

Abstract
Despite the clear need to control visceral leishmaniasis (VL), the existing diagnostic tests have serious shortcomings. Here, we introduce an innovative approach to directly identify Leishmania infantum antigens produced in vivo in humans with VL. We combined reverse-phase high-performance liquid chromatography (RP-HPLC) with mass spectrometry and categorized three distinct L. infantum proteins presumably produced in bone marrow/spleen/liver and excreted in the urine of patients with VL. The genes coding for these proteins (L. infantum iron superoxide dismutase, NCBI accession number XP_001467866.1; L. infantum tryparedoxin, NCBI accession number XP_001466642.1; and L. infantum nuclear transport factor 2, NCBI accession number XP_001463738.1) were cloned, and the recombinant molecules were produced in Escherichia coli. Antibodies to these proteins were produced in rabbits and chickens and were used to develop a capture enzyme-linked immunosorbent assay (ELISA) designed to detect these L. infantum antigens in the urine of VL patients. Specificity of the antibodies was confirmed by a Western blot analysis using both recombinant proteins and whole parasite extract. Importantly, a urinary antigen detection assay assembled with pairs of antibodies specific for each of these antigens identified 17 of 19 patients with VL. These results indicate that an improved antigen detection assay based on L. infantum proteins present in the urine of patients with VL may represent an important new strategy for the development of a specific and accurate diagnostic test that has the potential to both distinguish active VL from asymptomatic infection and serve as an important tool to monitor therapy efficacy.
Visceral leishmaniasis (VL) is endemic in 47 countries, with approximately 200 million people at risk of infection and an annual incidence estimated to be 500,000 cases (http://who.int/leishmaniasis/disease_epidemiology/en/index.html). The disease is caused by parasites of the Leishmania donovani complex (L. donovani and Leishmania archibaldi in the Old World and Leishmania infantum in Southern Europe, Africa, and South America). Notwithstanding the existence of antileishmanial drugs, global visceral leishmaniasis (VL) morbidity and mortality remain high and in many parts of the world are increasing due to coinfection with human immunodeficiency virus (HIV) (1, 2). In addition to being a human disease, VL caused by L. infantum is a zoonotic infection. Domestic dogs are the major vertebrate reservoirs of the parasite (41). Canine VL (CVL) is widely distributed in Latin America and Southern Europe (6, 19). In the United States, the potential for CVL to become a significant problem has recently been highlighted (7, 20, 22).
These alarming facts have been attributed in part to the absence of an efficacious VL vaccine. In addition, an accurate diagnostic test that can identify active VL versus asymptomatic disease remains a key component of measurements that aim to control this serious disease that is missing (11). Definitive diagnosis of active VL still relies primarily on the direct finding of the Leishmania parasites either in smears or in cultures from spleen or bone marrow aspirates, which are obtained using invasive procedures that are a risk to the patient's health. Importantly, the sensitivity of these tests is, in general, not high and varies enormously (14, 24, 28, 34, 51, 53). Alternatives to these procedures are a variety of nucleic acid amplification tests (3, 13, 29, 43). These tests are more sensitive than microscopic examination and parasite culture, but they remain restricted to referral hospitals and research centers despite efforts to simplify them (11).
Several conventional serological tests have been developed and are available for VL diagnosis. However, because of the overall principle of these tests, i.e., detection of antibody responses to parasite antigens, they have inherent limitations, particularly for the diagnosis of active VL. First, high serum antibody levels are present in both asymptomatic and active VL (5, 8, 9, 12, 16, 45). Second, serum anti-Leishmania antibodies remain present for several years after the patient has been cured, an outcome that complicates the diagnosis of relapsed VL (15, 25, 32). Third, a number of individuals from areas of VL endemicity with no history of VL do have antileishmanial antibodies, therefore complicating the specificity of these tests (21). Fourth, sensitivity of serological tests in VL/HIV-coinfected patients is poor, particularly if leishmaniasis occurs post-HIV infection (29, 47).
An interesting alternative approach to conventional serological tests is the direct identification of leishmanial antigens in the bodily fluids of humans with active VL. Indeed, we have previously used this premise to search for Mycobacterium tuberculosis proteins in the urine of patients with pulmonary tuberculosis. Using mass spectroscopy, we identified four unique peptides that have sequence homologies to the deduced amino acid sequences of proteins from M. tuberculosis in the urine samples of tuberculosis patients (31) and from mice infected with M. tuberculosis (36, 37). In addition, we confirmed the immunological and clinical validation of these molecules as candidates for the development of an antigen detection assay for active tuberculosis (39).
Here, we describe the use of this approach for the direct identification of L. infantum diagnostic candidate molecules in the urine of VL patients. Three parasite polypeptides could be clearly identified. These molecules have been extensively studied and used for the development of a promising antigen detection assay for VL diagnosis.
MATERIALS AND METHODS
Human samples.
A total of 25 urine samples from patients with VL were evaluated in this study. These samples were collected from patients diagnosed with VL based on the following criteria: a clinical course consistent with VL (e.g., fever, anemia, hepatosplenomegaly) and confirmatory laboratory findings (identification of Leishmania in bone marrow aspirates). A single urine sample was collected from each patient at the time of his/her first visit to the hospital for diagnosis and treatment of VL. Urine specimens were stored frozen at −20°C and subsequently shipped in dry ice to the Forsyth Institute, Cambridge, MA. The patients were from the Natan Portella Institute of Tropical Diseases, Federal University of Piauí, Teresina, PI, Brazil. In addition, urine samples were obtained from healthy subjects (n = 16) and from patients with cutaneous leishmaniasis (n = 10), Chagas' disease (n = 8), schistosomiasis (n = 14), and tuberculosis (n = 10). These samples were from patients from the area of Montes Claros city, MG, Brazil, and were collected and confirmed by the Department of Parasitology, Federal University of Minas Gerais, Belo Horizonte, MG, Brazil. Urine donation protocols were approved by the Investigational Review Board and the Ethics Committee of the Federal University of Piauí Medical School and Federal University of Minas Gerais, respectively.
Mass spectroscopy analysis.
Frozen individual samples (15 ml) from five patients with confirmed VL were thawed, centrifuged, and filtered over 0.2-μm filters. Urine specimens were then concentrated using Centricon P3 (3-kDa cutoff filters) to ∼200 to 300 μl. Equal volumes of concentrated urine specimens were mixed with electrophoresis sample buffer and then submitted to SDS-PAGE, followed by Coomassie staining. Bands ranging from ∼10 kDa to ∼75 kDa were, in general, excised from the gel and submitted for mass spectroscopy (MS) analysis at the Taplin Mass Spectrometry Facility, Harvard Medical School, Boston, MA. Figure 1 illustrates a typical pattern of bands obtained for the concentrated urine of a patient with VL. This pattern was highly analogous to those of all other VL urine samples as well as to that obtained for urine specimens of two healthy control subjects. Figure 1 also illustrates the positions of the bands that were excised from the gel for further analyses. For each urine sample, 8 to 10 bands were cut from the gel. Each band was then independently submitted to MS runs. Gel bands were trypsin digested into peptides. Peptides were analyzed by nanoscale liquid chromatography coupled to a tandem mass spectrometer. Eluted peptides first had their molecular masses measured and then were fragmented, and finally, the fragment masses were measured. The specific fragmentation pattern was computer searched against predicted tryptic peptides from all known proteins from genome sequencing projects of humans and Leishmania protozoa. The power of the technique is in its redundancy. Because many peptides are generated from the initial gel band, multiple matches to the protein of interest are detected. In this way, the protein identity is completely unambiguous. Peptide score cutoff values were chosen at cross-correlation (Xcorr) values of 1.8 for singly charged ions, 2.5 for doubly charged ions, and 3.0 for triply charged ions, along with the magnitude of predicted fragment ion values (deltaCN) of 0.1 and the final correlation score rank (RSP) values of 1. The cross-correlation values chosen for each peptide ensure a high confidence match for the different charge states, while the deltaCN cutoff ensures the uniqueness of the peptide hit. The RSP value of 1 ensured that the peptide matched the top hit in the preliminary scoring and that the peptide fragment file matched only one protein hit.
Fig 1.
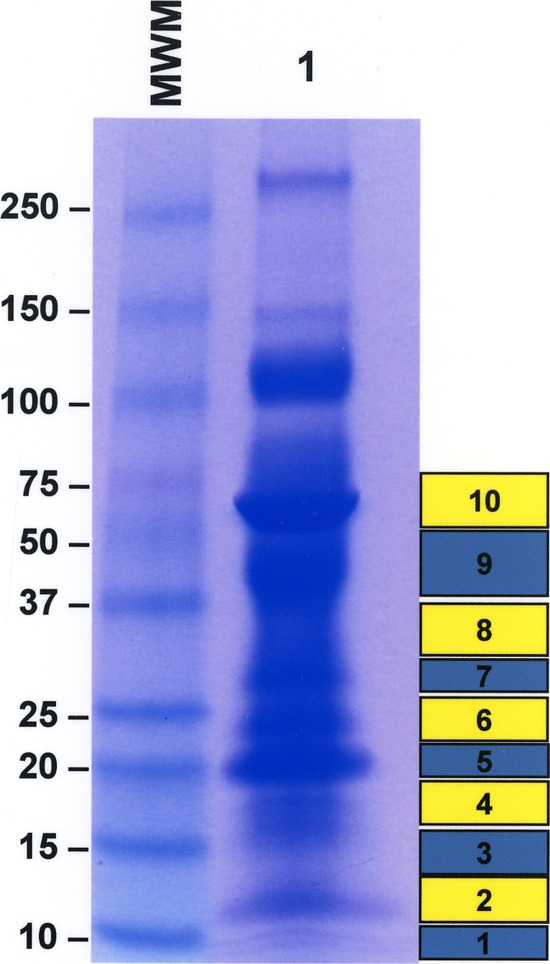
Illustration of the pattern of urine protein bands that were cut from polyacrylamide gels for mass spectroscopy analyses. Urine was from a patient with visceral leishmaniasis (VL). Urine was concentrated 10× using a Centricon P3 (3-kDa cutoff), mixed with sample buffer, and then submitted to SDS-PAGE (polyacrylamide gel with a 4 to 20% gradient), followed by Coomassie staining. Bands ranging from ∼10 kDa to ∼75 kDa were cut from the gel and submitted to mass spectroscopy analyses. Numbered boxes on the right side of the gel indicate the individual bands that were cut from the gel. Lane 1, 10× concentrated urine from VL patient; MM, molecular markers. Numbers on the left of the gel are the molecular masses of the markers.
Cloning of the Leishmania infantum gene, protein expression, and purification.
Oligonucleotide PCR primers were designed to amplify the full-length open reading frame of the target genes from genomic DNA of L. infantum. The forward primer contained an NdeI restriction site at the ATG initiation codon followed by sequences derived from the gene. The reverse primer included a BamHI restriction site followed by a stop codon and sequences from the gene. The resultant PCR products were digested with restriction enzymes and subcloned into the pET-14b expression vector, which was similarly digested for directional cloning. Alternatively, the DNA sequence was codon optimized for expression in Escherichia coli containing the same restriction enzyme sites (NdeI and BamHI). The DNA fragment was synthesized (Blue Heron, Bothell, WA), and the synthetic gene was subcloned into pET-14b as well. A ligated pET-14b vector was subsequently used to transform E. coli BL21(DE3)pLysS host cells (Novagen, Madison, WI) for expression. Recombinant proteins were obtained and purified from 100 ml of isopropyl-β-d-thiogalactopyranoside (IPTG)-induced batch cultures by affinity chromatography using QIAexpress Ni-NTA agarose matrix (Qiagen, Chatsworth, CA) as described previously (31, 37). The yields of recombinant proteins were 10 to 20 mg per liter of induced bacterial culture, and purity was assessed by SDS-PAGE followed by Coomassie blue staining. Endotoxin levels present in the purified recombinant protein preparations were measured by the Limulus amebocyte lysate assay (Lonza, Walkersville, MD) and shown to be <5 endotoxin units (EU)/ml.
Generation of specific polyclonal antibodies.
The purified recombinant protein (250 μg) was emulsified with an equal volume of incomplete Freund adjuvant (IFA) and injected at multiple subcutaneous (s.c.) sites into female New Zealand rabbits or New Hampshire Red chickens (Capralogics Inc., Hardwick, MA). The animals were given two s.c. boosters (250 μg of Ag in IFA) 3 weeks apart. One week after the final boost, the animals were bled and sera (from the rabbits) and yolks (from the chicken eggs) were collected. IgG (from rabbits) and IgY (from chickens) were purified by standard affinity chromatography or by using antigen immobilized on Sepharose 4B resin (cyanogen bromide [CNBr]-activated Sepharose 4B, GE Healthcare). A portion of the rabbit IgG was biotinylated with the EZ-Link Sulfo-NHS-LC biotinylation kit from Thermo Fischer Scientific (Pittsburgh, PA) according to the manufacturer's instructions. Between 1 and 2.5 molecules of biotin per IgG were invariably obtained for all the different batches.
Western blot.
Purified recombinant proteins (100 ng) and whole-antigen extract from Leishmania infantum were fractionated by SDS-PAGE (4 to 20% gradient gel) and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Medford, MA). Crude Leishmania infantum lysates were prepared from promastigote parasites cultured for 7 to 10 days in complete Schneider's medium at 26°C (promastigote lysate preparation). The blots were blocked overnight at 4°C with Tris-buffered saline with 0.1% Tween 20 (TBS-T) containing 1% bovine serum albumin (BSA) and subsequently probed with antigen-specific rabbit antisera or preimmune rabbit sera. After several rinses with TBS-T, goat anti-rabbit IgG labeled with horseradish peroxidase (Thermo Scientific Pierce, Rockford, IL) was added. After additional washings, bound conjugates were detected using the ECL enhanced chemiluminescence system (Amersham/GE Healthcare, Piscataway, NJ) and proteins were visualized by autoradiography (Kodak BioMax, Rochester, NY).
Capture ELISA.
A capture enzyme-linked immunosorbent assay (ELISA) antigen detection test was developed using purified IgG anti-L. infantum recombinant antigens obtained from antisera produced in two different rabbits or IgY produced in chickens. Briefly, wells of 96-well ELISA plates (high-binding EIA/RIA plates; Corning International, Corning, NY) were coated overnight at 4°C with 0.2 μg of purified IgG (or IgY) diluted in bicarbonate buffer, pH 9.0. Wells were washed with phosphate-buffered saline (PBS) and 0.1% Tween 20 (Sigma Chemical Co., St. Louis, MO) and blocked at room temperature with PBS with 1% BSA and 0.1% Tween 20 (PBS/BSA/Tween) for 2 h. After washing, human urine samples were added and incubated overnight at 4°C. Plates were washed, followed by incubation for 1 h with biotin-labeled IgG (obtained from the second immunized rabbit) or biotin-labeled IgY at 1/2,000, a dilution previously determined by a conventional sandwich ELISA. Following several rinses in PBS/BSA/Tween, peroxidase-labeled streptavidin at a 1/2,000 dilution (BD Bioscience, Franklin Lakes, NJ) was added for 30 min. The plates were then washed, and reactions were developed with 3,3′,5,5′-tetramethylbenzidine (TMB) substrate and read at 450 nm.
RESULTS
Isolation of three unique L. infantum protein antigens from the urine of patients with VL.
Urine was collected from five patients with active, parasitologically confirmed VL registered at the Natan Portella Institute of Tropical Medicine, Teresina, PI, Brazil. None of the enrolled patients had any clinical signs or symptoms or laboratory findings compatible with renal or urinary tract abnormalities. These exclusion criteria were important to rule out renal pathology, which can theoretically be a factor that would be biasing the finding of L. infantum antigens present in the patients' urine. None of the patients were under anti-Leishmania therapy at the time of urine collection. Individual urine samples were analyzed by mass spectrometry, generating a total of approximately 400 peptide sequences. Most sequences of the identified peptides had sequence homologies to those of human proteins. However, eight peptide sequences that had no known homologies with human proteins had sequence homologies to the deduced sequences of three different L. infantum proteins (Table 1). Two of these peptides (LNAAAESNSGLASK and GGGEPSGPLASAIVDSFGSFASFK) had sequences with perfect matching to the sequence of L. infantum iron superoxide dismutase (NCBI accession number XP_001467866.1) at positions 40 to 53 and 87 to 110, respectively. Four other peptides (QNDMVDMSSLSGK, MPWLSIPFEK, QYKVESIPTLIGLNADTGDTVTTR, and VESIPTLIGLNADTGDTVTTR) had sequences with perfect matching to the sequence of L. infantum tryparedoxin (NCBI accession number XP_001466642.1) at positions 17 to 29, 84 to 93, 103 to 126, and 106 to 126, respectively. Finally, two peptides (FANLGFTEAAFK and EQVQGVDAIMAR) had sequences with perfect and strong matching to the sequence of a putative L. infantum protein (nuclear transport factor 2; NCBI accession number XP_001463738.1) at positions 40 to 51 and 52 to 63, respectively. Importantly, most of these L. infantum peptides were identified in two out of five urine samples, thus strongly validating these findings (Table 2). Finally, no leishmanial matching peptides were found in urine specimens of two control subjects processed in a manner identical to that of the urine specimens of the VL patients (not shown).
Table 1.
L. infantum peptides identified by mass spectroscopy in individual urine samples of patients with visceral leishmaniasis
| Peptide | Xcorr value | Putative L. infantum peptide donor protein | NCBI annotation | Gene length (bp) | Molecular mass of protein (kDa) | pI |
|---|---|---|---|---|---|---|
| LNAAAESNSGLASK | 3.602 | Iron superoxide dismutase (Li-isd1) | XM_001467829 | 588 | 21.53 | 8.7 |
| GGGEPSGPLASAIVDSFGSFASFK | 5.274 | Iron superoxide dismutase (Li-isd1) | XM_001467829 | 588 | 21.53 | 8.7 |
| QNDMVDMSSLSGK | 2.813 | Tryparedoxin (Li-txn1) | XM_001466605 | 438 | 16.7 | 5.2 |
| MPWLSIPFEK | 2.789 | Tryparedoxin (Li-txn1) | XM_001466605 | 438 | 16.7 | 5.2 |
| QYKVESIPTLIGLNADTGDTVTTR | 3.630 | Tryparedoxin (Li-txn1) | XM_001466605 | 438 | 16.7 | 5.2 |
| VESIPTLIGLNADTGDTVTTR | 4.168 | Tryparedoxin (Li-txn1) | XM_001466605 | 438 | 16.7 | 5.2 |
| FANLGFTEAAFK | 4.0849 | Nuclear transport factor 2 (Li-ntf2) | XM_001463701 | 375 | 13.89 | 4.9 |
| EQVQGVDAIMAR | 3.1871 | Nuclear transport factor 2 (Li-ntf2) | XM_001463701 | 375 | 13.89 | 4.9 |
Table 2.
Occurrence of identified L. infantum peptides among the VL patients who donated urine for MS analyses
| Protein | Peptide | Presence of each peptide in each patient (gender, age [yr]) |
||||
|---|---|---|---|---|---|---|
| A (female, 2) | B (male, 7) | C (male, 29) | D (female, 2) | E (female, 6) | ||
| Li-isd1 | LNAAAESNSGLASK | + | ||||
| GGGEPSGPLASAIVDSFGSFASFK | + | |||||
| Li-txn1 | QNDMVDMSSLSGK | + | + | |||
| MPWLSIPFEK | + | + | ||||
| QYKVESIPTLIGLNADTGDTVTTR | + | + | ||||
| VESIPTLIGLNADTGDTVTTR | + | + | ||||
| Li-ntf2 | FANLGFTEAAFK | + | + | |||
| EQVQGVDAIMAR | + | |||||
Gene cloning and protein expression/purification and characterization of discovered L. infantum antigens.
The open reading frame of each of the full-length genes coding for each protein was amplified by PCR from L. infantum genomic DNA, followed by subcloning into the pET-14b expression vector. The recombinant proteins L. infantum iron superoxide dismutase (Li-isd1) and L. infantum tryparedoxin (Li-txn1) were readily obtained. However, this cloning strategy resulted in no expression of the L. infantum nuclear transport factor (Linft2) protein. Nevertheless, using a synthetic gene with an optimized sequence for E. coli, followed by subcloning in pET-14b, resulted in excellent overexpression of the recombinant protein. Recombinant proteins were next purified using Ni-nitrilotriacetic acid (NTA) agarose resin, and purity was assessed by SDS-PAGE with Coomassie blue staining and is illustrated in Fig. 2A. To validate the recombinant proteins as replicas of the native parasite molecules, a Western blot analysis was carried out using specific rabbit IgG antibodies. A crude lysate of the parasite and the purified recombinant proteins were immunoblotted and then probed with the specific polyclonal rabbit antisera (Fig. 2B). All three antisera recognized a single band in the crude parasite lysate that migrated at a molecular mass that was similar to that for the recombinant proteins. Therefore, the results support that the recombinant proteins are replicas of the native molecules produced by L. infantum. A second band of ∼45 kDa was also recognized by the anti-Li-ntf2 antiserum in the crude parasite lysate. It is possible that the Li-ntf2 protein aggregates with itself or that the band is part of a larger protein complex or a building block of a larger protein originated by posttranslational modification.
Fig 2.

Purification and characterization of the recombinant Li-isd1, Li-txn1, and Li-ntf2 proteins. Recombinant proteins containing His-tagged amino terminal residues were expressed in E. coli BL21(DE3)pLysS, followed by purification by affinity chromatography using Ni-NTA agarose matrix. Purity was evaluated by SDS-PAGE (polyacrylamide gel with a 4 to 20% gradient) and Coomassie blue staining (A). Lane 1, Li-isd1; lane 2, Li-txn1; lane 3, Li-ntf2. (B) Characterization of the proteins was done by a Western blot analysis. Fifty nanograms of purified recombinant proteins (lanes R) and 2 μg of crude antigenic preparation of promastigote forms of L. infantum (lanes N, for native proteins) were submitted to electrophoresis under reducing conditions in polyacrylamide gel with a 4 to 20% gradient and transferred to a PVDF membrane. Proteins were identified using specific rabbit anti-recombinant protein antisera. Reactivity was detected with goat anti-rabbit IgG labeled with horseradish peroxidase and a luminol-based analysis system, followed by autoradiography. Numbers on the left side indicate the molecular masses of the markers.
Recognition of recombinant Li-isd1, Li-txn1, and Li-ntf2 proteins by sera from patients with VL.
To evaluate if the discovered antigens were biologically active during disease, an ELISA was carried out using sera from seven well-characterized patients with active VL. All patients were bone marrow smear confirmed for VL. The patients and control sera were from the Natan Portella Institute of Tropical Medicine, Teresina, PI, Brazil. Figure 3 indicates that the patients' sera tested by ELISA reacted with the three antigens at higher titers than did control sera (n = 5). These results suggest that during disease, the L. infantum Li-isd1, Li-txn1, and Li-nft2 proteins are produced in sufficient quantities to sensitize the patient's immune system to produce specific antibodies, thus confirming them as biologically significant molecules produced in vivo during disease.
Fig 3.

Recognition of the recombinant proteins Li-isd1, Li-txn1, and Li-ntf2 by sera from patients with VL. Recognition of the antigens was determined by ELISA. Microtiter plates (96-well) were coated with 100 ng of antigen/well, blocked, and incubated with a 1/50 dilution of the sera from VL patients and healthy control subjects. Antibody reactivity was measured using peroxidase-labeled goat anti-human IgG. Reactions were developed after addition of the substrate (H2O2) and the chromophore TMB. Results are expressed as ODs read at 450 nm. Symbols show the results obtained for urine specimens of seven individual patients with VL (A) and five normal control subjects (B).
Optimization of a capture ELISA for Li-isd1, Li-txn1, and Li-ntf2.
Reagents were initially tested in order to obtain the highest sensitivity limit of detection (LOD) to identify the Leishmania antigens in human urine. Purified rabbit and chicken immunoglobulins (IgG/IgY) were initially titrated, and those antibodies that provided optical density (OD) readings arbitrarily higher than 0.5 above the background at the lowest concentrations were selected for further evaluations as capture reagents. The purified antibodies were then titrated as capture reagents in an antigen detection ELISA. Plates were coated with different concentrations of the antibodies, followed by incubation with a fixed concentration of corresponding antigens (5 ng/ml). Biotinylated antibody specific for each antigen was then added, followed by peroxidase-labeled streptavidin and TMB. This approach led to the selection of an antigen affinity-purified rabbit antibody specific for the antigen Li-txn1, a whole rabbit IgG fraction (affinity purified using Streptococcus protein G) specific for Li-ntf2, and an antigen affinity-purified chicken IgY specific for the antigen Li-isd1. The concentrations of antibody required to provide the highest OD signals above the background varied for each antigen/antibody capture ELISA and were 100 ng/well, 875 ng/well, and 2,000 ng/well for Li-isd1, Li-txn1, and Li-ntf2, respectively.
To determine the LOD, several concentrations of the antigens were tested in the presence and absence of human urine samples obtained from normal and healthy subjects. As can be seen in Fig. 4, as little as ∼4 pg/ml of the Li-isd1 antigen can be detected in the presence of either PBS or control human urine. The limits of detection for Li-txn1 and Li-ntf2 were ∼20 pg/ml and ∼5 pg/ml, respectively. Importantly, normal human urine did not interfere with the sensitivity of the antigen detection capture ELISAs specific for the three candidate antigens. These results are important in that they indicate that interference of urine should not occur with assays developed to diagnose VL using this human specimen.
Fig 4.

Determination of the limit of detection (LOD) of capture ELISAs assembled for the proteins Li-isd1, Li-txn1, and Li-ntf2, spiked in urine specimens of normal healthy subjects. Capture antibodies at previously determined concentrations were used to coat the ELISA plates. The following antibody concentrations were used: antigen affinity-purified IgY anti-Li-isd1, 100 ng/well (A); antigen affinity-purified rabbit anti-Li-txn1 antibodies, 875 ng/well (B); and purified rabbit IgG anti-Li-ntf2 antibody (2,000 ng/well) (C). Wells were then incubated with various concentrations of the antigen diluted either in saline plus 1% BSA or in urine from normal healthy subjects, followed by incubation with biotin-labeled anti-antigen secondary antibody. Reactions were developed after addition of peroxidase-labeled streptavidin, substrate (H2O2), and the chromophore TMB. Results are expressed as ODs read at 450 nm.
Detection of Li-isd1, Li-txn1, and Li-ntf2 in patients' urine specimens using a capture ELISA.
Given that the limit of antigen detection of the capture ELISAs assembled with different antibodies to the L. infantum antigens present in the urine of patients with VL was in the range of 5 to 50 pg/ml of protein and that human urine did not interfere with the assay sensitivity, we next investigated the possible utility of these assays for the development of an antigen detection assay for the diagnosis of active VL. This type of assay is in high demand to accurately diagnose VL and to distinguish active disease from asymptomatic infection. To validate the assay, 19 urine specimens from well-characterized VL patients from the Natan Portella Institute of Tropical Medicine (Teresina, PI, Brazil) and 16 urine specimens from healthy control individuals were initially tested. The patient population comprised 4 females aged 2 to 36 years old and 15 males aged 5 to 44 years old. Diagnosis of VL was based on the presence of the following signs and symptoms in all patients: chronic and irregular fever, abdominal pain, progressive emaciation, malaise, anemia, weight loss, hepatosplenomegaly (>2 cm from the costal margin), and finding of a Leishmania amastigote in bone marrow aspirates. All urine specimens were collected before the initiation of the anti-Leishmania therapy and stored frozen at −20°C. The urine specimens were then thawed, and the capture ELISA was performed without concentration or any specific manipulation of the urine specimens. Figure 5 shows the results and indicates that the majority of the VL patients' urine samples provided strong positive ELISA signals for the three antigens. The cutoffs to consider positive signals were established as the OD readings given by the mean of the readings of the 16 urine samples from healthy, normal individuals plus 3 standard deviations (SDs) of the mean. Specifically, 15 patients' samples provided strong positive ELISA signals for the antigen Li-isd1. In addition, an equal number of urine samples provided positive ELISA signals for the antigen Li-ntf2, and 9 patients' urine samples were positive for the antigen Li-txn1. Altogether, these results validate the use of these L. infantum proteins as important candidates for the development of a sensitive and specific antigen detection assay for the diagnosis of active VL.
Fig 5.

Antigen detection assays for the identification of the proteins Li-isd1, Li-txn1, and Li-ntf2 in urine of VL patients and controls. Urine specimens were from VL patients and normal, healthy control subjects. Assays were carried out using capture ELISAs assembled as described in the legend for Fig. 4. (A) Anti-Li-isd1. (B) Anti-Li-txn1. (C) Anti-Li-ntf2. Samples from VL patients (n = 19) were from Teresina, PI, Brazil. Control samples were from healthy individuals from countries where VL is endemic who were living in the United States. Dashed lines represent the cutoff values, which were calculated using the average of the ODs obtained from the urine specimens from 16 normal, healthy control subjects plus 3 SDs. Note that, compiling the results obtained for the three proteins, the antigen detection assay was positive in 17/19 VL patients. These are representative results of at least three experiments performed at different times with the same urine samples and same capture ELISA.
Specificity of the capture ELISA for VL.
To begin to evaluate the specificity of the capture ELISA for VL, urine samples were collected from patients with cutaneous leishmaniasis (CL), Chagas' disease, schistosomiasis, and tuberculosis (TB). These diseases were chosen for this initial specificity validation because they represent important pathologies caused by organisms that produce proteins that can potentially cross-react with the antibodies used in the VL antigen detection assay under study. Therefore, the capture ELISAs for Li-isd1, Li-txn1, and Li-ntf2 were performed simultaneously with the urine specimens from these patients and the urine specimens from VL patients. No cross-reaction was observed with any of the urine specimens from the four groups of patients tested (Fig. 6). Although the number of patients thus far studied is limited, these results suggest that the antigen detection assay under development should be highly specific for VL.
Fig 6.

Specificity of the capture ELISA for Li-isd1, Li-txn1, and Li-ntf2. Urine specimens were from patients with cutaneous leishmaniasis (CL) (n = 10), Chagas' disease (CD) (n = 8), schistosomiasis (SC) (n = 14), or tuberculosis (TB) (n = 10), from normal, healthy control subjects (HLT) (n = 16), or from patients with VL (n = 19). Assays were carried out using capture ELISAs assembled as described in the legend for Fig. 4. (A) Anti-Li-isd1. (B) Anti-Li-txn1. (C) Anti-Li-ntf2. Dashed lines represent the cutoff values, which were calculated using the average of the ODs obtained from the urine specimens from 16 normal, healthy control subjects plus 3 SDs. Note that no cross-reaction was observed with any of the 42 urine specimens with diseases other than VL. The box-and-whiskers diagram was used to highlight the differences and dispersions (the black components of the bars indicate skewness) of the ODs obtained for the urine specimens from VL patients compared to the ODs obtained for patients with CL, CD, SC, or TB and healthy control subjects.
DISCUSSION
We have recently described an interesting alternative approach for the direct identification of M. tuberculosis antigens in bodily fluids of mice infected with this organism as well as in urine specimens of patients with pulmonary tuberculosis. The identified antigens have been successfully tested as vaccine candidates as well as candidate tools for the development of an antigen diagnostic test for active tuberculosis (10, 31, 36, 37, 39).
The translation of this strategy for antigen discovery to patients with VL was readily achieved. Pooled urine specimens collected from patients with well-characterized VL yielded over 400 peptide sequences with homology to those of human proteins and nine peptide sequences with homology to those of three L. infantum proteins. Therefore, we reasoned that these molecules are interesting possible molecular markers of active disease and, consequently, good candidates for the development of an antigen detection assay for VL.
Expression and purification of the recombinant proteins were achieved with no major difficulties, and the validation as genuine L. infantum molecules was done by a Western blot analysis. The demonstration that the molecule was produced in vitro was done using rabbit or chicken antisera specific for the recombinant proteins. The antisera specific for the proteins Li-isd1 and Li-txn1 detected a single band of the expected molecular mass of L. infantum proteins in the parasite lysate. In contrast, the antiserum specific for Li-ntf2, in addition to a band that migrates at approximately the 14-kDa position, which coincides with the predicted molecular mass of the native Li-ntf2 (13.89 kDa), detects a second band that migrates at an ∼45-kDa molecular mass position. At this time, we cannot conclude whether the band of ∼45 kDa is a homo- or heteropolymer of Li-ntf2 or not. Nonetheless, it is unlikely that it represents a nonspecific reaction of the antiserum with an unrelated antigen present in L. infantum because the recognition of this band was much stronger than that of the band that matches the predicted molecular mass of the native molecule. However, further investigation will need to be carried out to clarify the nature of this 45-kDa molecule. Moreover, and as expected, all three recombinant molecules migrate slightly slower than the native molecules. This is indeed expected, as the recombinant proteins have a slightly higher molecular mass than the native molecules due to the presence of the 6His tag.
The concept of detecting microbial molecules in human bodily fluids of infected individuals for diagnosis purposes has a strong precedent. For example, molecules from numerous viruses, bacteria, such as Streptococcus pneumoniae or Legionella pneumophila, and parasites, such as Entamoeba histolytica, have long been described in various human samples (e.g., blood, mucous secretions, and feces) of patients suffering from the diseases caused by these microorganisms (23, 27, 30, 40, 49, 50). Interestingly, many of these molecules were successfully used either as vaccines (e.g., for hepatitis A and B) or as tools for the development of antigen detection-based diagnostics. Perhaps the most successful example of such tests is the commercially available test to detect Streptococcus pyogenes (group A) in patients with tonsillitis. This rapid test has been universally used as a routine diagnostic of S. pyogenes pharyngitis for more than 10 years (18, 26, 33, 38).
Paradoxically, although an antigen detection assay has the potential to discriminate active VL from asymptomatic infection and cured patients, development of such a test has only recently become a matter of interest. Indeed, a test that is based on detection of L. donovani polysaccharide antigens in the urine of VL patients is under clinical validation. Despite conflicting results regarding the sensitivity and specificity, a significant correlation between this assay and conventional serological and parasitological tests has been reported (4, 17, 42, 46, 48, 52). Therefore, the highly purified recombinant proteins described in the present studies are of great interest as candidates to either replace or complement the sensitivity and specificity of the previously described polysaccharide detection test. This possibility is supported by the findings that together, the protein detection test can detect the native antigens in the urine of 17/19 patients with active VL. Interestingly, no cross-reaction was observed with urine specimens of patients with CL, Chagas' disease, schistosomiasis, or TB, pointing to high specificity. Although the sensitivity is slightly below the ideal for an actual clinical test, we believe that the results are highly encouraging because the capture ELISA used to detect the antigens Li-txn1 and Lintf2 was assembled with whole IgG fractions from the immunized rabbits and not with antigen-purified antibody. We are currently in the process of preparing monoclonal antibodies specific for these antigens as well as large quantities of the recombinant molecule to facilitate the purification in large amounts of antibody to assemble a more sensitive capture ELISA. These reagents will also facilitate the development of other detection systems, e.g., tests based on luminescence or fluorescence readouts (44).
It is important to mention that because antigen detection assays are highly dependent on the antigen load present in the sample under analysis, they are theoretically useful tools to follow up therapy efficacy, particularly of infectious diseases. Therefore, it will be interesting to test VL capture ELISAs for the antigens Li-isd1, Li-txn1, and Li-ntf2 for this purpose. In addition, the capture ELISA should be useful for the diagnosis of VL in VL/HIV-coinfected patients, as the conventional serological diagnosis of VL in these patients is problematic and not sensitive (29, 47).
Finally, the genes coding for the identified L. infantum proteins are highly conserved among the organisms of the L. donovani complex. Therefore, the antibodies raised against the L. infantum recombinant proteins should recognize equally well the proteins produced by L. donovani, thus suggesting that the proposed test will be equally sensitive to detect VL caused by either L. donovani or L. infantum.
In conclusion, these results strongly support the premise of the approach used in these studies, confirming that we have developed a powerful and reliable antigen discovery strategy to directly identify L. infantum diagnostic candidate antigens in human bodily secretions. In addition, these results confirm our previous hypothesis (31, 35, 36, 39) that this approach is broadly applicable to several infectious diseases, particularly those caused by organisms that have their genome already completely sequenced.
ACKNOWLEDGMENTS
We thank Farrokh Modabber for his critical reading of the manuscript and suggestions.
C.A. is an employee of DetectoGen Inc. and has no ownership or ownership option in the company. A.C.-N. is a consultant for DetectoGen Inc.
This work was supported by NIH grant R43AI084259-01, awarded to C.A.
Footnotes
Published ahead of print 18 April 2012
REFERENCES
- 1. Alvar J, et al. 2008. The relationship between leishmaniasis and AIDS: the second 10 years. Clin. Microbiol. Rev. 21: 334–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alvar J, et al. 1997. Leishmania and human immunodeficiency virus coinfection: the first 10 years. Clin. Microbiol. Rev. 10: 298–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Antinori S, et al. 2007. Clinical use of polymerase chain reaction performed on peripheral blood and bone marrow samples for the diagnosis and monitoring of visceral leishmaniasis in HIV-infected and HIV-uninfected patients: a single-center, 8-year experience in Italy and review of the literature. Clin. Infect. Dis. 44: 1602–1610 [DOI] [PubMed] [Google Scholar]
- 4. Attar ZJ, et al. 2001. Latex agglutination test for the detection of urinary antigens in visceral leishmaniasis. Acta Trop. 78: 11–16 [DOI] [PubMed] [Google Scholar]
- 5. Badaro R, et al. 2006. Immunotherapy for drug-refractory mucosal leishmaniasis. J. Infect. Dis. 194: 1151–1159 [DOI] [PubMed] [Google Scholar]
- 6. Belazzoug S. 1992. Leishmaniasis in Mediterranean countries. Vet. Parasitol. 44: 15–19 [DOI] [PubMed] [Google Scholar]
- 7. Boggiatto PM, et al. 2011. Transplacental transmission of Leishmania infantum as a means for continued disease incidence in North America. PLoS Negl. Trop. Dis. 5: e1019 doi:10.1371/journal.pntd.0001019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Braz RF, et al. 2002. The sensitivity and specificity of Leishmania chagasi recombinant K39 antigen in the diagnosis of American visceral leishmaniasis and in differentiating active from subclinical infection. Am. J. Trop. Med. Hyg. 67: 344–348 [DOI] [PubMed] [Google Scholar]
- 9. Carvalho EM, et al. 1992. Immunologic markers of clinical evolution in children recently infected with Leishmania donovani chagasi. J. Infect. Dis. 165: 535–540 [DOI] [PubMed] [Google Scholar]
- 10. Cayabyab MJ, Kashino SS, Campos-Neto A. 2012. Robust immune response elicited by a novel and unique Mycobacterium tuberculosis protein using an optimized DNA/protein heterologous prime/boost protocol. Immunology 135: 216–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chappuis F, et al. 2007. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 5: 873–882 [DOI] [PubMed] [Google Scholar]
- 12. Costa CH, et al. 2002. Asymptomatic human carriers of Leishmania chagasi. Am. J. Trop. Med. Hyg. 66: 334–337 [DOI] [PubMed] [Google Scholar]
- 13. Costa JM, et al. 1996. PCR enzyme-linked immunosorbent assay for diagnosis of leishmaniasis in human immunodeficiency virus-infected patients. J. Clin. Microbiol. 34: 1831–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cota GF, de Sousa MR, Rabello A. 2011. Predictors of visceral leishmaniasis relapse in HIV-infected patients: a systematic review. PLoS Negl. Trop. Dis. 5: e1153 doi:10.1371/journal.pntd.0001153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Almeida Silva L, et al. 2006. Immunologic tests in patients after clinical cure of visceral leishmaniasis. Am. J. Trop. Med. Hyg. 75: 739–743 [PubMed] [Google Scholar]
- 16. de Gouvea Viana L, et al. 2008. Combined diagnostic methods identify a remarkable proportion of asymptomatic Leishmania (Leishmania) chagasi carriers who present modulated cytokine profiles. Trans. R. Soc. Trop. Med. Hyg. 102: 548–555 [DOI] [PubMed] [Google Scholar]
- 17. Diro E, et al. 2007. Field evaluation of FD-DAT, rK39 dipstick and KATEX (urine latex agglutination) for diagnosis of visceral leishmaniasis in northwest Ethiopia. Trans. R. Soc. Trop. Med. Hyg. 101: 908–914 [DOI] [PubMed] [Google Scholar]
- 18. Dominguez J, et al. 2001. Detection of Streptococcus pneumoniae antigen by a rapid immunochromatographic assay in urine samples. Chest 119: 243–249 [DOI] [PubMed] [Google Scholar]
- 19. Dujardin JC, et al. 2008. Spread of vector-borne diseases and neglect of Leishmaniasis, Europe. Emerg. Infect. Dis. 14: 1013–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eddlestone SM. 2000. Visceral leishmaniasis in a dog from Maryland. J. Am. Vet. Med. Assoc. 217: 1686–1688, 1659 [DOI] [PubMed] [Google Scholar]
- 21. Falqueto A, et al. 2009. Cross-sectional and longitudinal epidemiologic surveys of human and canine Leishmania infantum visceral infections in an endemic rural area of southeast Brazil (Pancas, Espirito Santo). Am. J. Trop. Med. Hyg. 80: 559–565 [PubMed] [Google Scholar]
- 22. Gaskin AA, et al. 2002. Visceral leishmaniasis in a New York foxhound kennel. J. Vet. Intern. Med. 16: 34–44 [DOI] [PubMed] [Google Scholar]
- 23. Grundy MS. 1982. Preliminary observations using a multi-layer ELISA method for the detection of Entamoeba histolytica trophozoite antigens in stool samples. Trans. R. Soc. Trop. Med. Hyg. 76: 396–400 [DOI] [PubMed] [Google Scholar]
- 24. Guerin PJ, et al. 2002. Visceral leishmaniasis: current status of control, diagnosis, and treatment, and a proposed research and development agenda. Lancet Infect. Dis. 2: 494–501 [DOI] [PubMed] [Google Scholar]
- 25. Hailu A. 1990. Pre- and post-treatment antibody levels in visceral leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 84: 673–675 [DOI] [PubMed] [Google Scholar]
- 26. Heiter BJ, Bourbeau PP. 1995. Comparison of two rapid streptococcal antigen detection assays with culture for diagnosis of streptococcal pharyngitis. J. Clin. Microbiol. 33: 1408–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Helbig JH, et al. 2003. Clinical utility of urinary antigen detection for diagnosis of community-acquired, travel-associated, and nosocomial Legionnaires' disease. J. Clin. Microbiol. 41: 838–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ho EA, Soong TH, Li Y. 1948. Comparative merits of sternum, spleen and liver punctures in the study of human visceral leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 41: 629–636 [DOI] [PubMed] [Google Scholar]
- 29. Houghton RL, et al. 1998. A cloned antigen (recombinant K39) of Leishmania chagasi diagnostic for visceral leishmaniasis in human immunodeficiency virus type 1 patients and a prognostic indicator for monitoring patients undergoing drug therapy. J. Infect. Dis. 177: 1339–1344 [DOI] [PubMed] [Google Scholar]
- 30. Karki BM, Parija SC. 1999. Co-agglutination test for the detection of circulating antigen in amebic liver abscess. Am. J. Trop. Med. Hyg. 60: 498–501 [DOI] [PubMed] [Google Scholar]
- 31. Kashino SS, Pollock N, Napolitano DR, Rodrigues V, Jr, Campos-Neto A. 2008. Identification and characterization of Mycobacterium tuberculosis antigens in urine of patients with active pulmonary tuberculosis: an innovative and alternative approach of antigen discovery of useful microbial molecules. Clin. Exp. Immunol. 153: 56–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kumar P, Pai K, Tripathi K, Pandey HP, Sundar S. 2002. Immunoblot analysis of the humoral immune response to Leishmania donovani polypeptides in cases of human visceral leishmaniasis: its usefulness in prognosis. Clin. Diagn. Lab. Immunol. 9: 1119–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Laubscher B, van Melle G, Dreyfuss N, de Crousaz H. 1995. Evaluation of a new immunologic test kit for rapid detection of group A streptococci, the Abbott Testpack Strep A plus. J. Clin. Microbiol. 33: 260–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Melby PC. 2002. Recent developments in leishmaniasis. Curr. Opin. Infect. Dis. 15: 485–490 [DOI] [PubMed] [Google Scholar]
- 35. Mukherjee S, Daifalla N, Liu C, Campos-Neto A. 2003. Alternative approach to express Mycobacterium tuberculosis proteins in Escherichia coli. Biotechniques 35: 34–36, 38, 40 [DOI] [PubMed] [Google Scholar]
- 36. Mukherjee S, et al. 2004. Potential serological use of a recombinant protein that is a replica of a Mycobacterium tuberculosis protein found in the urine of infected mice. Clin. Diagn. Lab Immunol. 11: 280–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mukherjee S, et al. 2005. Cloning of the gene encoding a protective Mycobacterium tuberculosis secreted protein detected in vivo during the initial phases of the infectious process. J. Immunol. 175: 5298–5305 [DOI] [PubMed] [Google Scholar]
- 38. Murdoch DR, et al. 2001. Evaluation of a rapid immunochromatographic test for detection of Streptococcus pneumoniae antigen in urine samples from adults with community-acquired pneumonia. J. Clin. Microbiol. 39: 3495–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Napolitano DR, Pollock N, Kashino SS, Rodrigues V, Jr, Campos-Neto A. 2008. Identification of Mycobacterium tuberculosis ornithine carboamyltransferase in urine as a possible molecular marker of active pulmonary tuberculosis. Clin. Vaccine Immunol. 15: 638–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pillai S, Mohimen A. 1982. A solid-phase sandwich radioimmunoassay for Entamoeba histolytica proteins and the detection of circulating antigens in amoebiasis. Gastroenterology 83: 1210–1216 [PubMed] [Google Scholar]
- 41. Quinnell RJ, Courtenay O. 2009. Transmission, reservoir hosts and control of zoonotic visceral leishmaniasis. Parasitology 136: 1915–1934 [DOI] [PubMed] [Google Scholar]
- 42. Riera C, et al. 2004. Evaluation of a latex agglutination test (KAtex) for detection of Leishmania antigen in urine of patients with HIV-Leishmania coinfection: value in diagnosis and post-treatment follow-up. Eur. J. Clin. Microbiol. Infect. Dis. 23: 899–904 [DOI] [PubMed] [Google Scholar]
- 43. Riera C, et al. 2005. Value of culture and nested polymerase chain reaction of blood in the prediction of relapses in patients co-infected with leishmania and human immunodeficiency virus. Am. J. Trop. Med. Hyg. 73: 1012–1015 [PubMed] [Google Scholar]
- 44. Rissin DM, et al. 2010. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 28: 595–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Romero HD, et al. 2009. Comparative study of serologic tests for the diagnosis of asymptomatic visceral leishmaniasis in an endemic area. Am. J. Trop. Med. Hyg. 81: 27–33 [PubMed] [Google Scholar]
- 46. Salam MA, Khan MG, Mondal D. 2011. Urine antigen detection by latex agglutination test for diagnosis and assessment of initial cure of visceral leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 105: 269–272 [DOI] [PubMed] [Google Scholar]
- 47. Santos-Gomes G, Gomes-Pereira S, Campino L, Araujo MD, Abranches P. 2000. Performance of immunoblotting in diagnosis of visceral Leishmaniasis in human immunodeficiency virus-Leishmania sp.-coinfected patients. J. Clin. Microbiol. 38: 175–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sarkari B, Chance M, Hommel M. 2002. Antigenuria in visceral leishmaniasis: detection and partial characterisation of a carbohydrate antigen. Acta Trop. 82: 339–348 [DOI] [PubMed] [Google Scholar]
- 49. Schaffner A, Michel-Harder C, Yeginsoy S. 1991. Detection of capsular polysaccharide in serum for the diagnosis of pneumococcal pneumonia: clinical and experimental evaluation. J. Infect. Dis. 163: 1094–1102 [DOI] [PubMed] [Google Scholar]
- 50. Socan M, Marinic-Fiser N, Kese D. 1999. Comparison of serologic tests with urinary antigen detection for diagnosis of Legionnaires' disease in patients with community-acquired pneumonia. Clin. Microbiol. Infect. 5: 201–204 [DOI] [PubMed] [Google Scholar]
- 51. Srivastava P, Dayama A, Mehrotra S, Sundar S. 2011. Diagnosis of visceral leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 105: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sundar S, Agrawal S, Pai K, Chance M, Hommel M. 2005. Detection of leishmanial antigen in the urine of patients with visceral leishmaniasis by a latex agglutination test. Am. J. Trop. Med. Hyg. 73: 269–271 [PubMed] [Google Scholar]
- 53. Sundar S, Rai M. 2002. Laboratory diagnosis of visceral leishmaniasis. Clin. Diagn. Lab Immunol. 9: 951–958 [DOI] [PMC free article] [PubMed] [Google Scholar]