

Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) have come under scrutiny because of the gastrointestinal, renal and cardiovascular toxicity associated with prolonged use of these drugs. The purpose of this study was to identify molecular targets for NSAIDs related to cellular toxicity with a view to optimize drug efficacy in clinic. Coronary artery smooth muscle cells (CASMC) and endothelial cells (HCAEC) were treated with low (clinically achievable) and high (typically used in preclinical studies) concentrations of celecoxib (CXB), NS398 (NS) and ibuprofen (IBU) for 24h. NSAIDs-induced gene expression changes were evaluated by microarray analysis and validated by real-time RT-PCR and western blotting. The functional significance of differentially expressed genes was evaluated by Ingenuity Pathway Analysis (IPA). At high concentrations, NSAIDs altered the expression of genes regulating cell proliferation and cell death. NSAIDs also altered genes associated with cardiovascular functions including inflammation, thrombosis, fibrinolysis, coronary artery disease and hypertension. The gene expression was most impacted by IBU, CXB and NS, in that order. This study revealed that NSAIDs altered expression of an array of genes associated with cardiovascular events and emphasizes the potential for fingerprinting drugs in preclinical studies to assess the potential drug toxicity and to optimize the drug efficacy in clinical settings.
Keywords: NSAIDs, HCAEC, CASMC, Microarray, Cardiovascular genes
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are a group of structurally diverse compounds that exert their anti-inflammatory and analgesic effects primarily by inhibiting cyclooxygenase enzymes. NSAIDs demonstrate a wide range of IC50 for inhibition of COX-1 and COX-2. Traditional nonspecific NSAIDs inhibit both COX-1 and COX-2 enzymes whereas coxibs have higher selectively for COX-2. During the last three decades NSAIDs received great attention also in cancer research as preclinical studies identified NSAIDs as potential chemo-preventive and anti-tumor agents either alone or in combination with other cancer therapies (1–2). In addition to inhibiting COX-2, NSAIDs interfere with a variety of cellular processes including signal transduction, transcription, DNA repair, and cell-cycle progression, modulate Bcl-2 family proteins and induce apoptosis (3–6). NSAIDs also inhibit angiogenesis (7–8). Since the anti-tumor effects of NSAIDs are typically seen at concentrations much higher than those required to inhibit prostaglandin synthesis, presumably, COX-2-dependent as well as -independent mechanisms are involved in the effects of NSAIDs (5, 9).
We have a long-standing interest in NSAIDs and coxibs as modifiers of radiation response (2, 10–11). To elucidate further the mechanisms of action of the drugs as well as to identify potential biomarkers of the drug action, we recently analyzed global gene expression changes in PC3 human prostate carcinoma cells treated with COX-2-specific NSAID NS-398 (NS), non-specific NSAID ibuprofen (IBU) and by silencing COX-2 expression with RNAi (12). This study demonstrated that 24h treatment with clinically achievable low concentrations of NSAIDS differentially expressed <3% of the total genes altered by NSAIDs. However, treatment with high concentrations, typically used in preclinical studies, resulted in differential expression of several COX-2-independent targets of NSAIDs. One of the interesting observations was the differential expression of genes related to cardiovascular functions by NSAIDs.
Long-term use of nonspecific NSAIDs that inhibit both COX-1 and COX-2 is associated with gastric and renal toxicity, attributed to the inhibition of COX-1-derived prostaglandins (13). COX-2-specific drugs were developed on the hypothesis that selective COX-2 inhibition at the site of inflammation would reduce pain and inflammation sparing normal tissue toxicity, and were expected to be safer alternatives to the traditional NSAIDs for use in the clinic. Indeed, the results from several large randomized controlled clinical studies and meta-analyses showed that the COX-2 specific inhibitors rofecoxib and celecoxib, while retaining anti-inflammatory properties, had fewer gastrointestinal side effects compared to the dual specific COX inhibitors (14–15). However, data analyses from several trials and epidemiologic studies demonstrated that although there were differences among the classes, COX-2 specific inhibitors increased the risk of cardiovascular events (14, 16–17). Current evidence suggests that prolonged use of both, coxibs and traditional nonspecific NSAIDs may increase cardiovascular risk (16, 18–19). A recent study indicated that even short-term treatment with most NSAIDs was associated with increased risk of death and recurrent myocardial infarction (MI) in patients with prior MI (20).
Building on our work on prostate cancer cells, the purpose of this study was to examine the effect of COX-2-specific and nonspecific NSAIDs on cardiovascular cells. We investigated the global gene expression profile of human coronary artery smooth muscle cells (CASMC) and human coronary artery endothelial cells (HCAEC) treated with low and high concentrations of COX-2 inhibitors celecoxib (CXB) (10 and 40 μmol/L), NS-398 (NS) (10 and 100 μmol/L) and a nonspecific NSAID ibuprofen (IBU) (0.1 and 1.5mmol/L). The low concentrations of CXB and IBU used are close to clinically relevant molar concentrations (21–22). The higher concentrations were used to enhance the cellular effects and to reveal the full spectrum of the pharmacological activities of NSAIDs. The microarray analysis showed that high concentrations NSAIDs significantly altered expression of genes regulating cell cycle, proliferation, signal transduction, DNA damage and repair, apoptosis, autophagy, inflammatory response as well as growth factors and cytokines. Significantly, Ingenuity Pathway Analysis (IPA) of the microarray data also identified genes regulating gastrointestinal disease, renal and urological disease, inflammation, and cardiovascular functions in health and disease. The gene expression was most impacted by IBU, CXB and NS, in that order. The present study focuses mainly on genes associated with cardiovascular functions.
Materials and Methods
Cells
Cryopreserved human coronary artery smooth muscle cells (CASMC), human coronary artery endothelial cells (HCAEC) and the media were purchased from Lonza Walkersville Inc (Walkersville, MD). Cells were thawed and maintained according to the supplier’s instructions. CASMC cells were grown in SmBM basal medium supplemented with fetal bovine serum and growth factors (SmGM-2 BulletKit CC-3182). HCAEC cells were grown in EBM-2 basal medium supplemented with fetal bovine serum and growth factors (EGM-2 MV BulletKit CC-3202).
NSAID Treatment
Celecoxib was purchased from LKT Labs (St. Paul, MN), dissolved in dimethyl sulfoxide (DMSO) and aliquots were stored at −20°C. NS-398 was purchased from Cayman chemicals (Ann Arbor, MI), dissolved in DMSO and aliquots were stored at −20°C. Ibuprofen (I1892; Sigma-Aldrich Co, St. Louis, MO) was prepared fresh as a 100 mmols/L stock in distilled water and filter sterilized before adding to cells. Cells were treated with CXB (10 and 40 μmol/L), NS (10 and 100 μmol/L) and IBU (0.1 and 1.5 mmols/L) for 24h. Control dishes were treated with equivalent amounts of DMSO (0.1%) or H2O.
Cell Growth
CASMC and HCAEC were treated with low and high concentrations of CXB (10 and 40 μmol/L), NS (10 and 100 μmol/L) and IBU (0.1 and 1.5mmol/L). Cells were harvested after 24h and counted in a hemocytometer with 0.4% trypan blue. Each data point represents the percentage of cells after 24h drug treatment in comparison to its respective control.
Microarray Analysis
CASMC and HCAEC cells were treated with CXB (10 and 40 μmol/L), NS (10 and 100 μmol/L) and IBU (0.1 and 1.5mmol/L) for 24h. Total RNA from 3 separate biological replicates was extracted using QIAshredder spin column (Qiagen, CA) and was purified with Qiagen RNeasy mini kit. The microarray analysis was done using CodeLink Human Whole Genome Bioarrays representing 55,000 probes and data analysis was done using GeneSpring software (Agilent Technologies) as described previously (12). To ensure that genes were reliably measured, ANOVA was used to compare the means of each condition (n=3). Cut off ratios greater than 2.0 and less than 0.5 and a p value <0.05 relative to the respective control group were selected for this study.
Real-Time RT-PCR
Alterations in selected differentially expressed genes from different gene ontology categories were validated by real-time PCR using TaqMan Custom Express Plate (Part # 4391528, Applied Biosystems, Foster City, CA) and ABI PRISM 7500 Sequence Detection System instrument equipped with the SDS version 1.4software. Each 96-well assay plate was designed to contain an 18S endogenous control and 31 individual Taqman Gene Expression Assays in triplicates in specified well locations (See Supplemental Digital content 1 for the Assay ID of genes validated by real time RT-PCR). Two μg of RNA was reverse transcribed to synthesize single-stranded cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems; Part. No.4368814). For analysis of target genes 40ng cDNA and for endogenous control 2.5ng cDNA was used. The final reaction volume in each well was 20μl which contained cDNA, TaqMan Gene expression Master Mix (Applied Biosystems; Part No. 4369016) and water to adjust the volume. The threshold cycle (Ct) of the endogenous control was used to normalize target gene expression (ΔCt). The relative change in gene expression (ΔΔCt) was used for comparison of the gene expression in drug treated samples versus vehicle control. Data were analyzed using DataAssist Software v3.0 (Applied Biosystems).
Western Blotting
Cell extracts were prepared after treating cells with NSAIDs for 24h and 25 μg proteins were separated on 4–12% gels as described previously (11). Protein bands were captured by digital CCD camera (Fuji, LAS 3000). The membranes were stripped and re-probed for actin. Signal intensities were quantified using Image J 1.44p software (National Institutes of Health, Bethesda, MD), normalized to the loading control actin, and expressed as fold change compared with vehicle-treated controls.
COX-2 polyclonal antibody was purchased from Cayman Chemicals (Ann Arbor, MI). Thrombomodulin antibody and horseradish peroxide-conjugated goat and rabbit antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-actin antibody was from Millipore (Hayward, CA).
Ingenuity Pathway Analysis (IPA)
The functional significance of differentially expressed genes perturbed by CXB, NS and IBU was evaluated using Ingenuity Pathway analysis (IPA) software (Ingenuity Systems Version 8.7-3203, Redwood City, CA). Genes with a minimal 2-fold change and a p value <0.05 were selected for network generation and pathway analyses implemented in IPA tools as described previously (23).
Cell Cycle
Cells were treated with CXB (10 and 40 μmol/L), NS (10 and 100 μmol/L) and IBU (0.1 and 1.5mmol/L) for 24–48h, harvested and fixed in 70% ethanol. Cells were processed for cell cycle analysis using Guava Cell Cycle Reagent (Millipore, Hayward, CA), according to the protocol provided by the manufacturer. Data were collected on Guava Cytosoft and changes in cell cycle distribution were analyzed by Modfit program.
Apoptosis
Cells were treated with NSAIDs and harvested as above and processed for apoptosis analysis using Guava Nexin Reagent Kit (Millipore, Hayward, CA). Percentages of viable, early apoptotic and late apoptotic/dead cells were determined using Guava Nexin Application (Version 2.5.6) software according to the protocol provided by the manufacturer.
Data Analysis
Each data point represents mean ± SEM of 3 experiments. Statistical differences between the groups were evaluated by ANOVA. A p value of <0.05 was considered statistically significant.
Statistical differences in the expression levels of genes validated by RT-PCR analyses shown in Suppl. Digital Content 2 were analyzed using DataAssist Software v3.0 (Applied Biosystems). This software calculated p-values by t-test using Benjamini-Hochberg False Discovery Rate.
Results
Cell Growth
Fig. 1 shows the effect of 24h treatment with CXB, NS and IBU on the cell growth of CASMC and HCAEC, normalized to the respective controls. High concentrations of NSAIDs inhibited cell growth by 20–30% compared to their respective controls. In HCAEC, the difference in cell growth between the low and high concentrations of IBU was significant.
Fig. 1.
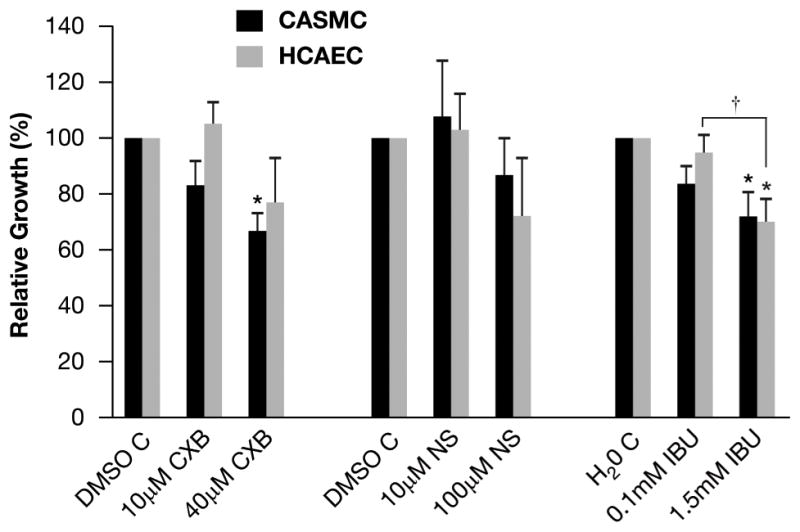
CASMC and HCAEC cell growth after treatment with NSAIDs. Cells were treated with CXB (10μmol/L and 40μmol/L), NS (10μmol/L and 100μmol/L) and IBU (0.1mmol/L and 1.5mmol/L) for 24h. Data points represent percent cell growth following NSAID treatment normalized to vehicle controls (mean ± SEM, n=3). The difference between low and high IBU was significant. †p<0.05
Global Gene Expression Changes
Of the total 55,000 genes represented in the CodeLink Bioarrays, 24h treatment with CXB, NS and IBU differentially expressed 2,824 genes in CASMC and HCAEC cells with high confidence (>2 fold change, p<0.05). In the CASMC 2,160 genes were differentially expressed by the 3 NSAIDs of which 2117 (98%) were altered by the high concentrations of the NSAIDs (Fig. 2A; a, b). Of these 966 (46%) were up-regulated (Fig. 2A, a) and 1151 (54%) were down-regulated (Figure. 1A, b). Only 43 (2%) genes were altered by low concentrations of the 3 NSAIDs (Figure 2A; c, d) and 81% of those were down-regulated (Fig. 2A, d). In the HCAEC 1,507 genes were differentially expressed by the 3 NSAIDs of which 1491 (99%) were altered by the high concentrations of the 3 NSAIDs (Fig. 2B, a, b). Of these 804 (53%) were up-regulated (Figure 1B, a) and 687 (46%) were down-regulated (Fig. 2B, b). Only 16 (1%) genes were altered by the low concentrations of the 3 NSAIDs in HCAEC (Fig. 2B, c, d).
Fig. 2.

Venn diagrams depicting the numbers of differentially expressed genes in (A) CASMC and (B) HCAEC after 24h treatment with NSAIDs. (a, c) genes up-regulated by high and low concentrations, respectively. (b, d) genes down-regulated by high and low concentrations, respectively.
Significant Functional Categories
The 2,824 differentially expressed genes (>2 fold change, p<0.05) by the 3 NSAIDs in CASMC and HCAEC were classified into functional categories by gene ontology classification (Table I). Enrichment of individual functional gene categories was determined by hypergeometric distribution p-values obtained from comparison of the 2,824 genes to the annotated genes in the category. The enrichment factor of the cell cycle-regulatory genes was the highest. The other significantly altered categories were DNA binding, DNA damage and DNA repair, stress response, signal transduction, apoptosis, growth factors, immune response and cytokines. Genes regulating cardiovascular functions were also significantly affected by the NSAIDs.
Table 1.
Enrichment of Genes Altered by NSAIDs in CASMC and HCAEC
| Category | Genes in category | CASMC | HCAEC | ||
|---|---|---|---|---|---|
| # of Genes | p-value | # of Genes | p-value | ||
| Cell cycle | 1143 | 180 | 5.33E-58 | 148 | 3.24E-56 |
| DNA replication | 282 | 60 | 4.66E-27 | 43 | 8.09E-20 |
| DNA repair | 285 | 54 | 5.52E-22 | 42 | 7.92E-19 |
| DNA binding | 3073 | 222 | 7.61E-19 | 173 | 1.98E-19 |
| Response to stress | 1114 | 105 | 1.62E-16 | 67 | 2.52E-09 |
| Cyclin | 152 | 33 | 8.14E-16 | 23 | 3.33E-11 |
| Proliferation | 950 | 90 | 1.79E-14 | 61 | 1.15E-09 |
| Apoptosis | 934 | 75 | 5.05E-09 | 50 | 7.00E-06 |
| DNA damage | 119 | 21 | 7.32E-09 | 15 | 9.64E-07 |
| Signal transduction | 2344 | 139 | 8.74E-07 | 83 | 1.06E-02 |
| Histone | 335 | 32 | 3.84E-06 | 24 | 2.15E-05 |
| Growth factor | 710 | 53 | 7.14E-06 | 26 | 8.36E-02 |
| Transcription factor | 1665 | 99 | 2.93E-05 | 87 | 9.66E-09 |
| Ubiquitin | 682 | 49 | 4.11E-05 | 34 | 6.87E-04 |
| Cytokine | 471 | 37 | 5.71E-05 | 14 | 4.13E-01 |
| Immune response | 865 | 55 | 3.45E-04 | 21 | 7.40E-01 |
| Oncogene | 301 | 24 | 8.62E-04 | 11 | 2.04E-01 |
| Cardiovascular | 1044 | 62 | 8.68E-04 | 39 | 3.27E-02 |
| Interferon | 193 | 16 | 4.00E-03 | 7 | 2.78E-01 |
| Inflammation | 217 | 16 | 1.18E-02 | 7 | 3.83E-01 |
Genes differentially expressed (> 2-fold change, p< 0.05) by CXB, NS and IBU in CASMC and HCAEC were classified into functional categories by gene ontology classification. Enrichment of individual functional gene categories was determined by hypergeometric distribution P values obtained from comparison of the number of genes differentially expressed to the number of annotated genes in each category.
Validation of microarray data
NSAID-induced changes in selected genes identified by microarray analysis were validated by real time RT-PCR. Supplemental Digital Content 2 shows the real time RT-PCR fold changes (upper row, bold numbers) and microarray fold changes (lower row) in selected genes from different categories. The RT-PCR data substantially validated the microarray data.
Heat maps
NSAID-induced fold changes in individual genes from selected functional categories (Table 1) were color-coded to demonstrate the expression patterns of individual genes within a category for each treatment. Fig. 3 shows the heat maps of genes from cell cycle (A), DNA damage (B), DNA repair (C), apoptosis (D) and DNA binding (E) categories.
Fig. 3.

Heat maps depicting color-coded NSAIDs-induced differential gene expression patterns of (A) cell cycle, (B) DNA damage, (C) DNA repair, (D) apoptosis and (E) DNA binding categories in CASMC (lanes 1–6) and HCAEC (lanes 7–12). Lanes 1&7: 10 μmol/L CXB, lanes 2&8: 40 μmol/L CXB; lanes 3&9: 10 μmol/L NS, lanes 4&10: 100 μmol/L NS, lanes 5&11: 0.1mmol/L IBU and lanes 6&12: 1.5mmol/L IBU. Yellow to red indicates up-regulated genes and blue indicates down-regulated genes.
Effect of NSAIDs on cell cycle regulatory genes
Seventy percent genes in this category were down-regulated by high concentration of CXB and IBU (Fig. 3A). In addition to the genes regulating cyclins, cyclin-dependent kinases, transcription factors and ubiquitination, genes involved in chromosome segregation and cytokinesis (PLK), microtubule formation (AURK), initiation of eukaryotic genome replication (MCM2 family) and components of DNA replication complex (POLD1, replication factor C, PCNA) were significantly down-regulated by high CXB and IBU. Down- regulation of selected cell cycle regulatory genes PLK1, CCNB1, RBL1, E2F-2, AUKA, CDC2 and TTK protein kinase was validated by real time RT-PCR (Supplemental Digital Content 2).
Effect of NSAIDs on DNA damage and repair genes
The majority of genes in these categories were down regulated (Fig. 3B and C). Microarray fold changes in DNA damage genes CHK1, GADD34 and DNA repair genes GADD45A, PARP and TOP2 were validated by real time RT-PCR (Supplemental Digital Content 2).
Effect of NSAIDs on genes regulating apoptosis and autophagy
Approximately 60 percent of genes in this category were up-regulated (Fig. 3D). Microarray fold changes in apoptosis related genes Caspase4, TNFAIP3, HMGB1, AIFM2 and TXNIP were validated by real time RT-PCR analysis (Supplemental Digital Content 2). High concentrations of IBU also up-regulated autophagy related genes MAP1A/1BLC3, ATG18, ATG8, DRAM, RAB24 and p62.
Effect of NSAIDs on DNA binding genes
Of the 284 differentially expressed genes in this category, roughly equal numbers were up and down-regulated by CXB and IBU (Fig. 3E). Expression of ATF3, MCM10, CEBPB, H2AFX, POLD1 and HDAC 9 was validated by real time RT-PCR (Supplemental Digital Content 2). IBU was more effective in modulating genes regulating ECM, proteases and angiogenesis compared to CXB (data not shown).
The heat maps and real time RT-PCR data showed that 1) low concentrations of NSAIDs altered very few genes, 2) IBU was the most effective NSAID compared to CXB and NS, and 3) at high concentrations the differential expression patterns of IBU and CXB were more similar than those of CXB and NS, the two coxibs.
Effect of NSAIDs on cell cycle
Since cell cycle was the topmost category affected by the NSAIDs (Table 1), effect of NSAIDs on cell cycle perturbation was evaluated (Table 2). The doubling times of CASMC and HCAEC are 38h and 26h, respectively. Low concentrations of NSAIDs did not alter cell cycle progression in CASMC or HCAEC.
Table 2.
Cell Cycle Perturbations in CASMC and HCAEC Cells Treated with NSAIDs
| CASMC | HCAEC | |||||
|---|---|---|---|---|---|---|
| G1 | S | G2 | G1 | S | G2 | |
| DMSO C | 69.3 ± 2.0 | 18.6 ± 2.1 | 12.1 ± 0.3 | 49.1 ± 2.2 | 25.0 ± 1.0 | 26.0 ± 0.7 |
| 10μM CXB | 67.2 ± 0.8 | 18.4 ± 2.0 | 14.3 ± 1.4 | 47.8 ± 4.1 | 23.6 ± 0.5 | 28.5 ± 3.1 |
| 40μM CXB | 73.1 ± 0.4† | 6.4 ± 0.4**†† | 20.5 ± 0.7**†† | 61.3 ± 1.4*† | 13.7 ± 1.6**†† | 25.0 ± 0.8 |
| DMSO C | 69.4 ± 1.3 | 19.2 ± 1.1 | 11.5 ± 1.5 | 47.5 ± 1.6 | 27.0 ± 1.1 | 25.5 ± 1.9 |
| 10μM NS | 65.9 ± 1.1 | 21.1 ± 1.6 | 13.0 ± 0.6 | 45.3 ± 1.0 | 22.6 ± 1.5 | 32.2 ± 0.6 |
| 100μM NS | 68.4 ± 0.7 | 18.3 ± 1.0 | 13.3 ± 1.7 | 44.8 ± 0.6 | 24.3 ± 1.6 | 30.9 ± 1.6 |
| H2O C | 69.0 ± 1.8 | 16.4 ± 0.7 | 13.6 ± 1.4 | 42.4 ± 1.9 | 24.8 ± 1.8 | 32.9 ± 1.8 |
| 0.1mM IBU | 71.1 ± 2.8 | 15.1 ± 1.6 | 13.9 ± 1.2 | 46.1 ± 2.9 | 23.7 ± 0.8 | 30.2 ± 3.0 |
| 1.5mM IBU | 59.7 ± 2.1*† | 24.5 ± 2.5*† | 15.9 ± 0.5 | 58.5 ± 0.9**† | 14.4 ± 4.0 | 27.0 ± 4.0 |
Cells were treated with CXB (10 & 40 μmol/L), NS (10 & 100 μmol/L) and IBU (0.1 & 1.5 mmol/L) for 24h (HCAEC) or 48h (CASMC) and percentages of cells in G1, S and G2M were determined. Each data point represents mean ± SEM (n=3).
Comparison between control and NSAIDs
p<0.05,
p<0.01
Comparison between low and high concentrations of NSAIDs
p<0.05,
p<0.01
In CASMC NSAIDs did not produce any detectable cell cycle changes at 24h (data not shown) but cell cycle perturbations were observed at 48h in cells treated with high concentrations of NSAIDs (Table 2). Compared to DMSO control the treatment with 40μmol/L CXB showed a slight increase in cells in G1 with a significant reduction in the percentage of cells in S phase, and an accumulation of cells in G2 phase. A comparison of cell cycle perturbations between the low and high concentrations of CXB indicated significant differences for G1, S and G2 compartments. NS had no significant effect on cell cycle progression. Compared to H2O control the treatment with high IBU showed a significant reduction in G1 and some accumulation of cells in S phase. The differences in cells in G1 and S phases following treatment with low and high concentrations of IBU were statistically significant.
In HCAEC, compared to their respective controls or low concentrations, high concentrations of CXB and IBU significantly increased the percentages of cells in G1 with a concomitant reduction in percentages of cells in S phase at 24h. NS had no significant effect on cell cycle progression.
Induction of apoptosis by NSAIDs
NSAIDs significantly altered the expression apoptosis regulatory genes (Table 1); therefore the effect of NSAIDs on induction of apoptosis was examined. Table 3 shows the percentages of viable, early apoptotic and late apoptotic/dead cells in CASMC and HCAEC after treatment with NSAIDs for 48h and 24h, respectively. In CASMC low or high concentrations of CXB did not produce any statistical differences in the percentages of viable or late apoptotic cells. There was a significant increase in early apoptotic cells after treatment with high CXB compared to low CXB. Treatment with NS398 did not result in any significant apoptotic changes. Treatment with high IBU significantly reduced the percentage of viable cells when compared to H2O control or low IBU, and increased the percentages of early and late apoptotic cells.
Table 3.
Percentages of Viable, Early Apoptotic and Late Apoptotic Cells in CASMC and HCAEC Treated with NSAIDs.
| CASMC | HCAEC | |||||
|---|---|---|---|---|---|---|
| Viable | Early Apoptotic | Late apoptotic | Viable | Early apoptotic | Late apoptotic | |
| DMSO C | 96.7 ± 0.4 | 1.4 ± 0.1 | 1.8 ± 0.3 | 72.1 ± 0.7 | 11.3 ± 0.6 | 16.6 ± 0.6 |
| 10μM CXB | 96.8 ± 0.1 | 1.1 ± 0.1* | 1.9 ± 0.1 | 70.3 ± 1.4 | 11.0 ± 0.6 | 18.7 ± 0.6 |
| 40μM CXB | 95.4 ± 1.2 | 1.5 ± 0.1† | 2.9 ± 1.1 | 72.8 ± 1.5 | 7.1 ± 0.5 | 20.1 ± 0.7** |
| DMSO C | 94.6 ± 2.1 | 1.4 ± 0.2 | 3.9 ± 2.0 | 68.9 ± 2.2 | 12.5 ± 0.9 | 18.6 ± 0.6 |
| 10μM NS | 94.7 ± 2.1 | 1.0 ± 0.1 | 4.1 ± 1.9 | 70.2 ± 1.8 | 11.8 ± 0.8 | 18.8 ± 0.6 |
| 100μM NS | 94.5 ± 1.2 | 1.4 ± 0.1 | 3.9 ± 1.2 | 62.0 ± 0.5† | 9.1 ± 0.6 | 28.8 ± 0.6**†† |
| H2O C | 94.4 ± 2.3 | 1.2 ± 0.3 | 4.3 ± 2.1 | 70.9 ± 0.4 | 11.3 ± 0.5 | 17.8 ± 0.7 |
| 0.1mM IBU | 94.6 ± 1.3 | 1.4 ± 0.1 | 3.8 ± 1.3 | 65.1 ± 1.2** | 12.3 ± 0.7 | 22.6 ± 0.2** |
| 1.5mM IBU | 84.6 ± 0.2*†† | 5.3 ± 0.3**†† | 10.0 ± 0.1† | 77.7 ± .3**†† | 6.8 ± 0.5*† | 15.5± 0.7*†† |
Cells were treated with CXB (10 & 40 μmol/L), NS (10 & 100 μmol/L) and IBU (0.1 & 1.5 mmol/L). NSAID-induced apoptotic changes were analyzed by Guava Nexin assay. Each data point represents mean ± SEM. CASMC: n=3; HCAEC: replicates of 2 separate experiments.
Comparison between control and NSAID
p<0.05,
p<0.01
Comparison between low and high concentrations of NSAIDs
p<0.05,
p<0.01
In HCAEC, high concentration of CXB significantly increased the percentage of late apoptotic cells compared to the control. Treatment with high NS resulted in significant reduction in viable cells compared to treatment with low NS. The increase in late apoptotic cells was significant when compared to the control or the low NS group. Low concentrations of IBU significantly reduced viable cells compared to the control. Compared to control or low IBU, there was significant increase in viable cells and reduction in early apoptotic cells after treatment with high IBU. Low concentration of IBU significantly increased the late apoptotic cells compared to control or high IBU.
Ingenuity Pathway Analysis
Genes differentially expressed by the NSAIDs were mapped to the functional networks in the IPA database and ranked by score. Functions associated with top 5 networks (score >10) of these genes are shown in Table 4 (CASMC) and in Table 5 (HCAEC). Low concentrations of NSAIDs altered very few genes (Fig. 2A, B) and there were only few networks to classify these genes (Table 4 and 5).
Table 4.
Functions Associated with Networks of Genes Differentially Expressed by NSAIDs in CASMC
| Treatment | Network ID | Score | Focus Molecules | Top Functions |
|---|---|---|---|---|
| 10μM CXB | 1 | 22 | 8 | Cell Cycle, Gene Expression, DNA Replication, Recombination, and Repair |
| 10μM NS | 1 | 29 | 11 | Cellular Movement, Cell-To-Cell Signaling and Interaction, Cellular Growth and Proliferation |
| 0.1mM IBU | 1 | 32 | 12 | Antigen Presentation, Cell-To-Cell Signaling and Interaction, Hematological System Development and Function |
| 40μM CXB | 1 | 60 | 33 | Cellular Assembly and Organization, DNA Replication, Recombination, and Repair, Cell Death |
| 2 | 43 | 27 | Cell Cycle, Cancer, Skeletal and Muscular Disorders | |
| 3 | 42 | 27 | Cell Cycle, Cellular Assembly and Organization, Amino Acid Metabolism | |
| 4 | 40 | 28 | Cell Cycle, Cancer, Genetic Disorder | |
| 5 | 38 | 25 | Cell Cycle, Cancer, Skeletal and Muscular Disorders | |
| 9 | 25 | 19 | Infection Mechanism, Antigen Presentation, Cellular Movement | |
| 13 | 21 | 18 | Cell Cycle, Cellular Movement, Cardiovascular System Development and Function | |
| 16 | 19 | 15 | Developmental Disorder, Renal and Urological Disease, Free Radical Scavenging | |
| 19 | 16 | 13 | Cellular Development, Hematological System Development and Function, Hematopoiesis | |
| 20 | 15 | 13 | Cellular Development, Connective Tissue Development and Function, Inflammatory Response | |
| 100μM NS | 1 | 32 | 15 | Cardiovascular System Development and Function, Cell Cycle, Infection Mechanism |
| 2 | 20 | 10 | Cellular Growth and Proliferation, Cellular Development, Cell Death | |
| 3 | 20 | 10 | Organismal Injury and Abnormalities, Cell Death, Cell Morphology | |
| 1.5mM IBU | 1 | 41 | 32 | Molecular Transport, RNA Trafficking, DNA Replication, Recombination, and Repair |
| 2 | 39 | 31 | Cell Cycle, Cellular Assembly and Organization, DNA Replication, Recombination, and Repair | |
| 3 | 37 | 31 | Cell Cycle, Cancer, Genetic Disorder | |
| 4 | 36 | 30 | Drug Metabolism, Cellular Function and Maintenance, Cellular Movement | |
| 5 | 35 | 29 | DNA Replication, Recombination, and Repair, Cell Cycle, Cancer | |
| 6 | 33 | 29 | Gastrointestinal Disease, Cardiovascular Disease, Cellular Compromise | |
| 8 | 31 | 28 | Cancer, Gastrointestinal Disease, Genetic Disorder | |
| 9 | 31 | 27 | Cancer, Reproductive System Disease, Cardiovascular Disease | |
| 14 | 28 | 26 | Cardiovascular System Development and Function, Cell-To-Cell Signaling and Interaction, Nervous System Development and Function | |
| 15 | 27 | 25 | Cell Death, Hematological System Development and Function, Hypersensitivity Response | |
| 21 | 23 | 23 | Gene Expression, Antimicrobial Response, Inflammatory Response |
Ingenuity pathway analysis of differentially expressed genes in CASMC treated with low and high concentrations of celecoxib, NS398 and ibuprofen. The network ID, score, number of focus molecules and the functions associated with top 5 networks with a score >10 are shown. In addition, the networks with score >10 and changes in molecules related to cardiovascular disease, gastrointestinal disease, renal and urological disease, inflammatory and immunological disease are also included. These categories and the hematological and cardiovascular system development and function categories are shown in bold.
Table 5.
Functions Associated with Networks of Genes Differentially Expressed by NSAIDs in HCAEC
| Treatment | Network ID | Score | Focus molecules | Top Functions |
|---|---|---|---|---|
| 10μM CXB | 1 | 32 | 12 | Cell Cycle, Cellular Development, Cellular Assembly and Organization |
| 10μM NS | 1 | 22 | 9 | Gene Expression, Cellular Development, Digestive System Development and Function |
| 0.1mM IBU | 1 | 27 | 10 | Cancer, Gene Expression, Drug Metabolism |
| 40μM CXB | 1 | 46 | 28 | Cell Cycle, Cancer, DNA Replication, Recombination, and Repair |
| 2 | 43 | 27 | Cellular Function and Maintenance, Gene Expression, Cellular Development | |
| 3 | 43 | 28 | Cell Cycle, DNA Replication, Recombination, and Repair, Cellular Growth and Proliferation | |
| 4 | 39 | 28 | Cell Cycle, DNA Replication, Recombination, and Repair, Cellular Assembly and Organization | |
| 5 | 39 | 26 | Cell Cycle, Cellular Movement, DNA Replication, Recombination, and Repair | |
| 9 | 27 | 20 | Hematological System Development and Function, Tissue Morphology, Cellular Development | |
| 12 | 22 | 17 | Cellular Development, Hematological System Development and Function, Hematopoiesis | |
| 16 | 20 | 16 | Infection Mechanism, Antimicrobial Response, Inflammatory Response | |
| 18 | 19 | 17 | Cell-To-Cell Signaling and Interaction, Hematological System Development and Function, Antigen Presentation | |
| 100μM NS | 1 | 63 | 26 | Cellular Growth and Proliferation, Tissue Morphology, Cell Cycle |
| 2 | 24 | 13 | Cell Cycle, Cancer, Gastrointestinal Disease | |
| 3 | 21 | 11 | Cellular Movement, Hematological System Development and Function, Immune Cell Trafficking | |
| 1.5mM IBU | 1 | 43 | 32 | Cell Cycle, Cellular Growth and Proliferation, Cardiovascular Disease |
| 2 | 39 | 31 | Cancer, Renal and Urological Disease, DNA Replication, Recombination, and Repair | |
| 3 | 38 | 30 | DNA Replication, Recombination, and Repair, Gene Expression, Cancer | |
| 4 | 37 | 30 | Carbohydrate Metabolism, Lipid Metabolism, Small Molecule Biochemistry | |
| 5 | 36 | 29 | Skeletal and Muscular System Development and Function, Cell Death, Immunological Disease | |
| 6 | 36 | 29 | Infection Mechanism, Cell Morphology, DNA Replication, Recombination, and Repair | |
| 12 | 30 | 26 | Gastrointestinal Disease, Genetic Disorder, Inflammatory Disease | |
| 16 | 26 | 25 | Cardiovascular System Development and Function, Skeletal and Muscular System Development and Function, Tissue Morphology | |
| 18 | 23 | 22 | Cell Cycle, Cancer, Hematological Disease | |
| 19 | 22 | 22 | Gene Expression, Antimicrobial Response, Inflammatory Response | |
| 24 | 18 | 19 | Inflammatory Disease, Ophthalmic Disease, Endocrine System Disorders | |
| 25 | 18 | 19 | Cell-mediated Immune Response, Cellular Movement, Immune Cell Trafficking |
Ingenuity pathway analysis of differentially expressed genes in HCAEC treated with low and high concentrations of celecoxib, NS398 and ibuprofen. In addition to top 5, the networks with score >10 and changes in molecules related to cardiovascular disease, gastrointestinal disease, renal and urological disease, inflammatory and immunological disease are also included. These categories and the hematological and cardiovascular system development and function categories are shown in bold.
IPA analysis of genes differentially expressed following treatment with high concentration of CXB and IBU generated more than 20 networks with score higher than 10. For both cell types cell cycle and DNA replication, recombination and repair were the main functional categories for high CXB and IBU. Cardiovascular disease was another significant category associated with high IBU. In addition to the top 5 networks, the networks with functional categories related to cardiovascular disease, cardiovascular system development and function, hematological system development and function, immune and inflammatory response, gastrointestinal, renal and urological disease are also included and highlighted (Table 4 and 5).
Genes related to cardiovascular functions
IPA analysis along with gene ontology search showed that NSAIDs altered the expression of 198 and 120 genes associated with cardiovascular functions in CASMC (Fig. 4A) and HCAEC (Fig. 4B), respectively; and the majority of them were altered by IBU. However, as seen in the heat map (Fig. 4C) several genes showed a similar upward or downward trend for CXB and IBU. Table 6 shows the toxicology functions of the cardiovascular genes identified by IPA analysis. High concentrations of IBU and CXB differentially expressed genes regulating inflammation, platelet activation, adhesion and chemotaxis, atherosclerosis, thrombosis, myocardial ischemia, infarction, tachycardia, arrhythmia and pericarditis, cardiac injury, damage and cell death, heart failure and cardiac hypertrophy. IPA data base which includes molecules identified by genome-wide association screening (GWAS) also revealed several genes related to coronary artery disease and hypertension. The microarray fold changes in selected cardiovascular genes were validated by real time RT-PCR (Supplemental Digital Content 2). A complete list of microarray analysis of the genes represented in the Fig. 4 is given in Supplemental Digital Content 3.
Fig. 4.

Venn diagrams depicting number of differentially expressed genes related to cardiovascular functions by CXB, NS and IBU in (A) CASMC and(B) HCAEC. (C) Heat map of cardiovascular genes identified by gene ontology classification and IPA in CASMC and HCAEC treated with low and high concentrations of NSAIDs. (See Supplemental Digital Content 3 for the microarray data for this figure).
Table 6.
Cardiovascular Functions of Genes Differentially Expressed by NSAIDs
| FUNCTIONS | GENE SYMBOLS |
|---|---|
| Inflammation, inflammatory response | RELB, KITLG, LAP (C/EBP), OSMR, THBD, PTX3, CCL20, CCL25,HMGB1, CXCL12, IL16, PRSS23, F2RL1, PLA2G4A, LP-PLA2 |
| Platelet activation | GP1BA,PLA2G4A, FGA, F2R |
| Chemotaxis, chemo-attraction, adhesion, | CCL20, CCL25,CCL2, DOCK2, CXCL12, IL16,THBS1 |
| Hyperlipidemia | PTGS2, TXNIP, VLDRLCH |
| Atherosclerotic lesion including fibrosis, growth, thrombosis | PTX3, PTGS2, ERN1,SERPINB8, HRG1, GP1BA, VLDLRCH, THBD,P2RY1,TFPI-2, FGA,, FN1, F2R, F2RL1, PLAT, ACAT2, CCL2, PDE4A, CD59, KPTN, PLA2G4A, LP-PLA2,PLG, THBS1, ANGPT2, PROC |
| Myocardial ischemia, heart failure | AMY2B, DUSP6, PTGS2, ADRA1B, IGF2R, MDM2, HTT, MYH7, PDE4A, BIRC5, APLN, CTGF, E2F1, NPR3PLG, |
| Ischemia, ischemic cardiomyopathy | DDIT3, PTGS2, ADORA2B, CXCL3, GP1BA, PPP1R15, NPR3, CXCL12, ENG, HMGB1, PARP1, F2R,PLG, |
| Myocardial infarction, infarct | CD55, ADORA2B, ADRA1B, IL1A, IRF1, NAMPT, PMAIP1, THBD, CAPN5, PTGS2BAT1, HMGB1, COL3A1, CXCL12, ACTA1, PLG, |
| Tachycardia, arrhythmia, atrial fibrillation | CHRNA7, CHRNE, KITLG, PTGS2, TAC1, ADORA2B, ADRA1B, UCN, KCNMA1,VCL, HMGB1 |
| Pericarditis | TUBA1A, TUBA4B, TUBB3, TUBB2C, TUBE1 |
| Dilation of heart | CCNA2, LMNA, PTGES, F2R |
| Cardiac damage, injury | ANXA1, ATF4, HMOX1,APLN |
| Cardiac necrosis/cell death | MDM2, NAMPT, TXNIP, UCN, GABRD,PARP1, TNFAIP3, BCL2L11, BIRC5, E2F1, FSTL1, ZYX, NRG |
| Cardiac hypertrophy | ATF3, ADAM17, PTGS2, DUSP1, HDAC9, FBXO32, IER3, MDM2, HMOX1, FABP3,SMTN, BIRC5, CXCL12, PBX1, PLA2G4A, F2R, CTSC, H2AFZ, IL33, NFATC4, PTGES, RGS4, S100A10, TNFAIP3, APLN, DES, MYH7, ANGPT2 |
| Hyperplasia, regeneration | CCNA2, HMGB1 |
| Stroke | FABP3, PTGS2, RRM2, SPATA13 |
Up-regulated genes are shown in bold and down-regulated genes are shown in italics. The expression pattern of the underlined genes PLG, THBS1, ANGPT2 and PROC was dependent on either the specific drug or the cell type.
At high concentrations, all the 3 NSAIDs up-regulated COX-2 expression in CASMC and HCAEC. The expression of thrombomodulin, a thrombin receptor was also up-regulated by the NSAIDs. Fig. 5 shows the upregulation of COX-2 and thrombomodulin proteins by western blot analysis in CASMC and HCAEC treated with NSAIDs, confirming the up-regulation of these genes.
Fig. 5.

Validation of selected genes by western blotting. (A) CASMC and (B) HCAEC cells were treated with NSAIDs for 24h. COX-2 (top panel) and thrombomodulin (TM) (lower panel) protein levels were analyzed by western blotting. Data shown are representative of 3 separate experiments. Δ Western: fold change by western blot analysis. Δ Microarray: fold change by microarray analysis.
Discussion
Long-term use of NSAIDs is associated with the normal tissue toxicity that could be a limiting factor in using these drugs to their maximum potential as analgesic, anti-inflammatory, chemopreventive or antitumor agents. Following our observation that expression of genes associated with cardiovascular functions was altered by NSAIDs in prostate cancer cells, we investigated whether these events would also be induced in cardiovascular cells and whether the clinical toxicity may have been anticipated. To identify the molecular targets of NSAIDs in cardiovascular cells we examined the differential gene expression patterns in human CASMC and HCAEC cells treated with low and high concentrations of CXB, NS, and IBU. Of particular note is the differential expression of genes regulating normal cardiovascular functions and cardiovascular disease, gastrointestinal disease, renal and urological disease and inflammation, as the apparent targets of NSAIDs. The gene expression was most impacted by IBU, CXB and NS, in that order. CXB showed more similarities to IBU compared to the other COX-2 inhibitor NS.
In the present study IBU differentially expressed many more genes compared to CXB and NS398, however, the gene expression and protein responses of CXB were more similar to IBU than to NS. NSAIDs are a group of structurally diverse compounds. The differences between NSAIDs are attributed to 1) the drugs exhibit a wide range of IC50 for inhibition of COX-1 and COX-2 and the ratio of IC50 inhibition of COX-1/COX-2 (COX-2 selectivity), and 2) differences in chemical structure and pharmacological properties. In in vitro studies the cellular response to NSAIDs could be influenced by serum concentration in media which may affect drug protein binding (24). The duration of exposure to the drug is also an important factor. In the present study, NS differentially expressed very few genes although NS is a more selective COX-2 inhibitor than CXB. One of the reasons could be that NS treatment appears to require longer exposure than other NSAIDs. Many studies suggest that the effect of NS398 appears to be more pronounced after longer exposure (25).
Exposure to high concentrations of NSAIDs for 24h inhibited cell growth by 20 to 30% in CASMC and HCAEC. CASMC cells did not show any significant cell cycle changes at 24h exposure, however, 48h treatment with high concentrations of CXB and IBU resulted in cell cycle perturbations. NSAIDs at high concentrations blocked HCAEC in G1 and inhibited cell progression into S phase at 24h. Previous reports on NSAID-induced vascular cell cycle perturbations are somewhat conflicting. Treatment with 50μM celecoxib arrested HUVECs in G1 stage, but had no effect on VSMCs (24). However, in choroid-retinal endothelial (RF/6A) cells celecoxib (25μM) inhibited cell proliferation by arresting cells in G2/M and S phase (26). Amongst the nonspecific NSAIDs, non-salicylate NSAIDs ibuprofen, sulindac, diclofenac and indomethacin inhibited A-10 rat aortic VSMC proliferation by arresting cells in G1, whereas salicylate NSAIDs aspirin and sodium salicylate failed to induce G1 block (27). NSAIDs affect various cell cycle regulatory proteins (28). The present study identified several cell cycle regulatory targets of NSAIDs not reported previously in cardiovascular cells. NSAID-induced cell cycle perturbations appear to be at least partially, due to drug-induced DNA damage and inhibition of DNA damage repair.
Microarray analysis indicated that expression of several DNA damage and repair genes that affect the process of apoptosis was altered by CXB and IBU in both cell types. Apoptosis was one of the significant categories affected by NSAIDs involving both pro and anti-apoptotic genes. Although IBU was more effective, CXB also altered several apoptosis-regulating genes in CASMC and HCAEC. There is extensive literature on NSAID-induced apoptosis in tumor cells; however, data on NSAID-induced apoptosis in endothelial cells are scanty. Treatment of HUVECs with 50μM celecoxib increased the number of sub G1 apoptotic cells but not in VSMCs (24). Treatment of SMC with salicylate also did not increase sub G1 cells (28). In the present study IBU increased apoptosis in CASMC at 48h. In HCAEC, all the 3 NSAIDs reduced the percentages of cells in early apoptosis and increased the percentages of cells in late apoptosis at 24h. It is apparent, that by 24h the HCAEC cells progressed beyond early stage into late apoptotic state.
One of the most interesting observations of this study was the differential expression of genes related to normal cardiovascular functions and cardiovascular disease. Inflammation and thrombosis play fundamental roles in the pathogenesis of atherosclerosis and there is extensive cross talk between the two systems (29–31). In vivo, cardiovascular events such as thrombosis and atherogenesis involve interactions between endothelial cells, vascular smooth muscle cells, platelets, macrophages and cytokines (29). Significantly, our microarray data revealed alterations in several important genes involved in the regulation of thrombosis, fibrinolysis, cell signaling and atherogenesis in CASMC and HCAEC treated with NSAIDs in vitro (Fig. 6, Table 6).
Fig. 6.

NSAIDs perturbed genes related to thrombosis, fibrinolysis, and PAR signaling. Thrombin cleaves Fibrinogen (FGA) to form insoluble fibrin monomers (Box 1). Fibronectin (FN1) and thrombospondin 1 (TSP1) bind to fibrinogen, and are positive regulators of blood coagulation. Glycoprotein 1b (GP1BA) functions as a receptor for von Willebrand factor. Fibrin and fibrinogen are further degraded by plasmin (Box 2, 3). Plasmin is derived from cleavage of proenzyme plasminogen (PLG) by tissue-type plasminogen activator (PLAT). Annexin 2 (ANAX2) forms a heterotetramer with S100 family protein p11 and serves as a cofactor for plasmin generation. Tissue factor pathway inhibitor (TFPI-2) and protein C (PROC) are key components of anticoagulant pathways. Thrombin and other coagulation proteases (factor Xa, VIIa) activate protease activator receptors PAR1 or PAR2 (Box 4) which initiate G protein-mediated intracellular signaling, leading to cell type dependent pro-inflammatory or protective responses in endothelial and smooth muscle cells. Genes down-regulated by NSAIDs are shown in light gray. Genes up-regulated by NSAIDs are shown in dark gray.
The process of clotting is initiated by thrombin cleaving fibrinogen (FGA), a soluble blood born glycoprotein, to form insoluble fibrin monomers, the most abundant component of blood clots (32). Fibrinogen and fibrin are further degraded by plasmin, a fibrinolytic enzyme. Plasmin is derived from plasminogen (PLG), a circulatory zymogen, by cleavage of the peptide bond by urokinase or tissue plasminogen activator (PLAT). Annexin 2 (ANAX2), a receptor for PLG and PLAT on endothelial cell surface, is a cofactor for plasmin generation (33). ANAX2 forms a heterotetramer with the S100family protein p11 (34). PLAT, FGAa chain, Annexin2 and S100A10 (p11) were down-regulated by IBU. PLAT is considered to be an important risk factor for coronary heart disease (35). Increased enzyme activity of PLAT causes hyperfibrinolysis, which manifests as excessive bleeding; decreased activity leads to hypofibrinolysis resulting in thrombosis. Molecules associated with FGA including FN1, TSP-1 were also down-regulated whereas GP1BA was up-regulated by IBU.
Activation of PARs, the protease activated receptors, in response to tissue injury and proteases initiates G protein mediated intracellular signaling, leading to cell type dependent pro-inflammatory and protective responses (29, 36–37). CASMC and HCAEC expressed PAR-1 and PAR-2 (also known as F2R and F2RL1). PAR-1 stimulation by thrombin results in platelet aggregation and PAR-1 inhibitors are potential anti-platelet agents. PAR-1 was down-regulated by CXB and IBU in HCAEC. PAR-2 was down-regulated by IBU in both cell types. PAR-2 is of importance in inflammatory conditions that induce endothelial PAR-2 expression and vasodilation (38). Purinergic receptor P2RY1 was down-regulated by IBU. P2RY1 functions as a receptor for extracellular ADP. Binding of ADP to this receptor in platelets leads to mobilization of intracellular calcium, resulting in a change in the shape of platelets and their aggregation (39).
Microarray analysis revealed that anticoagulant molecules protein C (PROC) and tissue factor pathway inhibitor (TFPI) were differentially expressed by NSAIDs. Protein C (PROC) is cleaved to its activated form, activated protein C (APC), by the thrombin-thrombomodulin complex. APC in turn acts as a serine protease to degrade the coagulation cascade cofactors Va and VIIIa, thereby exerting anticoagulant and protective responses (36). In the present study thrombin receptor thrombomodulin (THBD), an important inhibitor of blood coagulation, was up-regulated by CXB and IBU in both cell lines. Neither CXB nor IBU altered PROC in CASMC or HCAEC. However, PROC was down-regulated by NS in CASMC, even at low concentration. Tissue factor pathway inhibitor (TFPI) regulates coagulation by inactivating the TF/VIIa complex and factor Xa. TFPI-2 was inhibited by IBU in CASMC and was also inhibited to some extent by IBU and CXB in HCAEC. Thus IBU, and to a lesser extent CXB down-regulated the expression of some of the important genes regulating hemostasis, thrombosis and signaling in CASMC and HCAEC.
The increase in cardiovascular events associated with NSAIDs has been attributed to the disruption of the normal balance between the two products of COX prostaglandin synthesis, PGI2, a potent vasodilator and an inhibitor of platelet function and thromboxane A2, a vasoconstrictor and a potent stimulator of platelet aggregation. Inhibition of COX-2 by coxibs was thought to promote atherothrombosis by inhibition of PGI2(40). Rabausch et al demonstrated that COX-2-derived prostaglandins regulate the expression of functionally active thrombomodulin, an important inhibitor of blood coagulation, providing a platelet-independent mechanism to explain the prothrombotic effects of COX-2 inhibitors (41). In immunohistochemical studies of atherectomy tissues, SMCs frequently expressed COX-2 at thrombomodulin-positive sites (41). However, in the present study, high concentrations of NSAIDs up-regulated COX-2 and thrombomodulin expression at mRNA and protein level in CASMC and HCAEC. We have previously shown that high concentrations of NSAIDs up-regulate COX-2 in tumor cells also (12). The functional significance of NSAIDs-induced COX-2 is not clear and requires further investigation.
Normal tissue toxicity and complications related to NSAIDs are associated with prolonged use of these drugs. However, a recent study demonstrated that even short-term treatment with most NSAIDs was associated with increased and instantaneous cardiovascular risk in MI survivors (20). The present experimental design does not allow an extrapolation of the results reported on the long term effects of NSAIDs. Also, it is apparent that the observed alterations in the gene expression and their significance to cardiovascular events could be qualified by particular pathophysiological condition of the patients, including the presence or absence of malignancy. Cancer patients have an increased propensity for venous thromboembolism and patients with advanced cancer, in particular, are associated with hypercoagulable state (42–43). Therefore, the present experimental results could serve as the basis for further investigations to afford mechanistic insights into the biology of NSAID actions in patients with cardiovascular disease, rheumatoid disease as well as cancer. Both, the coxibs and the non selective NSAIDs, are a heterogeneous class of drugs with multiple mechanisms of actions. NSAIDs and other commonly prescribed drugs are currently being subjected to increasingly stricter scrutiny for adverse side effects. In addition to the cardiovascular genes, an interesting observation from IPA is the finding of gastrointestinal and renal pathway alterations, both of which are organ systems with clinical toxicity. This study highlights the potential for preclinical drug fingerprinting to identify drug targets with a view to optimize the drug efficacy in clinical settings.
Supplementary Material
Supplemental Digital Content 1: Assay ID of genes validated by real-time RT-PCR
Supplemental Digital Content 2: RT-PCR validation of selected genes differentially expressed by NSAIDs
Supplemental Digital Content 3: Microarray data of NSAID-induced differentially expressed genes associated with cardiovascular functions in CASMC and HCAEC
Acknowledgments
Sources of funding: This work was supported by the Intramural Research Program of National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Footnotes
Conflicts of Interest: None
References
- 1.Milas L, Mason KA, Crane CH, Liao Z, Masferrer J. Improvement of radiotherapy or chemoradiotherapy by targeting COX-2 enzyme. Oncology (Williston Park) 2003;17(5 Suppl 5):15–24. [PubMed] [Google Scholar]
- 2.Teicher BA, Bump EA, Palayoor S, Northey D, Coleman CN. Signal transduction inhibitors as modifiers of radiation therapy in human prostate carcinoma xenografts. Radiation Oncology Investigations. 1996;4(5):221–30. [Google Scholar]
- 3.Lin HP, Kulp SK, Tseng PH, Yang YT, Yang CC, Chen CS. Growth inhibitory effects of celecoxib in human umbilical vein endothelial cells are mediated through G1 arrest via multiple signaling mechanisms. Mol Cancer Ther. 2004;3(12):1671–80. [PubMed] [Google Scholar]
- 4.Raju U, Nakata E, Yang P, Newman RA, Ang KK, Milas L. In vitro enhancement of tumor cell radiosensitivity by a selective inhibitor of cyclooxygenase-2 enzyme: mechanistic considerations. Int J Radiat Oncol Biol Phys. 2002;54(3):886–94. doi: 10.1016/s0360-3016(02)03023-7. [DOI] [PubMed] [Google Scholar]
- 5.Tegeder I, Pfeilschifter J, Geisslinger G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J. 2001;15(12):2057–72. doi: 10.1096/fj.01-0390rev. [DOI] [PubMed] [Google Scholar]
- 6.Tseng WW, Deganutti A, Chen MN, Saxton RE, Liu CD. Selective cyclooxygenase-2 inhibitor rofecoxib (Vioxx) induces expression of cell cycle arrest genes and slows tumor growth in human pancreatic cancer. J Gastrointest Surg. 2002;6(6):838–43. doi: 10.1016/s1091-255x(02)00061-6. discussion 44. [DOI] [PubMed] [Google Scholar]
- 7.Jones MK, Wang H, Peskar BM, Levin E, Itani RM, Sarfeh IJ, Tarnawski AS. Inhibition of angiogenesis by nonsteroidal anti-inflammatory drugs: insight into mechanisms and implications for cancer growth and ulcer healing. Nat Med. 1999;5(12):1418–23. doi: 10.1038/70995. [DOI] [PubMed] [Google Scholar]
- 8.Wei D, Wang L, He Y, Xiong HQ, Abbruzzese JL, Xie K. Celecoxib inhibits vascular endothelial growth factor expression in and reduces angiogenesis and metastasis of human pancreatic cancer via suppression of Sp1 transcription factor activity. Cancer Res. 2004;64(6):2030–8. doi: 10.1158/0008-5472.can-03-1945. [DOI] [PubMed] [Google Scholar]
- 9.Grosch S, Maier TJ, Schiffmann S, Geisslinger G. Cyclooxygenase-2 (COX-2)-independent anticarcinogenic effects of selective COX-2 inhibitors. J Natl Cancer Inst. 2006;98(11):736–47. doi: 10.1093/jnci/djj206. [DOI] [PubMed] [Google Scholar]
- 10.Palayoor ST, Bump EA, Calderwood SK, Bartol S, Coleman CN. Combined antitumor effect of radiation and ibuprofen in human prostate carcinoma cells. Clin Cancer Res. 1998;4(3):763–71. [PubMed] [Google Scholar]
- 11.Palayoor ST, Burgos MA, Shoaibi A, Tofilon PJ, Coleman CN. Effect of radiation and ibuprofen on normoxic renal carcinoma cells overexpressing hypoxia-inducible factors by loss of von Hippel-Lindau tumor suppressor gene function. Clin Cancer Res. 2004;10(12 Pt 1):4158–64. doi: 10.1158/1078-0432.CCR-04-0005. [DOI] [PubMed] [Google Scholar]
- 12.John-Aryankalayil M, Palayoor ST, Cerna D, Falduto MT, Magnuson SR, Coleman CN. NS-398, ibuprofen, and cyclooxygenase-2 RNA interference produce significantly different gene expression profiles in prostate cancer cells. Mol Cancer Ther. 2009;8(1):261–73. doi: 10.1158/1535-7163.MCT-08-0928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med. 1999;340(24):1888–99. doi: 10.1056/NEJM199906173402407. [DOI] [PubMed] [Google Scholar]
- 14.Bombardier C, Laine L, Reicin A, Shapiro D, Burgos-Vargas R, Davis B, Day R, Ferraz MB, Hawkey CJ, Hochberg MC, Kvien TK, Schnitzer TJ. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343(21):1520–8. doi: 10.1056/NEJM200011233432103. 2 p following 8. [DOI] [PubMed] [Google Scholar]
- 15.Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, Whelton A, Makuch R, Eisen G, Agrawal NM, Stenson WF, Burr AM, Zhao WW, Kent JD, Lefkowith JB, Verburg KM, Geis GS. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA. 2000;284(10):1247–55. doi: 10.1001/jama.284.10.1247. [DOI] [PubMed] [Google Scholar]
- 16.Brophy JM. Cardiovascular risk associated with celecoxib. N Engl J Med. 2005;352(25):2648–50. doi: 10.1056/NEJM200506233522519. author reply -50. [DOI] [PubMed] [Google Scholar]
- 17.Solomon SD, Wittes J, Finn PV, Fowler R, Viner J, Bertagnolli MM, Arber N, Levin B, Meinert CL, Martin B, Pater JL, Goss PE, Lance P, Obara S, Chew EY, Kim J, Arndt G, Hawk E. Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials: the cross trial safety analysis. Circulation. 2008;117(16):2104–13. doi: 10.1161/CIRCULATIONAHA.108.764530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hippisley-Cox J, Coupland C. Risk of myocardial infarction in patients taking cyclo-oxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs: population based nested case-control analysis. BMJ. 2005;330(7504):1366. doi: 10.1136/bmj.330.7504.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332(7553):1302–8. doi: 10.1136/bmj.332.7553.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schjerning Olsen AM, Fosbol EL, Lindhardsen J, Folke F, Charlot M, Selmer C, Lamberts M, Bjerring Olesen J, Kober L, Hansen PR, Torp-Pedersen C, Gislason GH. Duration of treatment with nonsteroidal anti-inflammatory drugs and impact on risk of death and recurrent myocardial infarction in patients with prior myocardial infarction: a nationwide cohort study. Circulation. 2011;123(20):2226–35. doi: 10.1161/CIRCULATIONAHA.110.004671. [DOI] [PubMed] [Google Scholar]
- 21.Andrews J, Djakiew D, Krygier S, Andrews P. Superior effectiveness of ibuprofen compared with other NSAIDs for reducing the survival of human prostate cancer cells. Cancer Chemother Pharmacol. 2002;50(4):277–84. doi: 10.1007/s00280-002-0485-8. [DOI] [PubMed] [Google Scholar]
- 22.McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A. 1999;96(1):272–7. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.John-Aryankalayil M, Palayoor ST, Cerna D, Simone CB, 2nd, Falduto MT, Magnuson SR, Coleman CN. Fractionated radiation therapy can induce a molecular profile for therapeutic targeting. Radiat Res. 2010;174(4):446–58. doi: 10.1667/RR2105.1. [DOI] [PubMed] [Google Scholar]
- 24.Niederberger E, Manderscheid C, Grosch S, Schmidt H, Ehnert C, Geisslinger G. Effects of the selective COX-2 inhibitors celecoxib and rofecoxib on human vascular cells. Biochem Pharmacol. 2004;68(2):341–50. doi: 10.1016/j.bcp.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 25.Park MK, Kim MK, Kim JC, Sung YK. Pattern of apoptosis by NS398, a selective COX-2 inhibitor, in hepatocellular carcinoma cell lines. Cancer Res Treat. 2005;37(5):313–7. doi: 10.4143/crt.2005.37.5.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amrite AC, Kompella UB. Celecoxib inhibits proliferation of retinal pigment epithelial and choroid-retinal endothelial cells by a cyclooxygenase-2-independent mechanism. J Pharmacol Exp Ther. 2008;324(2):749–58. doi: 10.1124/jpet.107.128918. [DOI] [PubMed] [Google Scholar]
- 27.Baskar R, Sparatore A, Del Soldato P, Moore PK. Effect of S-diclofenac, a novel hydrogen sulfide releasing derivative inhibit rat vascular smooth muscle cell proliferation. Eur J Pharmacol. 2008;594(1–3):1–8. doi: 10.1016/j.ejphar.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 28.Marra DE, Simoncini T, Liao JK. Inhibition of vascular smooth muscle cell proliferation by sodium salicylate mediated by upregulation of p21(Waf1) and p27(Kip1) Circulation. 2000;102(17):2124–30. doi: 10.1161/01.cir.102.17.2124. [DOI] [PubMed] [Google Scholar]
- 29.Croce K, Libby P. Intertwining of thrombosis and inflammation in atherosclerosis. Curr Opin Hematol. 2007;14(1):55–61. doi: 10.1097/00062752-200701000-00011. [DOI] [PubMed] [Google Scholar]
- 30.Levi M, van der Poll T. Two-way interactions between inflammation and coagulation. Trends Cardiovasc Med. 2005;15(7):254–9. doi: 10.1016/j.tcm.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 31.Viles-Gonzalez JF, Fuster V, Badimon JJ. Links between inflammation and thrombogenicity in atherosclerosis. Curr Mol Med. 2006;6(5):489–99. doi: 10.2174/156652406778018707. [DOI] [PubMed] [Google Scholar]
- 32.Furie B, Furie BC. The molecular basis of blood coagulation. Cell. 1988;53(4):505–18. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]
- 33.Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol. 2005;129(3):307–21. doi: 10.1111/j.1365-2141.2005.05444.x. [DOI] [PubMed] [Google Scholar]
- 34.Kassam G, Le BH, Choi KS, Kang HM, Fitzpatrick SL, Louie P, Waisman DM. The p11 subunit of the annexin II tetramer plays a key role in the stimulation of t-PA-dependent plasminogen activation. Biochemistry. 1998;37(48):16958–66. doi: 10.1021/bi981713l. [DOI] [PubMed] [Google Scholar]
- 35.Lee CW, Ahn JM, Park DW, Kim YH, Hong MK, Song JK, Kim JJ, Park SW, Chi HS, Park SJ. Tissue plasminogen activator on admission is an important predictor of 30-day mortality in patients with acute myocardial infarction undergoing primary angioplasty. Atherosclerosis. 2008;196(1):327–32. doi: 10.1016/j.atherosclerosis.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 36.Cirino G, Vergnolle N. Proteinase-activated receptors (PARs): crossroads between innate immunity and coagulation. Curr Opin Pharmacol. 2006;6(4):428–34. doi: 10.1016/j.coph.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 37.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407(6801):258–64. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 38.Gudmundsdottir IJ, Megson IL, Kell JS, Ludlam CA, Fox KA, Webb DJ, Newby DE. Direct vascular effects of protease-activated receptor type 1 agonism in vivo in humans. Circulation. 2006;114(15):1625–32. doi: 10.1161/CIRCULATIONAHA.106.638478. [DOI] [PubMed] [Google Scholar]
- 39.Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J. 2010;74(4):597–607. doi: 10.1253/circj.cj-09-0982. [DOI] [PubMed] [Google Scholar]
- 40.FitzGerald GA. COX-2 and beyond: Approaches to prostaglandin inhibition in human disease. Nat Rev Drug Discov. 2003;2(11):879–90. doi: 10.1038/nrd1225. [DOI] [PubMed] [Google Scholar]
- 41.Rabausch K, Bretschneider E, Sarbia M, Meyer-Kirchrath J, Censarek P, Pape R, Fischer JW, Schror K, Weber AA. Regulation of thrombomodulin expression in human vascular smooth muscle cells by COX-2-derived prostaglandins. Circ Res. 2005;96(1):e1–6. doi: 10.1161/01.RES.0000153150.27690.f2. [DOI] [PubMed] [Google Scholar]
- 42.Fernandez PM, Patierno SR, Rickles FR. Tissue factor and fibrin in tumor angiogenesis. Semin Thromb Hemost. 2004;30(1):31–44. doi: 10.1055/s-2004-822969. [DOI] [PubMed] [Google Scholar]
- 43.Ruf W, Mueller BM. Thrombin generation and the pathogenesis of cancer. Semin Thromb Hemost. 2006;32 (Suppl 1):61–8. doi: 10.1055/s-2006-939555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content 1: Assay ID of genes validated by real-time RT-PCR
Supplemental Digital Content 2: RT-PCR validation of selected genes differentially expressed by NSAIDs
Supplemental Digital Content 3: Microarray data of NSAID-induced differentially expressed genes associated with cardiovascular functions in CASMC and HCAEC