

Abstract
Membrane proteins of cytotoxic T cells specifically reorganize to form an immunological synapse (IS) on interaction with their specific target. In this paper, we investigated the redistribution of Kv1.3 channels, which are the dominant voltage-gated potassium channels, in the plasma membrane of allogen-activated human cytotoxic T lymphocytes (CTLs) on interacting with their specific target cells. Kv1.3 channels bearing a FLAG epitope were expressed in the CTLs and the cell-surface distribution of fluorescently labeled ion channels was determined from confocal laser-scanning microscopy images. FLAG epitope-tagged Kv1.3 channels showed a patchy distribution in CTLs not engaged with target cells, whereas the channels were accumulated in the IS formed between CTLs and specific target lymphocytes. Localization of Kv1.3 channels in the IS might open an unrevealed possibility in the regulation of ion channel activity by signaling molecules accumulated in the IS.
Activated cytotoxic T lymphocytes (CTLs) play an essential role in the destruction of virally infected and tumor cells. Conjugate formation between the cytotoxic and target cells is guided by the T cell antigen receptor (TCR)–MHC–peptide interactions and is regulated by auxiliary cell-surface molecule interactions (1). Conjugate formation is followed by the delivery of the lethal hit specifically destroying the target cell by granule exocytosis-mediated or cell-surface death receptor-mediated pathways (2, 3).
Killing requires a transient interaction between the killer and the target cells. This interaction takes place at a specialized intercellular contact, called the immunological synapse (IS), where the encounter causes proteins to segregate into micrometer-scale domains (4). Studies of the IS between T cells and antigen-presenting cells reported the enrichment of the lymphocyte function-associated antigen 1 in the periphery and TCR/CD3, CD28, and CD2 membrane proteins in the center of the supramolecular activation clusters, accompanied by the selective recruitment of cytosolic protein talin in the periphery, and Lck or PKCθ in the center of the IS (5–9). The IS formed between cytotoxic (CTLs) and target cells has recently been characterized by using confocal and electron microscopy by Stinchcombe et al. (10). Similar to the IS described for CD4+ T lymphocytes, the IS of cytotoxic cells contains a central region with proteins involved in TCR signaling, such as Lck, and PKCθ, surrounded by a ring of adhesion molecules. The central region of the IS of cytotoxic cells, however, contains a segregated secretory domain where the exocytosis of the lytic granules takes place. The signaling domain provides the specificity of recognition, whereas the other one provides the lethal hit. A qualitatively similar IS is formed between natural killer (NK) cells and their respective target cells (11).
Activity of voltage-gated potassium channels has been shown to modulate the killing process mediated by both NK cells and CTLs (12–14). Blockers of voltage-gated potassium channels inhibit NK- and CTL-mediated cytotoxicity in doses comparable to those required to block ionic currents. Together with the enhanced Rb+ efflux accompanying CTL-mediated cytotoxicity (15), and the modulation of the cytotoxic response by the membrane potential of the effector cells (16), these studies indicated the involvement of K+ channel activity in killing.
Several recent studies (17, 18) have indicated that voltage-gated potassium channels segregate into specialized membrane microdomains modulating their activity. The predominant voltage-gated potassium channel of T lymphocytes is Kv1.3. We have shown that the activity of this channel is influenced by the cholesterol content of the membrane, thereby predicting the role of membrane rafts in the regulation of these K+ channels (19). Bock et al. (20) reported the redistribution of Kv1.3 channels between small sphingolipid- and cholesterol-enriched membrane rafts and large ceramide-enriched membrane platforms on increasing the ceramide content of the membrane. We have recently characterized the spatial distribution of Kv1.3 channels in the membrane of Jurkat T lymphocytes and the relationship between Kv1.3 channels and CD3 molecules (21). Electron microscopic investigations revealed that the distribution of Kv1.3 channels in the plasma membrane of Jurkat T cells was nonrandom. Confocal laser-scanning microscopy (CLSM) images confirmed that Kv1.3 channels and CD3 molecules accumulated in largely overlapping membrane areas, whereas flow cytometric fluorescence resonance energy transfer (FRET) measurements proved a molecular proximity between Kv1.3 channels and CD3 proteins (21).
Taking into account the critical role of molecular clustering of the receptors involved in T cell signaling (22), the importance of Kv1.3 channels in lymphocyte-mediated cytotoxicity, and the possibility of K+ channel regulation by interaction with T cell-signaling molecules, we investigated the distribution of Kv1.3 channels in the membrane of CTLs after their interaction with target cells. Here, we report that fluorescently tagged Kv1.3 channels are recruited into the contact area of the CTLs and target cells. Numerical analysis of CLSM images showed a significant colocalization of the labeled channels and the lipid raft marker cholera toxin B subunit (CTX), whereas negligible crosscorrelation was found between the transferrin receptor (excluded from the lipid raft) and the labeled channel or the raft marker. Our acceptor photobleaching energy transfer measurements indicated molecular proximity between CD3 and Kv1.3 in CTLs in the absence of an interacting target cell. In addition, these molecules were recruited into the IS on target cell-T cell contact and maintained a modest proximity.
Materials and Methods
Cells. Antigen-specific CD8+ human CTLs were generated from peripheral blood mononuclear cells by means of repetitive coculturing of the cells with JY lymphoblasts (23) in complete medium supplemented with 10 mM Hepes (pH 7.3) and 50 μM 2-mercaptoethanol (Serva). The stimulator JY cells were treated with 50 μg/ml mitomycin C for 40 min and washed thoroughly. Peripheral blood mononuclear cells depleted from adherent cells were stimulated with mitomycin C-treated JY cells on the first day and were cultured for 2 weeks. From the fourth day, the medium was supplemented with 20 units/ml human recombinant IL-2 (R & D Systems), and refreshed every 3 days. On the fourteenth day, CTLs were restimulated with mitomycin C-treated JY cells and the whole cycle was repeated. After at least 2 months of culturing, lymphocytes were frozen for storage. This procedure yielded a cell population containing >90% CD8+ cells. Stored CTLs were thawed, pulsed with stimulator JY cells again, and used for the experiments 3–5 days after the last stimulation. The JY cell line is a human Epstein–Barr virus-transformed B lymphoblastoid cell line, expressing a high density of HLA-A2, B7, DQw 1, 3, and DR in the plasma membrane, originally described by Terhorst et al. in 1976 (24). The NK cell-resistant Raji B cell line is identified as an HLA-A2(–), A24(–) cell line derived from a patient with Burkitt's lymphoma (American Type Culture Collection; ref. 25). The Daudi cell line (American Type Culture Collection) is negative for β2-microglobulin (26).
Electrophysiology and Current Recording. Whole-cell patch-clamp measurements were carried out by using an Axopatch-200A amplifier connected to a personal computer through Axon Instruments (Foster City, CA) digidata 1200 data acquisition hardware (27, 28). For data acquisition and analysis the pclamp8 software package (Axon Instruments) was used. Pipettes were pulled from GC 150 F-15 borosilicate glass capillaries in two stages and were fire-polished, resulting in electrodes having 2–3 MΩ resistance in the bath. The bath solution was (in mM): 145 NaCl/5 KCl/1 MgCl2/2.5 CaCl2/5.5 glucose/10 Hepes (pH 7.35). The internal solution contained (in mM): 140 KF, 2 MgCl2, 1 CaCl2, 10 Hepes, and 11 EGTA (pH 7.2). Cells were labeled with Alexa Fluor 546-tagged anti-human CD8 and CD8+ cells were identified for current recording by using a Nikon TE2000 fluorescence microscope.
CTL Transfection and CTL-Target Cell Engagement. The FLAG epitope was cloned into the gene encoding the human Kv1.3 potassium channel (Kv1.3/FLAG; ref. 21) and CTLs were transfected with this gene by using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. Twenty-four hours after transfection, CTLs were washed in RPMI medium 1640 solution (without phenol red) and 2.5 × 105 CTLs were mixed with equal amount of JY target cells in 1 ml of complete RPMI medium 1640 (without phenol red), were centrifuged (for 1 min at 200 × g), and were kept at 37°C for 5 min. The conjugated cells were stored on ice and were fixed in 1% formaldehyde in PBS for 1 h.
Labeling Procedures. Transfected, target-cell-engaged and formaldehyde-fixed cells were washed twice with Hanks' solution and labeled with anti-FLAG M2 primary antibody (mouse-anti-human; Sigma) followed by Alexa Fluor 546- or 568-tagged rabbit anti-mouse IgG (RAMIG) (Sigma). For labeling other cell-surface proteins, the following dye-conjugated mAbs were used: X-FITC-tagged W6/32 (29) against MHC class I, Alexa Fluor 488-tagged MEM75 against transferrin receptors, Alexa Fluor 647-tagged MEM31 against CD8 molecules, and Cy5-tagged MEM57 targeting CD3. MEM antibodies were kind gifts of V. Horejsí (Institute of Molecular Genetics, Prague). Alexa Fluor dyes and fluorescein were purchased from Molecular Probes, and Cy5 was bought from Amersham Pharmacia (Piscataway, NJ). Cells were incubated with 50 μg/ml of the primary antibodies in PBS containing 0.1% BSA on ice for 40 min. The lipid raft marker GM1 ganglioside was targeted by Cy5-tagged CTX (Sigma) at 4 μg/ml concentration, which was used similarly to the primary antibodies. After incubation with the antibodies cells were washed twice in ice-cold PBS and fixed in 1% formaldehyde for 1 h on ice. They were then mounted on polylysine-coated coverslips, with Mowiol (Calbiochem) to reduce photobleaching.
CLSM. Spatial localization of Kv1.3/FLAG ion channels, CD3, CD8, MHC I, transferrin receptor, and GM1 gangliosides in the plasma membrane of CTLs was studied by means of CLSM (Zeiss LSM 510). For the excitation of fluorescein and Alexa Fluor 488, the 488-nm line of an Argon ion laser, for Alexa Fluor 546 and Alexa Fluor 568, a 543-nm He-Ne laser, and for Cy5 and Alexa Fluor 647, a 633-nm He-Ne laser was used. Fluorescence was detected through 505- to 550-nm bandpass, 560- to 615-nm bandpass, and >650-nm longpass filters, respectively. The “multi-track” option of the microscope was used, scanning each laser line separately for the individual excitation of the dyes to exclude crosstalk. With a Plan-Apochromat ×63 oil-immersion objective (N.A. 1.4) image stacks with 0.8- to 1.5-μm thick optical slices were collected.
Correlation Analysis of Protein Colocalization at the Few-Hundred-Nanometer Distance Scale. Single optical slices or projection images created from 2–3 optical slices recorded from the bottom or the top of doubly labeled cells were evaluated to determine protein colocalization. This procedure was performed by calculating the crosscorrelation between the pixel intensities of image pairs by using a program written by G. Vereb in labview (National Instruments, Austin, TX; refs. 21 and 30). Perfectly identical spatial distribution patterns should yield a correlation coefficient of C = l, whereas unrelated or independent distributions should result in C = 0.
Pixel-by-Pixel FRET Measurements and Data Analysis. Molecular association/proximity of Kv1.3/FLAG and CD3 at the 1- to 10-nm distance scale was investigated by FRET measurements by using the acceptor photobleaching technique (31, 32). Kv1.3/FLAG was labeled by anti-FLAG mAbs followed by Alexa Fluor 568-tagged RAMIG serving as FRET donor, whereas CD3 was stained by Cy5-MEM57 mAbs used as FRET acceptor. A single optical slice of both the donor and acceptor intensity distributions (ID0ij and IA0ij) of the selected cell was recorded by using low excitation intensities (1–10% transmission of the laser line filter) of the 543- and 633-nm laser lines, and 560- to 615-nm bandpass and >650-nm longpass emission filters, respectively. Subsequently Cy5 dye molecules were bleached by consecutive scans with the 633-nm laser line at maximum laser intensity. The effectiveness of bleaching was near 100% as proven by postbleach acceptor images. After photobleaching, an image of the donor (ID1ij) was taken again. Images were lowpass-filtered, and the FRET efficiency, Eij, at pixel ij was calculated as
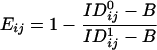 |
[1] |
where B is the mean background intensity determined from cell-free pixels of the image. The above calculation was performed for all pixels with intensities higher than the threshold, and the result was presented in the form of a pseudocolor FRET efficiency map and also as a frequency distribution histogram of Eij values.
Cytotoxicity Assay. Assessment of the specific killing of effector T lymphocytes was done by a flow cytometric assay based on a procedure described in ref. 33 with slight modifications. Briefly, 1 × 107 JY target cells were labeled by 20 μg/ml DiOC18(3) for 10 min and washed with complete medium. A quantity of 104 labeled JY target cells were mixed with effector CTLs in a 24-well plate with an elevating effector-to-target cell (E/T) ratio. Mixed cells were centrifuged at 200 × g for 1 min and incubated for 4.5 h. For the last 30 min of the incubation, 10 μg/ml propidium iodide was added to the cells. Samples were stored on ice until fluorescence measurements were made on a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Cell debris was excluded from data analysis based on forward and side light scatter gating. Membrane-labeled target cells were selected according to high FL1 green fluorescence intensity. Propidium iodide-positive dead target cells were identified based on the red FL2 histogram and the live target cell percentage was determined by subtracting the dead cell percentage from 100%. The specific killing percentage was calculated by the following equation: [(live target cells without effector cells in percent) – (actual live target cells with effector cells in percent)]/(live target cells without effector cells in percent) × 100. Potassium channel blocker margatoxin (MgTx) in specified concentrations was put directly into the medium of the CTL-target cell mixture. Every sample was prepared and analyzed in triplicate.
Results
HLA-A2-Specific Killing of Target Cells by Allogen-Activated CTLs. To assess the functional relevance of our model system, we tested the specific killing ability of allogen-activated CTLs on different target cells. Fig. 1 shows that allogen-activated polyclonal CTLs created by using our experimental procedures kill HLA-A2 homozygous JY cells in an E/T ratio-dependent manner. CTLs do not kill β2-microglobulin-deficient Daudi cells, even at a high E/T ratio, and hardly kill Raji cells, which do not express HLA-A2 proteins. Homogeneity of these cytotoxic lymphocytes was examined by flow cytometric immunophenotyping. All CTLs expressed high levels of CD3, CD5, and CD7 antigens, whereas 90% of these mature T cells expressed a high level of CD8 (Fig. 1 Inset) The remaining 10% of the cells expressed CD4. Cells showed homogeneous, but broad staining pattern for HLA-DR and all cells were CD13-, CD19-, CD20-, CD22-, and CD34-negative (data not shown).
Fig. 1.

Cytotoxic activity of human allospecific effector CD8+ T lymphocytes. The killing of specific target (human JY B cell line) and irrelevant cell lines (Raji and Daudi) were studied. Cytotoxicity was evaluated by DiOC18(3) – propidium iodide two-color flow cytometric measurement. DiOC18(3)-labeled target cells (10,000 cells per well) were incubated for 3.5 h in the presence of the indicated amounts of effector cells (E/T ratio). For the last 30 min, propidium iodide was added, and the percentage of the live target cells was measured. Specific lysis was calculated as described in Materials and Methods. Results shown represent four independent experiments. (Inset) The result of flow cytometric analysis of CD4/CD8 double-stained forward and side light scatter-gated lymphocytes.
To further justify our model system, we studied the effect of potassium channel blocker MgTx on cell-mediated cytotoxicity at different E/T ratios. Specific killing of the JY cells has dropped significantly in a dose-dependent manner at 5- and 20-nM MgTx concentrations, whereas the viability of the toxin-treated control JY cells and that of the effector T lymphocytes did not alter considerably (data not shown).
Activated CTLs Express a Large Number of Voltage-Gated Kv1.3 K+ Channels. To justify the appropriateness of epitope-tagged Kv1.3 channel transfection into CTLs for colocalization studies, we had to prove that Kv1.3 is the native voltage-gated K+ channel in allogen-activated CTLs. Fig. 2 shows whole-cell current traces evoked by depolarizing steps to different test potentials in a CTL. The currents are activated in a time- and membrane potential-dependent manner. The current–voltage relationship (Fig. 2 Inset) shows that the current is activated at membrane potentials more positive than –60 mV in this cell, the activation threshold ranged between –60 and –40 mV for five different CTLs studied. The midpoint and slope of the voltage dependence of steady-state activation of the whole-cell K+ conductance was –33.8 ± 4.8 mV and 10.7 ± 0.7 mV, respectively (n = 5). These values were obtained by fitting Boltzmann functions to the normalized whole-cell conductance curves. During prolonged depolarization, the current decayed with a single exponential time course, due to inactivation with a time constant of 180 ± 27 ms at +50 mV test potential (n = 9). Extracellular application of 10 mM tetraethylammonium increased this time constant by 56 ± 4% (n = 4). These biophysical parameters are in good agreement with those reported earlier for Kv1.3 channels (28, 34, 35). Pharmacological characterization of the current also supported this assumption: the sensitivity of the current to MgTx (dissociation constant, Kd = 50 ± 12 pM, n = 3), charybdotoxin (3.4 ± 0.6 nM, n = 3), and tetraethylammonium (14 ± 1 mM, n = 4), was similar to that of Kv1.3 expressed in lymphocytes (35–37). Based on the pharmacological and biophysical properties of the cells we concluded that CTLs express Kv1.3 channels. The average peak current at +50 mV test potential was 1.9 ± 0.3 nA in antigen-activated CTLs (n = 13), whereas in unstimulated CD8+ lymphocytes, 0.6 ± 0.09 nA was measured under identical recording conditions (n = 14, P < 0.05).
Fig. 2.

Whole-cell currents of an antigen-activated CTL. Potassium currents were elicited by depolarizing steps ranging from –70 to +50 mV in 10-mV increments. The depolarizing steps lasted for 2 s, of which the first 500 ms are shown for each trace. The cell was held at –120 mV for 50 s between the steps. Data were digitized at 2.5 kHz and filtered at 1 kHz. Every other trace was omitted from the figure for clarity. (Inset) The peak current-voltage relationship of this cell. Peak current was determined from each trace, corrected for ohmic leak, and plotted as a function of test potential.
Spatial Rearrangement of Kv1.3/FLAG in CTLs Engaged in IS. The spatial distribution of Kv1.3/FLAG in CTLs, and the effect of engagement with target (JY) cells were studied by CLSM. Fig. 3 shows 1.1-μm-thick optical slices of a CTL alone (Top) and those of CTLs conjugated with target cells (Middle and Bottom). As seen in the image, Kv1.3 channels showed a patchy distribution in the plasma membrane of CTLs not in contact with target cells. The hot spots were evenly distributed in the membrane, similar to MHC I- and CD8-rich domains (Fig. 3 Top, red, Kv1.3/FLAG labeled with anti-FLAG mAb followed by Alexa Fluor 546-RAMIG; green, MHC class I labeled with fluorescein-W6/32 mAb; and blue, CD8 tagged by Alexa Fluor 647-MEM31 mAb). The distribution of Kv1.3 channels is similar to observations made on Jurkat cells by us (21) and by others (20). On the other hand, in CTLs conjugated with target cells, a rearrangement of Kv1.3/FLAG was observed: in the majority of cases, ion channels were recruited into the CTL–target cell interface (Fig. 3 Middle). In the images seen in the middle row two target cells are conjugated with two CTLs, one of which is Kv1.3/FLAG-positive, whereas the other one did not express epitope-tagged channels. In some cases Kv1.3/FLAG surrounded the IS formed between the CTL and the target cell like a belt (Fig. 3 Bottom). MHC I molecules on target cells as well as CD8 coreceptors on CTLs, which are both directly involved in the formation of the IS, were also enriched in or around the contact area (Fig. 3 Middle and Bottom).
Fig. 3.

Enrichment of Kv1.3/FLAG in or around the IS. Distribution of Kv1.3/FLAG (red: anti-FLAG followed by Alexa Fluor 546-RAMIG), MHC class I (green: X-FITC-W6/32), and CD8 (Alexa Fluor 647-anti-CD8) molecules in a lone CTL (Top) and CTL-JY target cell conjugates (Middle and Bottom). The thickness of the optical slice was 1.1 μm. In lone CTLs, the channels are distributed in small patches throughout the membrane (Top). On CTL-target interaction Kv1.3 channels are redistributed and enriched in the IS in the majority of CTLs (Middle) or surround the synapse like a ring (Bottom). (Scale bar, 5 μm.)
Molecular Proximity of Kv1.3/FLAG and the CD3 Complex. Our previous FRET studies on Jurkat cells indicated that Kv1.3/FLAG molecules were in the molecular vicinity of CD3 (21). It was reported previously that CD3 molecules were also recruited into the IS formed between the CTL and the target cell (10). Therefore, we asked whether Kv1.3 channels and CD3 molecules were also colocalized in CTLs. We performed acceptor photobleaching energy transfer measurements by using CLSM to investigate the proximity of Kv1.3/FLAG and CD3 at the molecular level. To this aim, Kv1.3/FLAG epitopes were labeled by anti-FLAG mAbs followed by Alexa Fluor 568-RAMIG used as a FRET donor, and CD3 molecules were targeted by Cy5-MEM57 mAbs used as a FRET acceptor. A confocal section of a doubly labeled cell before photobleaching is presented in Fig. 4 A–C (red, Kv1.3/FLAG; blue, CD3; magenta, overlap). After eliminating the long-wavelength fluorescence (and absorption) of the acceptors by bleaching the sample with high-intensity 633-nm laser light, the donor intensity was measured again (data not shown), and the efficiency of FRET was calculated from the pre- and postbleach donor images on a pixel-by-pixel basis according to Eq. 1 as described in Materials and Methods. The spatial distribution of FRET efficiency is presented in a pseudocolor image (Fig. 4D), and also as a relative frequency distribution histogram of FRET efficiency values in the individual pixels (Fig. 4E). The mean FRET efficiency is significantly larger than 0 (<E> ≈6%), implying that at least a certain population of Kv1.3/FLAG is in the molecular proximity of CD3 complexes. FRET experiments were also performed on CTLs engaged in an IS (data not shown). The mean of the FRET efficiency histogram calculated in the contact area was 2–3%, indicating that the molecular association between Kv1.3/FLAG and CD3 was modestly maintained in the IS.
Fig. 4.

Molecular association of Kv1.3/FLAG and CD3. (A–C) Confocal sections showing the distribution of Kv1.3/FLAG (red) and CD3 (blue) on a CTL before acceptor photobleaching. Epitopes were labeled by anti-FLAG mAb followed by Alexa Fluor 546-RAMIG and Cy5-anti-CD3, respectively. The overlay image (C) is pink at pixels where the overlap is significant. (D) Pixel-by-pixel FRET efficiency map (pseudocolor) between Kv1.3/FLAG and CD3 calculated from the Kv1.3 images recorded before and after acceptor photobleaching (data not shown). The color code ranges between FRET efficiency values of 0 (black) to 100% (red). Pixels with fluorescence intensities lower than the background threshold are black. (E) The histogram gives the frequency distribution of FRET efficiencies calculated in the individual pixels. (Scale bar, 5 μm.)
Kv1.3/FLAG Is Partially Partitioned Into Lipid Rafts. The TCR, along with CD3, are known to be recruited to lipid rafts on activation (38). Considering the colocalization of Kv1.3 and CD3, we tested the distribution of Kv1.3 channels in CTL/target-cell conjugates with respect to lipid rafts by CLSM (see Fig. 5). In these experiments, Kv1.3/FLAG (Fig. 5A, red) was labeled by anti-FLAG followed by Alexa Fluor 546-RAMIG, whereas lipid rafts were visualized by labeling GM1 gangliosides (a lipid raft marker) with Cy5-tagged CTX (ref. 20 and Fig. 5B, blue). As a negative control, transferrin receptors, known to be excluded from lipid rafts, were marked by Alexa Fluor 488-MEM75 mAbs (Fig. 5C, green). CLSM measurements indicated a significant overlap between membrane domains rich in Kv1.3/FLAG and GM1 gangliosides. This domain-level colocalization at the resolution of the confocal microscope (≈200 nm) was numerically evaluated by crosscorrelation analysis (30) of the spatial intensity distributions of fluorescently labeled Kv1.3/FLAG and CTX, yielding a relatively high crosscorrelation coefficient: C ≈ 0.5. (In case of perfect colocalization, the value of C would be 1; however, in our experience, the highest crosscorrelation values, gained when labeling two different epitopes of the same cell surface molecule, e.g., MHC class I light and heavy chains, were usually around C ≈ 0.9, due to experimental noise in the images.) On the other hand, the distribution of transferrin receptors resulted in very low correlation coefficients either with images of cholera toxin B or those of Kv1.3/FLAG distributions (C ≈ 0.1; Fig. 5). Besides the colocalization of Kv1.3 and the lipid raft marker CTX, we also studied the relationship between Kv1.3 channels and other lipid raft markers. Our experiments showed significant colocalization of Kv1.3 with the membrane probe DiIC18, which is enriched in lipid rafts due to its saturated fatty acid tails (39). A similar, significant co-localization of Kv1.3 channels was found with the T cell raft marker CD48 (C≈0.3; data not shown).
Fig. 5.

Association of Kv1.3/FLAG with lipid rafts. Distribution of Kv1.3/FLAG (red), lipid rafts (blue), and transferrin receptors (TfR; green) in a CTL conjugated with a JY target cell. Epitopes were labeled with anti-FLAG followed by Alexa Fluor 546-RAMIG, Cy5-CTX (binding to GM1, a lipid raft marker), and Alexa Fluor 488-MEM75, respectively. A–E represent the topmost optical section of cells. The extent of colocalization of the labeled epitopes was characterized numerically by crosscorrelation coefficients of the fluorescence intensities in the individual pixels: C (FLAG, lipid raft) = 0.52, C (FLAG, transferrin receptor) = 0.09, and C (CTX, transferrin receptor) = 0.12 for the images shown. The high correlation between Kv1.3/FLAG and CTX implies that the ion channels are mainly localized in rafts. Transferrin receptors known to be excluded from lipid rafts served as a negative control. F was recorded around the equator of the cells to illustrate the formation of the IS. (The enrichment of Kv1.3 at the 6 o'clock position is probably the remnant of a disrupted IS.) (Scale bar, 5 μm.)
Discussion
It is generally accepted that membrane potential and the activity of potassium channels is important for generation and propagation of the mitogenic signal in lymphocytes (40), for cell volume regulation (41), and for cell-mediated cytotoxicity (12, 13). Previous studies (42, 43) reported a wide variety of voltageg-ated ion channels in the plasma membrane of human and rodent T lymphocytes. Human T lymphocytes express a unique voltage-gated ion channel encoded by the Kv1.3 gene (44). We have recently reported that Kv1.3 channels bearing the FLAG epitope were functional and the biophysical parameters of the mutant current were similar to those of the wild-type Kv1.3 (21). To use this epitope-tagged Kv1.3 channel for the localization of the channels in the membrane of activated CTLs, we had to prove that Kv1.3 is the native voltage-gated K+ channel in these cells. Based on biophysical and pharmacological characterization of the voltage-gated K+ current, we reported in this study that activated CTLs generated by using our experimental procedures expressed Kv1.3 channels in their plasma membrane. This result agrees with previous studies (13, 45, 46) where a voltage-gated potassium conductance was found in human cytotoxic T cells, in alloantigen-stimulated T cells, in CD8+ T effector memory cells, and in NK cells. Thus, transfection of Kv1.3/FLAG channels into CTLs leads to the expression of a native-like channel. Similar to other studies, we also found that the expression of Kv1.3 channels increased on activation of the cells (46, 47).
Based on the numerical analysis of the distribution of immunogold-labeled Kv1.3/FLAG channels, we have reported (21) that Kv1.3 channels formed clusters in the membrane of Jurkat T-cells: the number of channels per unit membrane area deviated significantly from the expected values predicted by a stochastic Poisson distribution. On a different hierarchical level, CLSM also revealed a nonuniform distribution of Kv1.3 channels in the membrane. Bock et al. (20) reported a similar nonrandom distribution of Kv1.3 channels in Jurkat cells, and the redistribution of the channels on stimulation of the Fas-ligand/ceramide pathway. Motivated by this knowledge, we studied the distribution of Kv1.3 channels in CTLs on their engagement with a specific target in the IS. We showed that Kv1.3 channels were enriched in the contact area formed between cytotoxic and target cells. We argue that this is a specific event associated with the formation of the IS for the following reasons: (i) we did not see polarized expression of Kv1.3 channels in the absence of target cells or when CTLs were mixed with nonrecognized target cells (Daudi and Raji cells, data not shown), (ii) the distribution of transferrin receptor, which is an irrelevant protein regarding the function of the IS, was not altered in the membrane on target cell engagement, and (iii) concurrent enrichment of CD8 coreceptors and Kv1.3/FLAG channels was seen in the contact area of interacting CTLs and target cells. The two different spatial arrangements of Kv1.3/FLAG with respect to the IS; i.e., the recruitment in the contact area or belt-like arrangement around it, might be correlated with the type of the synapse (early vs. mature synapse), which is similar to the rearrangement of other membrane proteins during the maturation of the IS (4, 6, 8).
The recruitment of Kv1.3/FLAG channels into the IS might be governed by several factors, including reorganization of the lipid rafts on activation of the cells (4). We have described that Kv1.3/FLAG channels and the CTX-labeled lipid rafts showed significant colocalization, whereas the nonraft transferrin receptor and Kv1.3/FLAG channels did not reside in the same membrane domain. This result agrees with the results of Bock et al. (20) who showed lipid raft association of Kv1.3 in Jurkat T cells. Besides lipid raft association of Kv1.3, we also demonstrated that Kv1.3 channels and CD3 molecules showed a molecular proximity as reported by pixel-by-pixel FRET calculated from microscopic images. This result is consistent with our previous observation (21) in Jurkat T-cells by using flow cytometric energy transfer measurements, and could be attributed to the raft localization of both TCR/CD3 and Kv1.3. Interestingly, the energy transfer efficiency between CD3 and Kv1.3 was lower in the cytotoxic cell-target cell-contact area than in nonengaged CTLs, indicating that the molecular rearrangements during the formation/maturation of the IS might affect the relationship between these molecules (4).
What can be the significance of the recruitment of Kv1.3 into the IS? The major physiological function of Kv1.3 channels is the control of the membrane potential of T cells (47). It has been shown earlier (12–14), as well as in this paper, that potassium channel blockers inhibit the cytotoxic activity of CTLs. CTL-mediated cytotoxicity is also regulated by membrane potential (16); however, we do not think that recruitment of Kv1.3 channels into the IS might result in a significantly different local membrane potential, as compared to the rest of the cell. Considering the small size of lymphocytes (≈7–10 μm) and the extremely high input resistance of the lymphocyte membrane (several GΩ) small ionic currents induce uniform changes in the membrane potential throughout the cell. On the other hand, protein kinases concentrated in the IS might phosphorylate Kv1.3 channels more efficiently by recruitment of the channels into the IS. Several lines of evidence suggest that protein kinases involved in TCR/CD3 signaling, e.g., Lck and PKC, regulate the biophysical properties and physiological functions of Kv1.3 channels (48–50). Based on our observations, it is highly likely that the activation process of the TCR/CD3 complex associated with the formation of the IS may modify the activity of a nearby ion channel, thereby modulating the function of the IS, and, as a consequence, the killing of the target cells (51).
Acknowledgments
This work was supported by Egészségügyi Tudományos Tanács Grants 010/2001, 013/2001, 031/2001, 117/2001, 222/2003, 602/2003, and 603/2003; Országos Tudományos Kutatási Alapprogramok Grants TS040773, F035251, F034487, T42618, T043087, and T043509; Felsöoktatási Kutatási és Fejlesztési Pályázat Grants 327/2000, 622/2000, and 0075/2002; a Bolyai Fellowship (to G.V., Z.B., and A.B.); and a Békésy fellowship (to Z.V.).
Abbreviations: CTL, cytotoxic T lymphocyte; IS, immunological synapse; TCR, T cell antigen receptor; NK, natural killer; RAMIG, rabbit anti-mouse IgG; FRET, fluorescence resonance energy transfer; E/T, effector-to-target cell; CLSM, confocal laser-scanning microscopy; CTX, cholera toxin B subunit.
References
- 1.Berke, G. (1994) Annu. Rev. Immunol. 12, 735–773. [DOI] [PubMed] [Google Scholar]
- 2.Lieberman, J. (2003) Nat. Rev. Immunol. 3, 361–370. [DOI] [PubMed] [Google Scholar]
- 3.Kagi, D., Vignaux, F., Ledermann, B., Burki, K., Depraetere, V., Nagata, S., Hengartner, H. & Golstein, P. (1994) Science 265, 528–530. [DOI] [PubMed] [Google Scholar]
- 4.Davis, D. M. (2002) Trends Immunol. 23, 356–363. [DOI] [PubMed] [Google Scholar]
- 5.van der Merwe, P. A. (2002) Curr. Opin. Immunol. 14, 293–298. [DOI] [PubMed] [Google Scholar]
- 6.Grakoui, A., Bromley, S. K., Sumen, C., Davis, M. M., Shaw, A. S., Allen, P. M. & Dustin, M. L. (1999) Science 285, 221–227. [DOI] [PubMed] [Google Scholar]
- 7.Monks, C. R., Freiberg, B. A., Kupfer, H., Sciaky, N. & Kupfer, A. (1998) Nature 395, 82–86. [DOI] [PubMed] [Google Scholar]
- 8.Bromley, S. K., Burack, W. R., Johnson, K. G., Somersalo, K., Sims, T. N., Sumen, C., Davis, M. M., Shaw, A. S., Allen, P. M. & Dustin, M. L. (2001) Annu. Rev. Immunol. 19, 375–396. [DOI] [PubMed] [Google Scholar]
- 9.Holdorf, A. D., Lee, K. H., Burack, W. R., Allen, P. M. & Shaw, A. S. (2002) Nat. Immunol. 3, 259–264. [DOI] [PubMed] [Google Scholar]
- 10.Stinchcombe, J. C., Bossi, G., Booth, S. & Griffiths, G. M. (2001) Immunity 15, 751–761. [DOI] [PubMed] [Google Scholar]
- 11.Vyas, Y. M., Mehta, K. M., Morgan, M., Maniar, H., Butros, L., Jung, S., Burkhardt, J. K. & Dupont, B. (2001) J. Immunol. 167, 4358–4367. [DOI] [PubMed] [Google Scholar]
- 12.Felzen, B., Lavy, R., Garcia, M., Berke, G., Gardner, P. & Binah, O. (1996) Circ. Res. 78, 253–261. [DOI] [PubMed] [Google Scholar]
- 13.Schlichter, L., Sidell, N. & Hagiwara, S. (1986) Proc. Natl. Acad. Sci. USA 83, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma, B. (1988) Immunology 65, 101–105. [PMC free article] [PubMed] [Google Scholar]
- 15.Russell, J. H. & Dobos, C. B. (1981) Eur. J. Immunol. 11, 840–843. [DOI] [PubMed] [Google Scholar]
- 16.Gray, L. S., Gnarra, J. R., Russell, J. H. & Engelhard, V. H. (1987) Cell 50, 119–127. [DOI] [PubMed] [Google Scholar]
- 17.Martens, J. R., Navarro-Polanco, R., Coppock, E. A., Nishiyama, A., Parshley, L., Grobaski, T. D. & Tamkun, M. M. (2000) J. Biol. Chem. 275, 7443–7446. [DOI] [PubMed] [Google Scholar]
- 18.Martens, J. R., Sakamoto, N., Sullivan, S. A., Grobaski, T. D. & Tamkun, M. M. (2001) J. Biol. Chem. 276, 8409–8414. [DOI] [PubMed] [Google Scholar]
- 19.Hajdu, P., Varga, Z., Pieri, C., Panyi, G. & Gaspar, R., Jr. (2003) Pflügers Arch. 445, 674–682. [DOI] [PubMed] [Google Scholar]
- 20.Bock, J., Szabo, I., Gamper, N., Adams, C. & Gulbins, E. (2003) Biochem. Biophys. Res. Commun. 305, 890–897. [DOI] [PubMed] [Google Scholar]
- 21.Panyi, G., Bagdany, M., Bodnar, A., Vamosi, G., Szentesi, G., Jenei, A., Matyus, L., Varga, S., Waldmann, T. A., Gaspar, R., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 2592–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vereb, G., Szollosi, J., Matko, J., Nagy, P., Farkas, T., Vigh, L., Matyus, L., Waldmann, T. A. & Damjanovich, S. (2003) Proc. Natl. Acad. Sci. USA 100, 8053–8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bacso, Z., Bene, L., Bodnar, A., Matko, J. & Damjanovich, S. (1996) Immunol. Lett. 54, 151–156. [DOI] [PubMed] [Google Scholar]
- 24.Terhorst, C., Parham, P., Mann, D. L. & Strominger, J. L. (1976) Proc. Natl. Acad. Sci. USA 73, 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Epstein, M. A., Achong, B. G., Barr, Y. M., Zajac, B., Henle, G. & Henle, W. (1966) J. Natl. Cancer Inst. 37, 547–559. [PubMed] [Google Scholar]
- 26.Klein, E., Klein, G., Nadkarni, J. S., Nadkarni, J. J., Wigzell, H. & Clifford, P. (1968) Cancer Res. 28, 1300–1310. [PubMed] [Google Scholar]
- 27.Hamill, O. P., Marty, A., Neher, E., Sakmann, B. & Sigworth, F. J. (1981) Pflügers Arch. 391, 85–100. [DOI] [PubMed] [Google Scholar]
- 28.Varga, Z., Panyi, G., Péter, M., Pieri, C., Csécsei, G., Damjanovich, S. & Gáspár, R. (2001) Biophys. J. 80, 1280–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barnstable, C. J., Bodmer, W. F., Brown, G., Galfre, G., Milstein, C., Williams, A. F. & Ziegler, A. (1978) Cell 14, 9–20. [DOI] [PubMed] [Google Scholar]
- 30.Vereb, G., Matkó, J., Vámosi, G., Ibrahim, S. M., Magyar, E., Varga, S., Szöllösi, J., Jenei, A., Gaspar, R., Jr., Waldmann, T. A., et al. (2000) Proc. Natl. Acad. Sci. USA 97, 6013–6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bastiaens, P. I., Majoul, I. V., Verveer, P. J., Soling, H. D. & Jovin, T. M. (1996) EMBO J. 15, 4246–4253. [PMC free article] [PubMed] [Google Scholar]
- 32.Vereb, G., Meyer, C. K. & Jovin, T. M. (1997) in Interacting Protein Domains, Their Role in Signal and Energy Transduction, NATO ASI Series, ed. Heilmeyer, L. M. G., Jr., (Springer, New York), pp. 49–52.
- 33.Nunez, R. (2001) in Current Protocols in Cytometry, ed. Robinson, J. P. (Wiley, New York), pp. 9.17.9–9.17.11.
- 34.Cahalan, M. D., Chandy, K. G., Decoursey, T. E. & Gupta, S. (1985) J. Physiol. (London) 358, 197–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grissmer, S. & Cahalan, M. D. (1989) Biophys. J. 55, 203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Calvo, M., Leonard, R. J., Novick, J., Stevens, S. P., Schmalhofer, W., Kaczorowski, G. J. & Garcia, M. L. (1993) J. Biol. Chem. 268, 18866–18874. [PubMed] [Google Scholar]
- 37.Rauer, H., Lanigan, M. D., Pennington, M. W., Aiyar, J., Ghanshani, S., Cahalan, M. D., Norton, R. S. & Chandy, K. G. (2000) J. Biol. Chem. 275, 1201–1208. [DOI] [PubMed] [Google Scholar]
- 38.Xavier, R., Brennan, T., Li, Q., McCormack, C. & Seed, B. (1998) Immunity 8, 723–732. [DOI] [PubMed] [Google Scholar]
- 39.Korlach, J., Schwille, P., Webb, W. W. & Feigenson, G. W. (1999) Proc. Natl. Acad. Sci. USA 96, 8461–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cahalan, M. D. & Chandy, K. G. (1997) Curr. Opin. Biotechnol. 8, 749–756. [DOI] [PubMed] [Google Scholar]
- 41.Deutsch, C. & Chen, L.-Q. (1993) Proc. Natl. Acad. Sci. USA 90, 10036–10040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fukushima, Y., Hagiwara, S. & Henkart, M. (1984) J. Physiol. (London) 351, 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee, S. C., Levy, D. I. & Deutsch, C. (1992) J. Gen. Physiol. 99, 771–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Douglass, J., Osborne, P. B., Cai, Y. C., Wilkinson, M., Christie, M. J. & Adelman, J. P. (1990) J. Immunol. 144, 4841–4850. [PubMed] [Google Scholar]
- 45.Chandy, K. G., Decoursey, T. E., Cahalan, M. D., McLaughlin, C. & Gupta, S. (1984) J. Exp. Med. 160, 369–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wulff, H., Calabresi, P. A., Allie, R., Yun, S., Pennington, M., Beeton, C. & Chandy, K. G. (2003) J. Clin. Invest. 111, 1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cahalan, M. D., Wulff, H. & Chandy, G. (2002) J. Clin. Immunol. 21, 235–252. [DOI] [PubMed] [Google Scholar]
- 48.Szabo, I., Gulbins, E., Apfel, H., Zhang, X., Barth, P., Busch, A. E., Schlottmann, K., Pongs, O. & Lang, F. (1996) J. Biol. Chem. 271, 20465–20469. [DOI] [PubMed] [Google Scholar]
- 49.Chung, I. & Schlichter, L. C. (1997) J. Membr. Biol. 156, 73–85. [DOI] [PubMed] [Google Scholar]
- 50.Hanada, T., Lin, L., Chandy, K. G., Oh, S. S. & Chishti, A. H. (1997) J. Biol. Chem. 272, 26899–26904. [DOI] [PubMed] [Google Scholar]
- 51.Matko, J. (2003) Trends Pharmacol. Sci. 24, 385–389. [DOI] [PubMed] [Google Scholar]