |
| CL (l h−1 kg−1) | 4.63 (3.20–5.85) | 3.86 (2.29–10.20) | 0.166 | 1.55 (1.23–2.14) | 1.07 (0.68–1.48) | 0.001 |
| V (l) | 40.9 (27.2–208.0) | 33.1 (17.0–165.0) | 0.143 | 112.0 (77.7–196) | 71.1 (40.5–130.0) | 0.016 |
| V (l kg−1) | 0.97 (0.54–4.15) | 0.68 (0.39–3.36) | 0.104 | 2.33 (1.85–3.92) | 1.49 (0.84–2.54) | 0.005 |
| t1/2 (h) | 0.12 (0.11–0.54) | 0.12 (0.11–0.61) | 0.552 | 1.12 (0.78–1.27) | 0.98 (0.65–1.46) | 0.285 |
| AUC(0,∞) (ng ml−1 h) | 856 (684–1 240) | 446 (395–1 730) | 0.219 | 1890 (1370–2410) | 2670 (2010–4340) | 0.001 |
| AUC(0,∞)/dose (ng ml−1 h/mg kg−1) | 216 (171–314) | 259 (98–437) | 0.166 | 645 (467–815) | 932 (677–1460) | 0.001 |
| Back ext. AUC (%) | 70.1 (32.3–75.7) | 70.9 (61.2–76.6) | 0.501 | 10.3 (7.45–15.7) | 9.90 (5.33–14.2) | 0.607 |
| Ext. AUC (%) | 0.04 (0.03–0.28) | 0.06 (0.02–0.18) | 0.663 | 1.20 (0.27–2.24) | 0.91 (0.15–2.80) | 0.874 |
| (b) oral administration | ||||||
|---|---|---|---|---|---|---|
| ARS | DHA | |||||
| Mildly unwell (n = 7) | Moderately unwell (n = 13) | P value | Mildly unwell (n = 7) | Moderately unwell (n = 12) | P value | |
| Body weight (kg) | 50.0 (42.0–59.0) | 48.0 (40.0–64.0) | 0.605 | 50.0 (42.0–59.0) | 47.0 (40.0–64.0) | 0.641 |
| Dose (mg kg−1) | 4.00 (3.87–4.03) | 3.99 (3.98–4.09) | 0.300 | 2.93 (2.87–2.98) | 2.98 (2.88–3.03) | 0.234 |
| Number of points lambda | 3 (3–4) | 4 (3–5) | 0.035 | 4 (3–5) | 4 (3–5) | 0.154 |
| Cmax(ng ml−1) | 170 (86.4–1 240) | 226 (30.4–637) | 0.552 | 975 (635–2110) | 1180 (706–2000) | 0.353 |
| Cmax/dose (ng ml−1/(mg kg−1) | 43.9 (21.5–310) | 57.3 (7.78–157) | 0.552 | 329 (219–712) | 388 (234–664) | 0.398 |
| tmax (h) | 1.00 (0.22–1.07) | 1.00 (0.25–1.55) | 0.775 | 1.00 (0.50–3.05) | 1.50 (0.50–2.00) | 0.602 |
| CL/F (l h−1) | 1 000 (634–1 400) | 870 (545–1 540) | 0.405 | 85 (72–128) | 70 (35–101) | 0.023 |
| CL/F (l h−1 kg−1) | 17.0 (12.7–29.2) | 18.1 (10.3–34.2) | 0.663 | 1.8 (1.4–2.6) | 1.4 (0.9–2.5) | 0.028 |
| V/F (l) | 372 (102–907) | 176 (176–5 420) | 0.501 | 128 (74–295) | 85 (50–123) | 0.014 |
| V/F (l kg−1) | 8.3 (2.0–15.4) | 10.3 (3.9–113.0) | 0.362 | 2.6 (1.6–5.9) | 1.7 (1.1–2.6) | 0.018 |
| t1/2 (h) | 0.26 (0.09–0.79) | 0.36 (0.22–4.32) | 0.322 | 1.02 (0.67–1.59) | 0.84 (0.69–1.07) | 0.447 |
| AUC(0,∞) (ng ml−1 h) | 233 (137–312) | 207 (117–392) | 0.663 | 1610 (1140–2040) | 2180 (1190–3400) | 0.023 |
| AUC(0,∞)/dose (ng ml−1 h/mg kg−1) | 58 (35–78) | 52 (30–100) | 0.663 | 542 (386–687) | 721 (394–1130) | 0.028 |
| Ext. AUC (%) | 1.21 (0.21–21.70) | 0.95 (0.37–40.60) | 0.905 | 2.60 (0.52–3.72) | 1.41 (0.59–3.94) | 0.612 |
| F (%) | 26.8 (12.6–37.7) | 21.7 (13.5–75.0) | 0.501 | 77.0 (71.1–107.0) | 78.7 (42.3–129.0) | 0.800 |
All estimates are given as median (range). P values are given using the two-sample Wilcoxon rank-sum (Mann-Whitney) test. Number of points lambda number of observations used in the log-linear regression in the terminal elimination phase, C0 initial predicted plasma concentration after i.v. administration, Cmax maximum observed plasma concentration after oral administration, tmax observed time to reach Cmax, CL clearance, V volume of distribution, t1/2 terminal elimination half-life, AUC(0,∞) predicted area under the plasma concentration–time curve after the last dose from zero time to infinity, Back ext. AUC percentage of AUC(0,∞) back-extrapolated from the first observation to time zero, Ext. AUC percentage of AUC(0,∞) extrapolated from the last observation to infinity and F (%) oral bioavailability.
Figure 3.
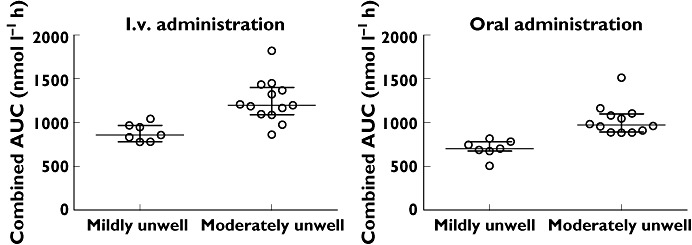
Combined drug exposure of ARS and DHA to illustrate antimalarial effect concentrations in pregnant women treated with intravenous and oral artesunate who were mildly or moderately unwell with uncomplicated malaria
Efficacy and adverse effects
Of the 20 treated women eight remained free of parasites by day 63 of follow-up (median days 42 (7–68) days). The remainder had recurrence of malaria, although this was mainly P. vivax malaria: 10 P. vivax, one P. falciparum and one mixed (P. falciparum and P. vivax) infection. The median time to reappearance for P. falciparum (and mixed) infection was 35 (28–42) days and for P. vivax was 31 (21–56) days. Overall four of the 20 women had a reappearance of P. falciparum before day 63 but three of these women had a P. vivax infection before their P. falciparum infection. PCR genotyping of these P. falciparum infections confirmed that two were recrudescent and two were novel infections. Using survival analysis the cure rate by day 63 of ARS (4 mg kg−1 day−1 for 7 days) for P. falciparum infection was 86.9% (95% CI 69.8, 100) and for any reappearance of malaria was 29.6% (95% CI 5.1, 54.1).
Fever clearance [median (range)] in the 10 women febrile on admission was rapid and similar for both groups: group 1 (i.v. first): 1 (1–2) vs. group 2 (oral first): 1(1) day, P = 0.414. Parasite clearance was also rapid and not significantly different: group 1, 2 (1–4) days vs. group 2, 1 (1–2) days, P = 0.084. The parasite clearance time was 1.5 (1–4) days in the 16 women without recrudescence vs. 2.5 days (1–4) in the two women with PCR confirmed recrudescence, P = 0.587. There were no suspected drug related adverse events. Symptoms on admission (headache 85.0%, joint pain, dizziness and fatigue 75% and anorexia 60.0%) all resolved rapidly with treatment. The treatment was very well tolerated and no significant side effects were reported. One patient was transfused on day 7 for severe anaemia as her haematocrit fell from 25% on admission to 18% on day 6. After excluding women with haematocrit values taken on or after a reappearance or transfusion, the median (range) difference in haematocrit was significantly lower than baseline at day 7, −2.2 (−7.0 to 8.0)%, P = 0.019, but was significantly higher than baseline by day 35, +2.0 (0 to 5.0)%, P = 0.042.
White blood cell and platelet count [median (range) (×103/µl)] improved significantly from day 0 to day 14: 6.2 (1.5–9.7) vs. 9.6 (6.8–17.1), P = 0.001; 130 (39–278) vs. 284 (24–492), P = 0.002. No adverse effect on biochemical parameters was observed following treatment (data not shown).
Pregnancy outcome
Three women were lost to follow-up before the outcome of pregnancy was known. The remaining 17 women all delivered live born, normal, singleton babies at a median gestation of 39.2 (35.6–41.5) weeks. There were 11.8% (2/17) premature births. Most of these infants 59% (10/17) were born at home. All 17 babies were weighed but only seven were weighed within 72 h of birth. They had a median birth weight of 2680 (2300–3530) g and 29% (2/7) were of low birth weight (<2500 g). No congenital abnormalities were detected by newborn examination. One infant went back to Myanmar after delivery, 12 of the remaining 16 were followed up to 1 year and four were lost to follow-up after 4, 6, 10 and 11 months. All infants had achieved normal developmental milestones at their last assessment.
Discussion
This is the most detailed examination of the pharmacokinetic properties of ARS in pregnancy conducted to date and it shows that the effects of malaria on oral drug disposition are greater than those of pregnancy. Following oral administration of ARS plasma concentrations of ARS and its main biologically active metabolite, DHA, were higher in pregnant women with acute malaria compared with those in the same women post partum in a healthy state. This was because of increased oral bioavailability, which confirms that acute malaria substantially reduces the pre-systemic biotransformation of ARS. Oral ARS is hydrolyzed rapidly in the stomach to the biologically active metabolite DHA and this is inactivated by glucuronidation. DHA concentrations were increased as well suggesting a disease related reduction in first-pass glucuronidation. This was reinforced by the significant reduction in plasma concentrations on day 6 observed in acute malaria (when there had been considerable recovery) but not in the same subjects studied in the post partum period when healthy (Table 3). This argues for a disease effect and not an effect related to autoinduction of metabolism, although in this study precise separation of the effects of disease and pregnancy was not possible. The pharmacokinetic properties of i.v. ARS overall were similar in pregnancy and in the same women post partum in a healthy state and neither pregnancy nor disease had a major effect on the disposition of ARS or DHA. However assessment of pharmacokinetic variables by disease severity showed a significantly higher drug exposure in the group of women who were more unwell indicating that disease severity does reduce drug clearance. This is reassuring for the treatment of severe malaria in pregnancy with parenteral ARS as there had been concern, based on earlier studies, that drug concentrations and therefore therapeutic responses might be reduced and doses might have to be increased.
From a therapeutic standpoint ARS can be considered as a pro-drug contributing only a small proportion of the overall parasiticidal effect. ARS is readily hydrolyzed at acid and neutral pH to the active metabolite DHA, and the active metabolite is inactivated by pre-systemic glucuronidation. Concentrations of both parent compound and metabolite are increased in acute malaria [10, 17]. A large portion of orally administered ARS is hydrolyzed in the stomach and absorbed as DHA. In this study the plasma exposure of ARS following oral administration was approximately four times lower than that following i.v. administration. Elimination of ARS from the blood following oral administration is determined by the rate of absorption. This reflected in an approximately three fold longer terminal elimination half-life after oral administration (i.e. flip-flop kinetics). However, this difference in terminal elimination half-life might be an artifact of more data below the limit of quantification after i.v. administration compared with oral administration biasing the log-linear regression. This might also explain the lack of a significant difference in ARS volume of distribution in pregnant and non-pregnant women after oral administration. Following i.v. administration in women post partum in a healthy state, median clearance of ARS was 213 l h−1 while the liver blood flow was only approximately 90 l h−1 (and is reduced in acute malaria [18]). This suggests that the majority of clearance is through de-esterification and degradation in the blood. However in terms of therapeutic responses it is the DHA concentrations which contribute the majority of effect and these were relatively unaffected.
This study did not confirm previous reports that plasma concentrations of the artemisinin derivatives were substantially reduced in pregnant women with acute malaria [3, 4]. Both of the previous reports compared concentrations in uncomplicated malaria in pregnancy to non-pregnant adult concentrations. Furthermore drug concentrations were quantified with a different method from that in this study. It is possible therefore that methodological problems related to drug analysis may have contributed to the differences between the earlier studies and the results of the present study. Plasma concentrations of both ARS and dihydroartemisinin may be apparently reduced in acute malaria if there are haemolytic blood products (which are usual in malaria) in the sample and the analytical method fails to compensate for ex vivo decomposition of the peroxides [12]. The data from the present study are reassuring regarding current dosing recommendations for ARS in pregnancy. This study identifies the cause of altered ARS pharmacokinetics in acute malaria in pregnancy as a disease related reduction in pre-systemic metabolism.
Acknowledgments
We sincerely thank the pregnant women for their co-operation in completing this study. We thank also the medical, nursing, midwifery, laboratory, logistic and administrative staff from SMRU and Mahidol Oxford Research Unit (MORU) in Bangkok.
Competing Interests
The Wellcome Trust is a UK based medical research charity and is independent from all drug companies. It has no financial links with the manufacturers of either the diagnostic tests or the drugs used in this study. There are no competing interests to declare.
Funding: The assays were supported by a grant from the Malaria in Pregnancy Consortium which is funded through a grant from the Bill & Melinda Gates Foundation to the Liverpool School of Tropical Medicine. The study site, Shoklo Malaria Research Unit, is part of the Faculty of Tropical Medicine in Bangkok, funded by the Wellcome-Trust Mahidol University Oxford Tropical Medicine Research Programme funded by the Wellcome-Trust of Great Britain. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Ward SA, Sevene EJ, Hastings IM, Nosten F, McGready R. Antimalarial drugs and pregnancy: safety, pharmacokinetics, and pharmacovigilance. Lancet Infect Dis. 2007;7:136–44. doi: 10.1016/S1473-3099(07)70025-7. [DOI] [PubMed] [Google Scholar]
- 2.Tarning J, McGready R, Lindegardh N, Ashley EA, Pimanpanarak M, Kamanikom B, Annerberg A, Day NP, Stepniewska K, Singhasivanon P, White NJ, Nosten F. Population pharmacokinetics of lumefantrine in pregnant women treated with artemether-lumefantrine for uncomplicated Plasmodium falciparum malaria. Antimicrob Agents Chemother. 2009;53:3837–46. doi: 10.1128/AAC.00195-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGready R, Stepniewska K, Lindegardh N, Ashley EA, La Y, Singhasivanon P, White NJ, Nosten F. The pharmacokinetics of artemether and lumefantrine in pregnant women with uncomplicated falciparum malaria. Eur J Clin Pharmacol. 2006;62:1021–31. doi: 10.1007/s00228-006-0199-7. [DOI] [PubMed] [Google Scholar]
- 4.McGready R, Stepniewska K, Ward SA, Cho T, Gilveray G, Looareesuwan S, White NJ, Nosten F. Pharmacokinetics of dihydroartemisinin following oral artesunate treatment of pregnant women with acute uncomplicated falciparum malaria. Eur J Clin Pharmacol. 2006;62:367–71. doi: 10.1007/s00228-006-0118-y. [DOI] [PubMed] [Google Scholar]
- 5.White NJ. Antimalarial drug resistance. J Clin Invest. 2004;113:1084–92. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li ZL. [Teratogenicity of sodium artesunate] Zhong Yao Tong Bao. 1988;13:42–4. 63–4. [PubMed] [Google Scholar]
- 7.White NJ. Clinical pharmacokinetics and pharmacodynamics of artemisinin and derivatives. Trans R Soc Trop Med Hyg. 1994;88(Suppl. 1):S41–3. doi: 10.1016/0035-9203(94)90471-5. [DOI] [PubMed] [Google Scholar]
- 8.Morris CA, Onyamboko MA, Capparelli E, Koch MA, Atibu J, Lokomba V, Douoguih M, Hemingway-Foday J, Wesche D, Ryder RW, Bose C, Wright L, Tshefu AK, Meshnick S, Fleckenstein L. Population pharmacokinetics of artesunate and dihydroartemisinin in pregnant and non-pregnant women with malaria. Malar J. 2011;10:114. doi: 10.1186/1475-2875-10-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Onyamboko MA, Meshnick SR, Fleckenstein L, Koch MA, Atibu J, Lokomba V, Douoguih M, Hemingway-Foday J, Wesche D, Ryder RW, Bose C, Wright LL, Tshefu AK, Capparelli EV. Pharmacokinetics and pharmacodynamics of artesunate and dihydroartemisinin following oral treatment in pregnant women with asymptomatic Plasmodium falciparum infections in Kinshasa DRC. Malar J. 2011;10:49. doi: 10.1186/1475-2875-10-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newton P, Suputtamongkol Y, Teja-Isavadharm P, Pukrittayakamee S, Navaratnam V, Bates I, White NJ. Antimalarial bioavailability and disposition of artesunate in acute falciparum malaria. Antimicrob Agents Chemother. 2000;44:972–7. doi: 10.1128/aac.44.4.972-977.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lefevre G, Looareesuwan S, Treeprasertsuk S, Krudsood S, Silachamroon U, Gathmann I, Mull R, Bakshi R. A clinical and pharmacokinetic trial of six doses of artemether-lumefantrine for multidrug-resistant Plasmodium falciparum malaria in Thailand. Am J Trop Med Hyg. 2001;64:247–56. doi: 10.4269/ajtmh.2001.64.247. [DOI] [PubMed] [Google Scholar]
- 12.Lindegardh N, Hanpithakpong W, Kamanikom B, Singhasivanon P, Socheat D, Yi P, Dondorp AM, McGready R, Nosten F, White NJ, Day NP. Major pitfalls in the measurement of artemisinin derivatives in plasma in clinical studies. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;876:54–60. doi: 10.1016/j.jchromb.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 13.Nosten F, ter Kuile F, Maelankirri L, Decludt B, White NJ. Malaria during pregnancy in an area of unstable endemicity. Trans R Soc Trop Med Hyg. 1991;85:424–9. doi: 10.1016/0035-9203(91)90205-d. [DOI] [PubMed] [Google Scholar]
- 14.Hanpithakpong W, Kamanikom B, Dondorp AM, Singhasivanon P, White NJ, Day NP, Lindegardh N. A liquid chromatographic-tandem mass spectrometric method for determination of artesunate and its metabolite dihydroartemisinin in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;876:61–8. doi: 10.1016/j.jchromb.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 15.Brockman A, Paul RE, Anderson TJ, Hackford I, Phaiphun L, Looareesuwan S, Nosten F, Day KP. Application of genetic markers to the identification of recrudescent Plasmodium falciparum infections on the northwestern border of Thailand. Am J Trop Med Hyg. 1999;60:14–21. doi: 10.4269/ajtmh.1999.60.14. [DOI] [PubMed] [Google Scholar]
- 16.Haataja L, McGready R, Arunjerdja R, Simpson JA, Mercuri E, Nosten F, Dubowitz L. A new approach for neurological evaluation of infants in resource-poor settings. Ann Trop Paediatr. 2002;22:355–68. doi: 10.1179/027249302125002029. [DOI] [PubMed] [Google Scholar]
- 17.Barradell LB, Fitton A. Artesunate. A review of its pharmacology and therapeutic efficacy in the treatment of malaria. Drugs. 1995;50:714–41. doi: 10.2165/00003495-199550040-00009. [DOI] [PubMed] [Google Scholar]
- 18.Pukrittayakamee S, White NJ, Davis TM, Looareesuwan S, Supanaranond W, Desakorn V, Chaivisuth B, Williamson DH. Hepatic blood flow and metabolism in severe falciparum malaria: clearance of intravenously administered galactose. Clin Sci. 1992;82:63–70. doi: 10.1042/cs0820063. [DOI] [PubMed] [Google Scholar]