

Abstract
The role of telomeres and telomerase as a target for cancer therapeutics is an area of continuing interest. This review is intended to provide an update on the field, pointing to areas in which our knowledge remains deficient and exploring the details of the most promising areas being advanced into clinical trials. Topics that will be covered include the role of dysfunctional telomeres in cellular aging and how replicative senescence provides an initial barrier to the emergence of immortalized cells, a hallmark of cancer. As an important translational theme, this review will consider possibilities for selectively targeting telomeres and telomerase to enhance cancer therapy. The role of telomerase as an immunotherapy, as a gene therapy approach using telomerase promoter driven oncolytic viruses and as a small oligonucleotide targeted therapy (Imetelstat) will be discussed.
Keywords: telomerase, telomeres, cancer therapy, senescence, immortality
Targeting telomerase-expressing cancer cells
Telomerase is the enzyme responsible for the maintenance of telomeres, which cap and protect the ends of chromosomes. Telomeres are made of tandem copies of a simple DNA repeat, (TTAGGG)n, which the enzyme telomerase synthesizes. In cells that lack telomerase, telomeres shorten with each round of cell division and this attrition eventually limits cellular lifespan [1]. After a finite number of cell divisions, cells without telomerase undergo a growth arrest termed replicative senescence. During cancer development, this limit poses an obstacle that tumour cells must overcome to become malignant. In the great majority of cancers, overcoming replicative senescence is achieved initially by inactivating important cell cycle checkpoints and eventually by up-regulation of telomerase expression once telomeres have become critically short [2]. This reactivation of telomerase stabilizes the telomeres and extends cellular lifespan. To escape senescence and gain cellular immortality, the great majority of cancers up-regulate telomerase. As a consequence, telomerase is detected in more than 85% of all cases of cancer [3, 4], making the enzyme an attractive target for cancer therapeutics. In cancer cells, telomerase inhibition has been shown to lead to telomere shortening and, after sufficient cell divisions, this attrition can cause an uncapping of telomeres and induction of apoptosis [5, 6]. In this review, we discuss the biological and biochemical properties of telomerase, its use as a diagnostic and prognostic marker for cancers and its value as a target for cancer therapeutics.
Human telomeres act as a mitotic clock
Telomeres protect the ends of chromosomes from degradation, interchromosomal fusions and other forms of inadequate recombination [7, 8]. A second vital function of telomeres is to hide the ends of chromosomes from DNA damage-sensing mechanisms, which would otherwise detect the ends as double-stranded DNA breaks (ds-DNA breaks). This protective function is mediated by the activities of telomere-associated protein complexes. Telomeric DNA repeats serve as anchor for the recruitment of DNA-binding factors telomeric repeat factor (TRF)1, TRF2 and protection of telomeres 1 (POT1) in combination with other telomere associated proteins, which together form a protective capping complex originally termed telosome and more recently shelterin [8, 9]. A second protective feature is the T-loop [10]. Although most of the telomere is made of duplex telomeric DNA, all telomeres end with a G-rich single-stranded 3′-overhang of 50 to 300 bases. Evidence suggests that this extension is sequestered into a large looping structure, termed a T-loop. Formation of this structure involves the looping of the telomere and the insertion of its 3′-telomeric overhang into upstream duplex telomeric DNA [8, 10]. It has been proposed that T-loops are especially well adapted to shield the ends of chromosomes from DNA damage-sensing mechanisms and other activities. However, for these protective features to be in place, telomeres may have to be of a minimum required size. Yet, because they lack telomerase, most normal human cells will shorten their telomeres with cell divisions until the shortest telomere has become uncapped (and perhaps ‘unlooped’), giving rise to a persistent DNA damage signal [2, 11, 12].
Telomeres shorten because of problems associated with the replication of linear DNA molecules, the so-called ‘end replication problems’[13]. When replication forks reach a telomere, problems are experienced at the levels of both the leading and lagging strand synthesis. On the lagging strand, internal priming by the last Okazaki fragment and removal of the RNA primer creates a gap of unreplicated DNA [1, 2]. On the leading strand, post-replication excision by the Apollo nuclease is needed to produce a single-stranded 3′-telomeric overhang that can serve as substrate for POT1 binding and T-loop formation [14, 15]. The net result is a loss of telomeric DNA repeats each time cells divide [1, 2]. As cells divide and telomeric DNA repeats are lost, telomeres become dysfunctional, lose their protective features and become uncapped (i.e. recognized as ds-DNA breaks by the DNA damage sensing and DNA repair machinery). The response of human cells to uncapped telomeres varies depending on whether these cells have functional DNA damage and cell cycle checkpoints (Fig. 1). In an intact cell, an uncapped telomere will be recognized as a ds-DNA break by DNA damage sensing mechanisms, the activation of which will induce ATM kinase activity, phosphorylation of p53 and up-regulation of cell cycle inhibitor p21WAF1[11, 12, 16, 17]. A late response involving the up-regulation of p16INK4a and activation of downstream retinoblastoma susceptibility protein (RB) can also be observed [16–18]. The net result is an irreversible cell cycle arrest and establishment of the senescent state. In cancer cells lacking functional checkpoints, these DNA damage signals will be ignored and the cell will continue to divide and shorten its telomeres [2]. Eventually, the uncapped telomeres can serve as substrate for non-homologous end joining (NHEJ) and through this process, can become fused to other dysfunctional telomeres [7, 8, 19]. At anaphase, the dicentric chromatids generated by these fusions will fail to segregate properly and, as the cells continue to divide, there will be recurrent cycles of anaphase-bridge, breakage and fusion. Invariably, such cycles lead to a loss of genomic integrity and to a state of crisis characterized by p53-independent apoptosis [6, 20]. During cancer development, the induction of senescence or crisis poses an obstacle that tumour cells must overcome to become malignant. In the great majority of cancers, overcoming these limits to cellular lifespan is achieved by means of telomerase expression [4].
Fig 1.
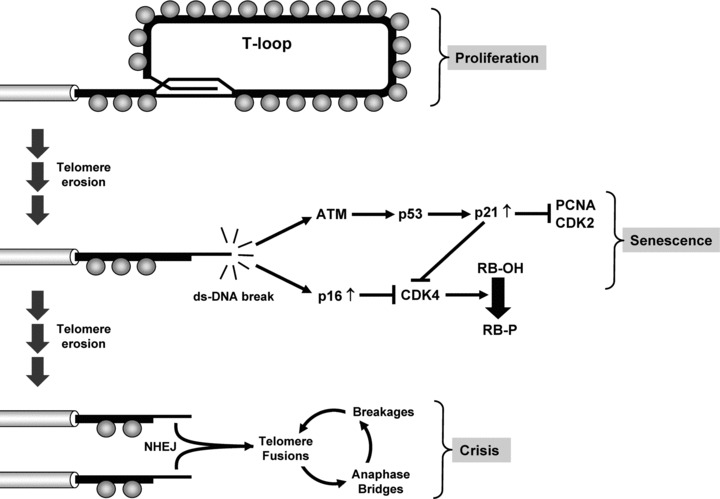
Induction of senescence and crisis as a function of telomere attrition. As cells divide and telomeric DNA repeats are lost, telomeres lose their protective shelterins (grey spheres), become unfolded (loss of the T-loop) and are recognized as ds-DNA breaks. In cells with intact checkpoints, the uncapped telomere leads to the activation of the ATM kinase, phosphorylation of p53 and up-regulation of the p21WAF1 gene (ATM/p53/p21 cascade). Once induced, p21WAF1 inhibits PCNA and cyclin-dependent kinases CDK2 and CDK4, thereby blocking the cell cycle. A late response involving the up-regulation of the p16INK4a gene is also observed. Once induced, p16INK4a blocks CDK4, the activity of which is required for inactivation of RB, a powerful inhibitor of the cell cycle (p16/CDK4/RB cascade). Acting in concert, these signalling pathways cause the cells to enter an irreversible state of growth arrest (Senescence). In cancer cells that lack components of these pathways (most commonly p53, p16 or RB), the DNA damage signals emanating from uncapped telomeres will be ignored and the cells will bypass senescence and with division, progressive telomere erosion will continue. When many telomeres become uncapped, the very short telomeres will serve as substrate for NHEJ and through this process, will become associated and/or fused to other dysfunctional telomeres. At anaphase, the dicentric chromatids produced by these fusions will fail to segregate properly (anaphase bridges), will break (breakages) and will again be involved in fusion events with other dysfunctional chromosomes (fusions). As cells continue to divide, these recurrent cycles of anaphase bridges, breakages and fusions will lead to a state of crisis characterized by p53-independent apoptosis (Crisis). Only a very rare human cell can bypass crisis and when a cell accomplishes this, a mechanism to maintain telomeres must be engaged. PCNA: proliferating cell nuclear antigen.
Telomerase expression and cellular lifespan
Telomerase compensates for the effects of the end replication problems by the synthesis of new telomeric DNA repeats [21]. In human beings, the telomerase complex has an estimated mass of over 1000 kD and may function as a homodimer. The enzyme telomerase is composed of several subunits, two of which are essential for its activity: the protein hTERT (human telomerase reverse transcriptase) and the small nuclear RNA hTR (human telomerase RNA) [22–25]. The latter contains a short sequence (5′-CUAACCCUAA-3′) that serves as a template for the synthesis of telomeric DNA repeats and hTERT provides catalytic activity (Fig. 2). The enzyme associates with its substrate, the 3′-telomeric overhang. The template hybridizes and aligns itself with the overhang, after which hTERT elongates the overhang while copying the template into DNA [21]. Each step adds a 6-base telomeric DNA repeat to the overhang, thereby elongating the telomere. The enzyme is generally but not always processive and is capable of adding many more repeats to the same telomere before falling off its substrate. To detect and measure the activity of telomerase, the Telomeric Repeat Amplification Protocol (TRAP) is most commonly used. In the TRAP assay, products of the telomerase reaction are quantified following PCR amplification [26, 27]. The assay is very sensitive and incorporates an internal standard (ITAS) with which to normalize the signals for variations in PCR efficiency. Telomerase activity is calculated as the ratio of the intensity of the telomeric products over that of the ITAS. Using the TRAP assay, the activity of the enzyme can be measured in a wide variety of samples, from very small tissue biopsies, bodily fluids to cell pellets [28].
Fig 2.
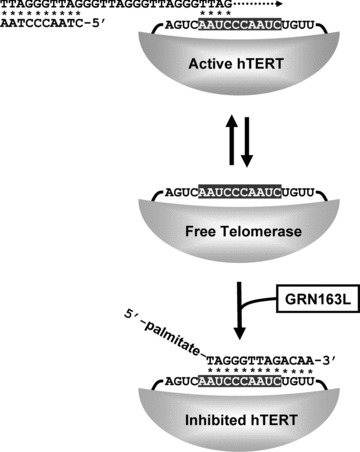
Telomerase activity and its inhibition with GRN163L. The main pathway that cells use to bypass crisis is to activate the ribonucleoprotein telomerase complex. When telomerase binds to a telomere, its RNA template region (shaded) hybridizes (asterisk) to the end of the 3′-telomeric overhang. Next, the hTERT subunit acts as a reverse transcriptase and copies the RNA template into DNA (dotted arrow). GRN163L (Imetelstat) is a N3′-P5′ thio-phosphoramidate oligonucleotide complementary to sequence of the RNA template (sequence shown) and which has been modified to carry a palmitate group at its 5′-end. When GRN163L hybridizes (asterisk) to the RNA template, the active site is blocked and the enzyme is inhibited.
At the early stages of human development, telomerase activity is ubiquitously present throughout the embryo [29, 30]. At these stages, telomerase is needed to compensate for the massive numbers of cell divisions needed to complete embryogenesis [31]. At the later stages, hTERT expression is repressed and telomerase activity becomes undetectable in most tissues, the exact timing of repression varying depending on the tissue. At birth, the activity is absent from most human cells with the exception of dividing male germ cell lineages and of rare proliferative cells of the blood, skin and gastrointestinal tract [29, 30]. In tissues that divide rapidly, tissue homeostasis is maintained by the continuous proliferation and differentiation of adult stem cells. To support the proliferation of these stem cells throughout a lifetime, it is believed that some telomerase activity is needed to slow down but not completely compensate for the rate of telomere loss [31]. The mechanism for partial telomere maintenance in proliferative stem cells is not well understood but in embryonic stem cells, telomerase completely compensates for telomere loss at each cell division. Indeed, much remains to be learned about the regulation of telomerase in normal stem cells.
A notable exception to the almost universal absence of telomerase in human tissues is cancer. A survey of the published literature established telomerase as one of the best markers of cancer cells [4]. Although most normal tissues lack the activity, telomerase is detected in more than 85% of human cancers, irrespective of the tumour type [2–4]. In most cases, the activity is localized to the tumour and the activity is absent from the surrounding normal tissues. Human cancer cells commonly possess mechanisms that compensate for telomere attrition and provide them with cellular immortality, a hallmark of cancer [32]. In the vast majority of cancers, this mechanism is the up-regulation of telomerase activity. Malignancies that develop from telomerase-negative tissues are almost always positive for the activity. Even in cancer cells originating from telomerase-positive precursors, telomerase is almost always up-regulated [4]. This ability to maintain telomeres is a fundamental property of cancer cells, which normal somatic human cells do not possess. This lack of telomere attrition provides cancer cells with cellular immortality, a capacity for unlimited number of cell divisions. This conversion to cellular immortality is needed to allow the many stages of carcinogenesis to proceed unimpeded by telomere attrition, giving the cancer cells the ability to invade without being halted by the induction of senescence or crisis [2, 16]. In some cases, however, telomerase is not detected in a tumour due to the engagement of an alternative telomere lengthening mechanism or because the tumour is self-limiting [33, 34]. In stage IVS (4S) neuroblastoma, for example, telomeres are extremely short and there is no detectable telomerase activity [34]. Amazingly, these tumours almost always regress spontaneously. This indicates that telomerase is not essential for cancer development as these children with neuroblastoma are clearly born with an advanced cancer. However, and perhaps more importantly, these results indicate that to have a sustained tumour that can progress, a mechanism to maintain telomeres must be engaged. Because of examples such as neuroblastoma where telomerase negative tumours regress, and because most cancer cells are immortal due to telomerase reactivation, targeting telomerase is an attractive strategy for cancer therapy. In the following sections, we will discuss three approaches, each using an entirely different strategy to target telomerase-expressing cancer cells. The first of these approaches consists of blocking the activity of the enzyme to limit the lifespan of telomerase-expressing cancer cells.
Telomerase inhibition to block tumour growth
Inhibiting telomerase in telomerase-expressing cancer cells leads to telomere shortening and when sufficient telomere erosion has taken place, one of two anti-proliferative barriers will be observed: either senescence or crisis [2, 35]. The two barriers are different and whether a telomerase-inhibited cancer cell experiences one or the other is dictated by the functionality of its DNA damage response and cell cycle checkpoints (Fig. 1). Senescence is a viable state of irreversible cell cycle arrest. Cells that possess functional checkpoints, such as primary human cells, undergo senescence as soon as the shortest telomere becomes uncapped [2, 35]. Crisis, which represents the preferred outcome, is a form of p53-independent apoptosis that generally leads to the death of the cancer cell [5, 6]. Because they lack functional checkpoints, most often because of mutations in the p53 and RB pathways, cancer cells will typically ignore these signals and will continue to shorten their telomeres until crisis is induced [2, 35, 36]. Because sufficient telomere shortening must occur before senescence or crisis are induced, a delay should be expected before the effects of telomerase inhibition are observed [35, 37]. This delay may preclude the use of these inhibitors as a primary line of treatment, but to block the incidence of recurrence following conventional therapy, these inhibitors are expected to be especially well suited (Fig. 3). After conventional therapy, residual cancer cells may survive and after multiple rounds of cell divisions, these cells can give rise to a new tumour. In the presence of a telomerase inhibitor, these residual cells would be predicted to shorten their telomeres as they divide, causing some to experience crisis before detectable tumour formation. For these reasons, telomerase inhibitors are now being tested in combination with conventional cancer therapy and in maintenance clinical trials aimed at reducing recurrence after surgery, chemotherapy or radiation therapy.
Fig 3.
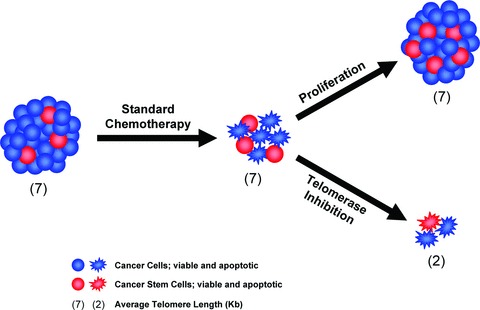
Combining standard chemotherapy with telomerase inhibition. Standard therapies have been optimized to kill bulk tumour cells, not necessarily the cancer stem cells which tend to express multidrug resistance. After standard chemotherapy, the residual cancer cells that survive may have become enriched in cancer stem cells. After multiple rounds of cell divisions, these cancer stem cells will give rise to a new tumour (arrow pointing up) and the heterogeneity of the original tumour is re-established. If the patient is treated with a telomerase inhibitor following chemotherapy (arrow pointing down), these residual cancer stem cells would be predicted to lose telomeric DNA repeats as they divide, forcing them to enter crisis after a limited number of cell divisions. The hope is that using telomerase inhibitors in a cancer maintenance setting may lead to more durable responses.
For inhibiting telomerase, the template region of the human telomerase RNA (hTR) presents an accessible target for oligonucleotide-based inhibitors [38–41]. Oligonucleotides that can hybridize to the template region of hTR, as shown previously, can be used to inhibit telomerase (Fig. 2). N3′-P5′thio-phosphoramidate oligonucleotides complementary to the hTR template have been found to be especially good telomerase inhibitors [42]. These compounds are water soluble, acid stable, resistant to nucleases and demonstrate high thermal stability of duplexes formed with their complementary RNA strands [42–46]. One compound, GRN163 (Geron Corporation, Menlo Park, CA, USA) caused telomerase inhibition and subsequent telomere shortening in a number of cancer cell lines [45–50]. As with most anionic oligonucleotides, repeated transfection of GRN163 with cationic lipophilic carriers was required for efficient intracellular uptake, because naked oligonucleotides are poorly internalized. A second-generation of GRN163 analogues, modified by lipidation, was made to facilitate cellular uptake. One such compound, GRN163L (formally known as Imetelstat), is a GRN163 oligonucleotide modified to carry a 5′-terminal palmitoyl (C16) moiety conjugated to the N3′-P5′ thio-phosphoramidate backbone [49] (5′-palmitate-TAGGGTTAGACAA-NH2-3′). GRN163L is lipid soluble and does not rely on any membrane transporter for cellular uptake. When compared to GRN163, GRN163L displayed greatly improved uptake and telomerase inhibition [46, 48, 49].
The effects of GRN163L have been investigated in cancer cells lines and in mice bearing human tumour xenografts. In a wide variety of cancer cell lines, GRN163L inhibited telomerase with IC50 in the nanomolar range [49]. In cancer cells chronically exposed to GRN163L, telomeres shorten with cell divisions and after a delay, a state of crisis is induced with evidence of chromosomal fusions, anaphase bridges and widespread apoptosis [45]. This inhibition of cellular growth by GRN163L has been reported in cancer cell lines of diverse origins, including multiple myeloma and tumours of the bladder, breast, liver, lung and stomach [45, 48, 51–54]. In mice bearing human tumour xenografts, GRN163L was determined to be well tolerated and efficacious. The maximum tolerated dose was 1000 mg/kg and telomerase inhibition could readily be detected with just 5 mg/kg [45]. Even when administered as a single agent, GRN163L could block the growth of implanted tumour cells. In mice carrying liver cancer xenografts, GRN163L alone inhibited tumour growth, in addition to sensitizing the tumour cells to conventional chemotherapy [48]. In a xenograft model of lung cancer metastasis, administration of a single dose of GRN163L was sufficient to prevent the growth of lung metastases [45]. There are several possibilities for this rapid response to GRN163L treatment. First, most primary cancers have very short telomeres, which could include some that may already be critically short. This would lead to a rapid induction of apoptosis. However, in some instances what is observed in vitro and in vivo does not concur. Thus, it is possible that GRN163L may also be exerting telomerase-independent effects on tumour growth through mechanisms that remains poorly characterized. However, on the basis of these and other pre-clinical studies, GRN163L has entered clinical trials in patients afflicted with different forms of cancer.
Phase I trials of GRN163L have just been completed in patients with chronic lymphocytic leukaemia, multiple myeloma and solid tumours (e.g. breast cancer and non-small cell lung cancer (NSCLC) [55]. From these trials, the bioavailability, pharmacokinetics, pharmacodynamics and tolerability over multiple cycles have been determined. Administered by intravenous infusions, GRN163L was found to have good safety profiles and excellent pharmacokinetics and biodistribution properties. At doses of 7.5 mg/kg and above, the level of exposure to GRN163L was higher than the exposure that was required in xenograft models to inhibit telomerase and tumour growth (Dr. Mark J. Ratain, AACR-NCI-EORTC 2009 International Conference, Boston, MA, USA). Dose escalation proceeded to 11.7 mg/kg but due to haematological toxicities, this dose was considered to exceed the maximum tolerated dose. Based on these and other considerations, 9.4 mg/kg was chosen as the recommended dose for phase II clinical trials. In combination trials, the only notable toxicities observed at this dose were reversible anaemia, thrombocytopenia and neutropenia, and it may be that some of these toxicities were due to concurrent use of standard chemotherapy. Starting at 9.4 mg/kg, phase II trials are now being initiated for NSCLC to evaluate GRN163L in a maintenance setting, with GRN163L given on days 1 and 8 of a 21 day cycle. It is well established that the standard first line induction therapy (debulking) for patients with advanced NSCLC produces partial responses in up to 35% of patients. Thus, 1–4 weeks after completing four to six cycles of induction chemotherapy, patients will be treated with Imetelstat (GRN163L), Bevacizumab (Avastin, an angiogenesis inhibitor) or observation (e.g. no maintenance therapy). Combinations of Imetelstat and Bevacizumab may be permitted in some individuals as well as cross-over from one to the other maintenance therapy. Approximately 100 patients will be enrolled in these studies with the main end-point being objective and progression-free survival. In addition to standard biomarkers, this trial will collect other tissues (e.g. peripheral blood mononuclear cells, circulating tumour cells, DNA/RNA and serum) to assay for a variety of other predictive markers. There will be approximately 25 cancer sites involved in the United States, Canada and Germany (Geron Corporation; personal communications, 2010). In other phase II trials, GRN163L will also be tested in combination with paclitaxel in patients with advanced breast cancer with the primary end-point being progression free survival. Finally, GRN163L will be tested as a single therapeutic agent for multiple myeloma, with one of the end-points being reduction in myeloma progenitor cells and essential thrombocythemia, where haematological and molecular responses will be assessed.
Mounting evidence suggests that, in many cases, recurrences are the products of residual cancer cells that possess properties of stem cells. Therefore, an important question will be whether GRN163L can target these cancer stem cells efficaciously. According to a consensus definition, a tumour cell is a cancer stem cell if it can self-renew, initiate the formation of new tumours, and regenerate all of the heterogeneous lineages of cancer cell types that comprise a tumour [56]. Rare cells that fit this definition have been identified in a growing number of diseases, from haematological malignancies to brain, breast and pancreatic cancer [57–59]. Conventional therapies have been optimized to kill bulk tumour cells, not necessarily the cancer stem cells which tend to express multidrug resistance pumps [59, 60]. When these cancer stem cells divide in the presence of a telomerase inhibitor, their telomeres should shorten and this attrition would be expected to compromise the ability of these cells to self-renew (Fig. 3). In recent studies, these predictions have been verified in the context of multiple myeloma as well as prostate, brain, breast and pancreatic cancer [61–64]. Hence, cancer stem cells isolated from multiple myeloma specimens and cell lines showed reduced proliferation with 100-fold fewer colonies after 5 weeks of exposure to GRN163L [61]. Likewise, telomerase was inhibited and telomeres shortened in cancer stem cells after treatment of prostate cancer cells with GRN163L [64]. In addition, the abundance of cancer stem cells was reduced following GRN163L treatment of prostate, pancreatic and breast cancer cell lines [62, 64]. Finally, a key characteristic of cancer stem cells isolated from brain and breast cancer is their ability to give rise to free-floating spheres, respectively called neurospheres and mammospheres. Treatment of breast cancer cell lines or primary brain tumour cells with GRN163L reduced the number of mammospheres and neurospheres that cancer stem cells could form [62, 63]. Taken together, these studies demonstrate broad anti-cancer stem cell activity of GRN163L, thereby confirming the rationale for using this drug to block disease recurrence after conventional cancer therapy.
Telomerase-targeted immunotherapy
A different approach to target telomerase-expressing cancer cells is to use a vaccine to stimulate the patient’s own immune system to attack tumour cells if they display hTERT peptides at their surface through major histocompatibility complex (MHC) presentation. Peptides displayed by MHC presentation are generated from within the cells by the limited proteolysis of cellular proteins, capture of the resulting peptides by MHC class I molecules, their transport to the plasma membrane and presentation at the surface by MHC class I molecules [65]. In telomerase-targeted immunotherapy, immune cells are exposed to a vaccine that consists of antigen-presenting cells that were either exposed to high levels of an immunogenic hTERT peptide or modified to overexpress an immunogenic fragment of hTERT. Once administered to a patient, the antigen-presenting cells elicit the activation and expansion of telomerase-specific cytotoxic T lymphocytes, which then can recognize and kill the telomerase-expressing tumour cells. Basic studies in human and murine systems have reported evidence of cytotoxic T cells that recognize dominant epitopes from hTERT and can kill tumour cells displaying these peptides [66, 67]. hTERT-based vaccines have also been reported to generate cytotoxic T cells against modified, low-affinity cryptic hTERT epitopes and these T cells protected animals against tumour challenges. In phase I clinical trials, tumour vaccines against hTERT have resulted in the induction of T-cell immune responses with minimal toxicities [68–70]. A frequent problem that limits the broad applicability of cancer vaccines is the heterogeneous expression of the tumour antigen within a tumour. However, in the case of telomerase, the antigen is both universally expressed in cancers and critical for maintenance of the malignant phenotype [71]. Already, several hTERT-based vaccines have been developed but the most advanced vaccines are the GV1001 (GemVax) and GRNVAC1 (Geron vaccine).
GV1001 is a 16-mer peptide from the active site of human TERT (aa 611–626) [68, 69]. Developed by KAEL-GemVax (Gwangju, South Korea) as an injectable formulation, the vaccinating GV1001 peptide is eventually trimmed and processed for presentation by MHC class I and class II molecules, allowing it to elicit the activity of telomerase-specific cytotoxic (CD8+) and helper (CD4+) T cells [72]. In phase I/II clinical trials for NSCLC, hepatocellular carcinoma and non-resectable pancreatic carcinoma, GV1001 has been well tolerated and shown to induce hTERT-specific T-cell responses [68–70]. GV1001 is currently in a randomized phase III clinical trial in patients with locally advanced or metastatic pancreatic cancer (ClinicalTrials.gov Identifier: NCT00425360). Patients will receive doublet therapy with gemcitabine and capecitabine either with or without GV1001 vaccination, with overall survival as the main end-point. The trial is lead by the National Cancer Research Institute in England and will enrol over 1100 pancreatic cancer patients across England. Results are expected in 2012.
GRNVAC1 is a preparation of antigen-presenting cells transduced with mRNAs encoding a near full-length hTERT protein [71]. In this case, the antigen-presenting cells are autologous dendritic cells isolated from the patients’ own tissues. Dendritic cells are a professional antigen-presenting cell whose main function is to process and display antigens [65]. In these cells, the hTERT fragment should be processed into a multitude of epitopes to elicit a polyclonal T-cell response, which could potentially recognize any hTERT peptide displayed by the patient’s tumour [71]. Currently, GRNVAC1 has completed phase II trials in patients with acute myelogenous leukaemia and metastatic prostate cancer (ClinicalTrials.gov Identifier: NCT00510133 and NCT01153113, respectively). So far, results show that GRNVAC1 is safe and well tolerated, with hTERT-specific immune responses detected in a large fraction of patients. Relapse-free survival in some patients with AML at high risk of relapse is encouraging. Results from these and other similar trials will determine if hTERT has potential has a target for broad-spectrum cancer immunotherapy.
Targeting telomerase-expressing cells with oncolytic viruses
The hTERT promoter is very tightly regulated and the promoter is generally inactive in cells that lack telomerase. Hence, a third approach to target telomerase-expressing cancer cells is to use the hTERT promoter to drive the expression of a suicide gene and/or control the replication of a lytic virus. In the first strategy, the hTERT promoter drives the expression of a pro-apoptotic protein, such as TRAIL (tumour necrosis factor-related apoptosis-inducing ligand) or prodrug-activating enzyme [73–78]. Viruses carrying the suicide gene are injected in a tumour and telomerase-expressing cancer cells are then killed by the administration of a prodrug, which the activating enzyme converts to a toxin. A second strategy, currently in clinical trials, is to use the hTERT promoter as a switch that controls the replication of a lytic virus [79]. Viral proteins E1A and E1B are required for replication of the adenovirus, and if the E1 gene is redesigned to be controlled by the hTERT promoter, the modified virus can only replicate in cells expressing telomerase [80–83]. One such virus, telomelysin (OBP-301), is currently being used in phase I clinical trials for various types of tumours [84]. In pre-clinical studies, this and other similarly engineered adenovirus showed great promises for the selective and efficacious targeting of telomerase-expressing cells. In vitro, the modified viruses could replicate in a wide variety of human cancer cell lines, effectively inducing cytolysis [80–83]. In normal human cells lacking telomerase, replication and cytotoxicity were greatly attenuated. In mice implanted with human lung, prostate or liver cancer cells, intratumoral injection of the virus effectively retarded tumour growth and extended the survival of mice, including mice with large tumour burden [80–83]. When injected into large xenografts, the virus could reduce the size of the tumours with formation of a massive ulceration at the site of injection [81]. Viruses were not detected in tissues outside of the injected tumours, except for the presence of circulating viruses in the blood as well as spreading of the virus to other distant tumour sites [81]. Viral particles released after cytolysis can infect other nearby cells or spread to the rest of the body via blood or lymphatic flow, potentially allowing the virus to reach other tumour sites, including distant metastases [79]. Based on these and other studies, a phase I clinical trial was conducted in patients with advanced solid tumours to assess the clinical safety of the OBP-301 virus, also known as telomelysin [84]. Sixteen patients with a variety of solid tumours were enrolled and given a single intratumoral injection of the virus, after which safety, response and pharmacodynamics were evaluated. The virus was well tolerated and evidence of antitumour activity was reported. One patient had a partial response at the injected malignant lesion and seven patients demonstrated stable disease 56 days after treatment. These studies demonstrated the feasibility of targeting telomerase-expressing cancer cells with oncolytic viruses. Second-generation viruses that combine the two strategies by using the hTERT promoter to control both viral replication and the expression of a suicide gene have also been designed [85, 86]. Studies of these other second-generation viruses should determine if these oncolytic viruses have potential in the clinics for broad-spectrum cancer therapy.
Concluding remarks
The maintenance of telomeres by the cellular ribonucleoprotein enzyme telomerase is of well-documented importance for cancer [2, 87–90]. Telomerase, a cellular reverse transcriptase that adds DNA to the ends of chromosomes, is reactivated or up-regulated in the vast majority of advanced malignancies, and is thus an almost universal target for the treatment of human cancers. Most human tumours not only express telomerase but also have very short telomeres, whereas telomerase activity is absent or at lower levels in normal tissues which also have longer telomeres. This relationship between activation of telomerase activity and short telomeres in human malignancies makes the inhibition of telomerase a valuable target for cancer therapeutics. Importantly, the mode of action of telomerase inhibitors predicts minimal side effects on normal stem cells that can express telomerase. In this review, we summarized the role of telomeres and telomerase in cancer and reviewed the current status of ongoing telomerase clinical trials. But many questions still remain, including: What are the key safety concerns, such as the effects of telomerase inhibitors on normal stem cells? Is telomerase activity regulated by the same mechanisms in cancer stem cells as it is in normal stem cells? How can telomerase inhibitors affect cancer stem cells if they are more quiescent than the more differentiated bulk tumour cells? Will human cancers become resistant to telomerase inhibitors? Clearly more basic research will be required as initial results from clinical trials emerge.
In summary, the multitude of studies on human telomeres and telomerase highlight the complex interplay between signals emanating from dysfunctional telomeres and tumour suppressor pathways that regulate stem cell biology, aging and cancer. A more complete understanding of these relationships should lead to more effective clinical trials.
Acknowledgments
This study was supported in part by grants P50CA127297 and P50CA70907.
Conflict of interest
The authors confirm that there are no conflicts with the exception that they have conducted collaborative research with Geron Corporation, Menlo Park, CA, USA.
References
- 1.Harley CB. Telomere loss: mitotic clock or genetic time bomb. Mutat Res. 1991;256:271–82. doi: 10.1016/0921-8734(91)90018-7. [DOI] [PubMed] [Google Scholar]
- 2.Shay JW, Wright WE. Telomerase activity in human cancer. Curr Opin Oncol. 1996;8:66–71. doi: 10.1097/00001622-199601000-00012. [DOI] [PubMed] [Google Scholar]
- 3.Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–5. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 4.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–91. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 5.Hahn WC, Stewart SA, Brooks MW, et al. Inhibition of telomerase limits the growth of human cancer cells. Nat Med. 1999;5:1164–70. doi: 10.1038/13495. [DOI] [PubMed] [Google Scholar]
- 6.Zhang X, Mar V, Zhou W, et al. Telomere shortening and apoptosis in telomerase-inhibited human tumor cells. Genes Dev. 1999;13:2388–99. doi: 10.1101/gad.13.18.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Lange T. Protection of mammalian telomeres. Oncogene. 2002;21:532–40. doi: 10.1038/sj.onc.1205080. [DOI] [PubMed] [Google Scholar]
- 8.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–10. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 9.Liu D, O’Connor MS, Qin J, et al. Telosome, a mammalian telomere-associated complex formed by multiple telomeric proteins. J Biol Chem. 2004;279:51338–42. doi: 10.1074/jbc.M409293200. [DOI] [PubMed] [Google Scholar]
- 10.de Lange T. T-loops and the origin of telomeres. Nat Rev Mol Cell Biol. 2004;5:323–9. doi: 10.1038/nrm1359. [DOI] [PubMed] [Google Scholar]
- 11.Karlseder J, Broccoli D, Dai Y, et al. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321–5. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 12.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–56. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 13.Watson JD. Origin of concatemeric T7 DNA. Nat New Biol. 1972;239:197–201. doi: 10.1038/newbio239197a0. [DOI] [PubMed] [Google Scholar]
- 14.Wu P, van Overbeek M, Rooney S, et al. Apollo contributes to G overhang maintenance and protects leading-end telomeres. Mol Cell. 2010;39:606–17. doi: 10.1016/j.molcel.2010.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lam YC, Akhter S, Gu P, et al. SNMIB/Apollo protects leading-strand telomeres against NHEJ-mediated repair. EMBO J. 2010;29:2230–41. doi: 10.1038/emboj.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ouellette MM, Choi KH. Telomeres and telomerase in ageing and cancer. Encyclopedia of life sciences http://wwwelsnet/). Chichester, UK: John Wiley & Sons Ltd; 2007.
- 17.Shay JW, Pereira-Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res. 1991;196:33–9. doi: 10.1016/0014-4827(91)90453-2. [DOI] [PubMed] [Google Scholar]
- 18.Robles SJ, Adami GR. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene. 1998;16:1113–23. doi: 10.1038/sj.onc.1201862. [DOI] [PubMed] [Google Scholar]
- 19.Smogorzewska A, Karlseder J, Holtgreve-Grez H, et al. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr Biol. 2002;12:1635–44. doi: 10.1016/s0960-9822(02)01179-x. [DOI] [PubMed] [Google Scholar]
- 20.Macera-Bloch L, Houghton J, Lenahan M, et al. Termination of lifespan of SV40-transformed human fibroblasts in crisis is due to apoptosis. J Cell Physiol. 2002;190:332–44. doi: 10.1002/jcp.10062. [DOI] [PubMed] [Google Scholar]
- 21.Collins K, Mitchell JR. Telomerase in the human organism. Oncogene. 2002;21:564–79. doi: 10.1038/sj.onc.1205083. [DOI] [PubMed] [Google Scholar]
- 22.Feng J, Funk WD, Wang SS, et al. The RNA component of human telomerase. Science. 1995;269:1236–41. doi: 10.1126/science.7544491. [DOI] [PubMed] [Google Scholar]
- 23.Harrington L, Zhou W, McPhail T, et al. Human telomerase contains evolutionarily conserved catalytic and structural subunits. Genes Dev. 1997;11:3109–15. doi: 10.1101/gad.11.23.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyerson M, Counter CM, Eaton EN, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–95. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura TM, Morin GB, Chapman KB, et al. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–9. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- 26.Herbert BS, Hochreiter AE, Wright WE, et al. Nonradioactive detection of telomerase activity using the telomeric repeat amplification protocol. Nat Protoc. 2006;1:1583–90. doi: 10.1038/nprot.2006.239. [DOI] [PubMed] [Google Scholar]
- 27.Wright WE, Shay JW, Piatyszek MA. Modifications of a telomeric repeat amplification protocol (TRAP) result in increased reliability, linearity and sensitivity. Nucleic Acids Res. 1995;23:3794–5. doi: 10.1093/nar/23.18.3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hess JL, Highsmith WE., Jr Telomerase detection in body fluids. Clin Chem. 2002;48:18–24. [PubMed] [Google Scholar]
- 29.Wright WE, Piatyszek MA, Rainey WE, et al. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18:173–9. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 30.Ulaner GA, Giudice LC. Developmental regulation of telomerase activity in human fetal tissues during gestation. Mol Hum Reprod. 1997;3:769–73. doi: 10.1093/molehr/3.9.769. [DOI] [PubMed] [Google Scholar]
- 31.Forsyth NR, Wright WE, Shay JW. Telomerase and differentiation in multicellular organisms: turn it off, turn it on, and turn it off again. Differentiation. 2002;69:188–97. doi: 10.1046/j.1432-0436.2002.690412.x. [DOI] [PubMed] [Google Scholar]
- 32.Hahn WC, Counter CM, Lundberg AS, et al. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–8. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 33.Henson JD, Neumann AA, Yeager TR, et al. Alternative lengthening of telomeres in mammalian cells. Oncogene. 2002;21:598–610. doi: 10.1038/sj.onc.1205058. [DOI] [PubMed] [Google Scholar]
- 34.Hiyama E, Hiyama K, Yokoyama T, et al. Correlating telomerase activity levels with human neuroblastoma outcomes. Nat Med. 1995;1:249–55. doi: 10.1038/nm0395-249. [DOI] [PubMed] [Google Scholar]
- 35.Ouellette MM, Lee K. Telomerase: diagnostics, cancer therapeutics and tissue engineering. Drug Discov Today. 2001;6:1231–7. doi: 10.1016/s1359-6446(01)02052-9. [DOI] [PubMed] [Google Scholar]
- 36.Wright WE, Shay JW. The two-stage mechanism controlling cellular senescence and immortalization. Exp Gerontol. 1992;27:383–9. doi: 10.1016/0531-5565(92)90069-c. [DOI] [PubMed] [Google Scholar]
- 37.Shay JW, Wright WE. Telomerase: a target for cancer therapeutics. Cancer Cell. 2002;2:257–65. doi: 10.1016/s1535-6108(02)00159-9. [DOI] [PubMed] [Google Scholar]
- 38.Shay JW, Wright WE. Mechanism-based combination telomerase inhibition therapy. Cancer Cell. 2005;7:1–2. doi: 10.1016/j.ccr.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 39.White LK, Wright WE, Shay JW. Telomerase inhibitors. Trends Biotechnol. 2001;19:114–20. doi: 10.1016/s0167-7799(00)01541-9. [DOI] [PubMed] [Google Scholar]
- 40.Hamilton SE, Pitts AE, Katipally RR, et al. Identification of determinants for inhibitor binding within the RNA active site of human telomerase using PNA scanning. Biochemistry. 1997;36:11873–80. doi: 10.1021/bi970438k. [DOI] [PubMed] [Google Scholar]
- 41.Norton JC, Piatyszek MA, Wright WE, et al. Inhibition of human telomerase activity by peptide nucleic acids. Nat Biotechnol. 1996;14:615–9. doi: 10.1038/nbt0596-615. [DOI] [PubMed] [Google Scholar]
- 42.Herbert BS, Pongracz K, Shay JW, et al. Oligonucleotide N3′–>P5′ phosphoramidates as efficient telomerase inhibitors. Oncogene. 2002;21:638–42. doi: 10.1038/sj.onc.1205064. [DOI] [PubMed] [Google Scholar]
- 43.Asai A, Oshima Y, Yamamoto Y, et al. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res. 2003;63:3931–9. [PubMed] [Google Scholar]
- 44.Gryaznov S, Pongracz K, Matray T, et al. Telomerase inhibitors–oligonucleotide phosphoramidates as potential therapeutic agents. Nucleosides Nucleotides Nucleic Acids. 2001;20:401–10. doi: 10.1081/NCN-100002314. [DOI] [PubMed] [Google Scholar]
- 45.Dikmen ZG, Gellert GC, Jackson S, et al. In vivo inhibition of lung cancer by GRN163L: a novel human telomerase inhibitor. Cancer Res. 2005;65:7866–73. doi: 10.1158/0008-5472.CAN-05-1215. [DOI] [PubMed] [Google Scholar]
- 46.Gellert GC, Dikmen ZG, Wright WE, et al. Effects of a novel telomerase inhibitor, GRN163L, in human breast cancer. Breast Cancer Res Treat. 2006;96:73–81. doi: 10.1007/s10549-005-9043-5. [DOI] [PubMed] [Google Scholar]
- 47.Chen Z, Koeneman KS, Corey DR. Consequences of telomerase inhibition and combination treatments for the proliferation of cancer cells. Cancer Res. 2003;63:5917–25. [PubMed] [Google Scholar]
- 48.Djojosubroto MW, Chin AC, Go N, et al. Telomerase antagonists GRN163 and GRN163L inhibit tumor growth and increase chemosensitivity of human hepatoma. Hepatology. 2005;42:1127–36. doi: 10.1002/hep.20822. [DOI] [PubMed] [Google Scholar]
- 49.Herbert BS, Gellert GC, Hochreiter AE, et al. Lipid modification of GRN163, an N3’–>P5’ thio-phosphoramidate oligonucleotide, enhances the potency of telomerase inhibition. Oncogene. 2005;24:5262–8. doi: 10.1038/sj.onc.1208760. [DOI] [PubMed] [Google Scholar]
- 50.Perry PJ, Arnold JR, Jenkins TC. Telomerase inhibitors for the treatment of cancer: the current perspective. Expert Opin Investig Drugs. 2001;10:2141–56. doi: 10.1517/13543784.10.12.2141. [DOI] [PubMed] [Google Scholar]
- 51.Shammas MA, Qazi A, Batchu RB, et al. Telomere maintenance in laser capture microdissection-purified Barrett’s adenocarcinoma cells and effect of telomerase inhibition in vivo. Clin Cancer Res. 2008;14:4971–80. doi: 10.1158/1078-0432.CCR-08-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dikmen ZG, Wright WE, Shay JW, et al. Telomerase targeted oligonucleotide thio-phosphoramidates in T24-luc bladder cancer cells. J Cell Biochem. 2008;104:444–52. doi: 10.1002/jcb.21635. [DOI] [PubMed] [Google Scholar]
- 53.Shammas MA, Koley H, Bertheau RC, et al. Telomerase inhibitor GRN163L inhibits myeloma cell growth in vitro and in vivo. Leukemia. 2008;22:1410–8. doi: 10.1038/leu.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hochreiter AE, Xiao H, Goldblatt EM, et al. Telomerase template antagonist GRN163L disrupts telomere maintenance, tumor growth, and metastasis of breast cancer. Clin Cancer Res. 2006;12:3184–92. doi: 10.1158/1078-0432.CCR-05-2760. [DOI] [PubMed] [Google Scholar]
- 55.Molckovsky A, Siu LL. First-in-class, first-in-human phase I results of targeted agents: highlights of the 2008 American society of clinical oncology meeting. J Hematol Oncol. 2008;1:20–8. doi: 10.1186/1756-8722-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 57.Allan AL, Vantyghem SA, Tuck AB, et al. Tumor dormancy and cancer stem cells: implications for the biology and treatment of breast cancer metastasis. Breast Dis. 2006;26:87–98. doi: 10.3233/bd-2007-26108. [DOI] [PubMed] [Google Scholar]
- 58.Farnie G, Clarke RB. Breast stem cells and cancer. Ernst Schering Found Symp Proc. 2006;5:141–53. doi: 10.1007/2789_2007_049. [DOI] [PubMed] [Google Scholar]
- 59.Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–23. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 60.Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 61.Brennan SK, Wang Q, Tressler R, et al. Telomerase inhibition targets clonogenic multiple myeloma cells through telomere length-dependent and independent mechanisms. PLoS One. 2010;5:e12487. doi: 10.1371/journal.pone.0012487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Joseph I, Tressler R, Bassett E, et al. The telomerase inhibitor imetelstat depletes cancer stem cells in breast and pancreatic cancer cell lines. Cancer Res. 2010;70:9494–504. doi: 10.1158/0008-5472.CAN-10-0233. [DOI] [PubMed] [Google Scholar]
- 63.Marian CO, Cho SK, McEllin BM, et al. The telomerase antagonist, imetelstat, efficiently targets glioblastoma tumor-initiating cells leading to decreased proliferation and tumor growth. Clin Cancer Res. 2010;16:154–63. doi: 10.1158/1078-0432.CCR-09-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marian CO, Wright WE, Shay JW. The effects of telomerase inhibition on prostate tumor-initiating cells. Int J Cancer. 2010;127:321–31. doi: 10.1002/ijc.25043. [DOI] [PubMed] [Google Scholar]
- 65.Van den Eynde BJ, van der Bruggen P. T cell defined tumor antigens. Curr Opin Immunol. 1997;9:684–93. doi: 10.1016/s0952-7915(97)80050-7. [DOI] [PubMed] [Google Scholar]
- 66.Vonderheide RH, Hahn WC, Schultze JL, et al. The telomerase catalytic subunit is a widely expressed tumor-associated antigen recognized by cytotoxic T lymphocytes. Immunity. 1999;10:673–9. doi: 10.1016/s1074-7613(00)80066-7. [DOI] [PubMed] [Google Scholar]
- 67.Vonderheide RH. Telomerase as a universal tumor-associated antigen for cancer immunotherapy. Oncogene. 2002;21:674–9. doi: 10.1038/sj.onc.1205074. [DOI] [PubMed] [Google Scholar]
- 68.Bernhardt SL, Gjertsen MK, Trachsel S, et al. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer. 2006;95:1474–82. doi: 10.1038/sj.bjc.6603437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brunsvig PF, Aamdal S, Gjertsen MK, et al. Telomerase peptide vaccination: a phase I/II study in patients with non-small cell lung cancer. Cancer Immunol Immunother. 2006;55:1553–64. doi: 10.1007/s00262-006-0145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Greten TF, Forner A, Korangy F, et al. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer. 2010;10:209–13. doi: 10.1186/1471-2407-10-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer. 2008;8:167–9. doi: 10.1038/nrc2275. [DOI] [PubMed] [Google Scholar]
- 72.Kyte JA. Cancer vaccination with telomerase peptide GV1001. Expert Opin Investig Drugs. 2009;18:687–94. doi: 10.1517/13543780902897631. [DOI] [PubMed] [Google Scholar]
- 73.Katz MH, Spivack DE, Takimoto S, et al. Gene therapy of pancreatic cancer with green fluorescent protein and tumor necrosis factor-related apoptosis-inducing ligand fusion gene expression driven by a human telomerase reverse transcriptase promoter. Ann Surg Oncol. 2003;10:762–72. doi: 10.1245/aso.2003.01.021. [DOI] [PubMed] [Google Scholar]
- 74.Liu J, Zou WG, Lang MF, et al. Cancer-specific killing by the CD suicide gene using the human telomerase reverse transcriptase promoter. Int J Oncol. 2002;21:661–6. [PubMed] [Google Scholar]
- 75.Majumdar AS, Hughes DE, Lichtsteiner SP, et al. The telomerase reverse transcriptase promoter drives efficacious tumor suicide gene therapy while preventing hepatotoxicity encountered with constitutive promoters. Gene Ther. 2001;8:568–78. doi: 10.1038/sj.gt.3301421. [DOI] [PubMed] [Google Scholar]
- 76.Plumb JA, Bilsland A, Kakani R, et al. Telomerase-specific suicide gene therapy vectors expressing bacterial nitroreductase sensitize human cancer cells to the pro-drug CB1954. Oncogene. 2001;20:7797–803. doi: 10.1038/sj.onc.1204954. [DOI] [PubMed] [Google Scholar]
- 77.Schepelmann S, Ogilvie LM, Hedley D, et al. Suicide gene therapy of human colon carcinoma xenografts using an armed oncolytic adenovirus expressing carboxypeptidase G2. Cancer Res. 2007;67:4949–55. doi: 10.1158/0008-5472.CAN-07-0297. [DOI] [PubMed] [Google Scholar]
- 78.Zhou JH, Tang B, Liu XL, et al. hTERT-targeted E. coli purine nucleoside phosphorylase gene/6-methylpurine deoxyribose therapy for pancreatic cancer. Chin Med J. 2007;120:1348–52. [PubMed] [Google Scholar]
- 79.Kyo S, Takakura M, Fujiwara T, et al. Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci. 2008;99:1528–38. doi: 10.1111/j.1349-7006.2008.00878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Irving J, Wang Z, Powell S, et al. Conditionally replicative adenovirus driven by the human telomerase promoter provides broad-spectrum antitumor activity without liver toxicity. Cancer Gene Ther. 2004;11:174–85. doi: 10.1038/sj.cgt.7700666. [DOI] [PubMed] [Google Scholar]
- 81.Kawashima T, Kagawa S, Kobayashi N, et al. Telomerase-specific replication-selective virotherapy for human cancer. Clin Cancer Res. 2004;10:285–92. doi: 10.1158/1078-0432.ccr-1075-3. [DOI] [PubMed] [Google Scholar]
- 82.Lanson NA, Jr, Friedlander PL, Schwarzenberger P, et al. Replication of an adenoviral vector controlled by the human telomerase reverse transcriptase promoter causes tumor-selective tumor lysis. Cancer Res. 2003;63:7936–41. [PubMed] [Google Scholar]
- 83.Wirth T, Zender L, Schulte B, et al. A telomerase-dependent conditionally replicating adenovirus for selective treatment of cancer. Cancer Res. 2003;63:3181–8. [PubMed] [Google Scholar]
- 84.Nemunaitis J, Tong AW, Nemunaitis M, et al. A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol Ther. 2010;18:429–34. doi: 10.1038/mt.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liao Z, Huang C, Zhou F, et al. Radiation enhances suicide gene therapy in radioresistant laryngeal squamous cell carcinoma via activation of a tumor-specific promoter. Cancer Lett. 2009;283:20–8. doi: 10.1016/j.canlet.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 86.Zhang JF, Wei F, Wang HP, et al. Potent anti-tumor activity of telomerase-dependent and HSV-TK armed oncolytic adenovirus for non-small cell lung cancer in vitro and in vivo. J Exp Clin Cancer Res. 2010;29:52–7. doi: 10.1186/1756-9966-29-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shay JW. Telomerase in human development and cancer. J Cell Physiol. 1997;173:266–70. doi: 10.1002/(SICI)1097-4652(199711)173:2<266::AID-JCP33>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 88.Langford LA, Piatyszek MA, Xu RS, et al. Telomerase activity in ordinary meningiomas predicts poor outcome. Human Pathol. 1997;28:416–20. doi: 10.1016/s0046-8177(97)90029-0. [DOI] [PubMed] [Google Scholar]
- 89.Wright WE, Shay JW. Time, telomeres and tumors. Is cellular senescence more than an anticancer mechanism. Trends Cell Biol. 1995;5:293–7. doi: 10.1016/s0962-8924(00)89044-3. [DOI] [PubMed] [Google Scholar]
- 90.Shay JW. Aging and Cancer – are telomeres and telomerase the connection. Mol Med Today. 1995;1:378–84. doi: 10.1016/s1357-4310(95)93872-9. [DOI] [PubMed] [Google Scholar]