

Abstract
We recently reported the isolation of a mutant of Pyrococcus furiosus, COM1, that is naturally and efficiently competent for DNA uptake. While we do not know the exact nature of this mutation, the combined transformation and recombination frequencies of this strain allow marker replacement by direct selection using linear DNA. In testing the limits of its recombination efficiency, we discovered that marker replacement was possible with as few as 40 nucleotides of flanking homology to the target region. We utilized this ability to design a strategy for selection of constructed deletions using PCR products with subsequent excision, or “pop-out,” of the selected marker. We used this method to construct a “markerless” deletion of the trpAB locus in the GLW101 (COM1 ΔpyrF) background to generate a strain (JFW02) that is a tight tryptophan auxotroph, providing a genetic background with two auxotrophic markers for further strain construction. The utility of trpAB as a selectable marker was demonstrated using prototrophic selection of plasmids and genomic DNA containing the wild-type trpAB alleles. A deletion of radB was also constructed that, surprisingly, had no obvious effect on either recombination or transformation, suggesting that its gene product is not involved in the COM1 phenotype. Attempts to construct a radA deletion mutation were unsuccessful, suggesting that this may be an essential gene. The ease and speed of this procedure will facilitate the construction of strains with multiple genetic changes and allow the construction of mutants with deletions of virtually any nonessential gene.
INTRODUCTION
Hyperthermophilic archaea are of special interest because of their evolutionary history and unique physiology, as well as several important biotechnology applications associated with their thermostable enzymes (4, 31). The development of genetic methods for this diverse group has presented many challenges, in part because of their extreme growth requirements. Recent progress has been made, however, in the ability to transform a variety of species by taking advantage of the fact that some are highly recombinogenic and/or able to take up DNA via natural competence. Sulfolobus sp., Thermococcus kodakaraensis, and Pyrococcus furiosus can all be transformed by linear DNA fragments (8, 12, 19, 20, 28, 29), but the length of the homologous flanking region needed for marker replacement varies. In T. kodakaraensis, which is naturally competent, more than 100 bp of homologous flanking region are required for homologous recombination (28), but in Sulfolobus acidocaldarius, which is transformed via electroporation, 10 to 30 bp of homology is sufficient (19). Several methods that rely on homologous recombination have also been developed to construct mutants in mesophilic archaea, including in Halobacterium sp. (23, 36), Haloferax volcanii (2, 3), and Methanosarcina acetivorans C2A (25).
The isolation of a mutant of Pyrococcus furiosus, previously designated COM1 (strain GLW101 [COM1 ΔpyrF]), that is efficiently competent for DNA uptake was recently reported (20). This strain is transformed by exogenous DNA without any chemical or physical treatment, as opposed to the wild-type P. furiosus, in which transformants were not obtained under the same conditions. Subsequently, this strain has enabled the construction of replicating shuttle vectors based on the chromosomal replication origin (9), as well as the production of strains that lack some key metabolic enzymes (5) and that overproduce affinity-tagged versions of the native (6) and a subcomplex form (15) of the cytoplasmic hydrogenase.
In this study, we show that 1,000 bp efficiently direct integration into the chromosome and as few as 40 bp allow efficient homologous recombination in the P. furiosus GLW101 chromosome using linear DNA fragments. Using this ability of GLW101 to recombine short segments of homologous DNA, a strategy was developed for generating deletion mutants by PCR amplification (without cloning) to select marker replacement events with subsequent excision, or “pop-out,” of the selected marker. An important feature of this method is that it allows direct selection of targeted mutants. We used this approach to generate a markerless deletion of trpAB, generating a strain (ΔtrpAB ΔpyrF) that allows simultaneous nutritional selection of both markers, as demonstrated using a recently constructed replicating plasmid for P. furiosus (9). To examine the role of genes predicted to be important for recombination, we used this method to attempt to delete radA and radB, both homologues in P. furiosus of the eukaryotic recombinase rad51. Surprisingly, deletion of radB had no apparent effect on either recombination or DNA transformation in the GLW101 (COM1 ΔpyrF) background. Mutants with a deletion of radA were not obtained, suggesting that deletion of this gene may be lethal. Further analysis of the GLW101 strain suggests that the natural competence phenotype does not result from uracil starvation or the loss of a restriction enzyme as a barrier to DNA transformation, but the exact nature of this mutation remains unknown.
MATERIALS AND METHODS
P. furiosus strains, media, and growth conditions.
P. furiosus DSM3638 (10) wild type, GLW101 (COM1 ΔpyrF) (20), and other strains (Table 1) were grown anaerobically in a defined medium with cellobiose as the carbon source (20) at 90°C for 16 to 18 h in 100-ml serum bottles containing 50 ml of liquid medium or on medium solidified with Phytagel (1% wt/vol) for 64 h. For growth of GLW101 and other uracil auxotrophic strains, the defined medium contained 20 μM uracil. Transformation of GLW101 was performed as described previously (20). The GLW104 strain was generated by transforming GLW101 with a PCR product containing the wild-type pyrF allele and selecting for uracil prototrophy. Transformation of the JFW02 strain was performed similarly, but tryptophan prototrophic selection was performed on a medium with or without 20 μM uracil and lacking tryptophan. The transformation efficiencies reported here were calculated as the number of transformant colonies per μg of DNA added and do not take into account plating efficiencies, which are typically 1 to 5%. Transformation frequencies were calculated as the proportions of transformant colonies to total cells and do take into account cell viability.
Table 1.
P. furiosus strains used and constructed in this study
| Strain | Genotype | Parent strain | Reference or source |
|---|---|---|---|
| DSM3638 | Wild type | 11 | |
| GLW101 | COM1 ΔpyrF | DSM 3638 | 21 |
| GLW102 | COM1 ΔpyrF trpF::Pgdh-pyrF | GLW101 | This work |
| GLW103 | COM1 ΔpyrF trpD::Pgdh-pyrF | GLW101 | This work |
| GLW104 | COM1 ΔpyrF::pyrF | GLW101 | This work |
| JFW01 | COM1 ΔpyrF trpAB::Pgdh-pyrF | GLW101 | This work |
| JFW02 | COM1 ΔpyrF ΔtrpAB | JFW01 | This work |
| JFW03 | COM1 ΔpyrF radB::Pgdh-pyrF | GLW101 | This work |
| JFW04 | COM1 ΔpyrF ΔradB | JFW03 | This work |
Purification of intermediate strains was performed by plating 10−3 dilutions of transformant cultures onto selective plate medium (without uracil) and picking isolated colonies into selective liquid medium. JFW01 and JFW03 were both purified to homogeneity in two rounds of colony purification. We were never able to obtain a pure marker replacement mutant for radA. Pop-out recombination was accomplished by growing strains with the marker replacement cassette inserted into the chromosome in defined cellobiose medium containing 20 μM uracil from a 1% inoculum (∼7 generations) and then plating onto defined medium containing 20 μM uracil and 3 mM 5-fluoroorotic acid (5-FOA).
Restriction endonuclease assays.
Cell-free extracts (CFE) were prepared from 1-liter cultures as described previously (7). Endonuclease assays were performed in 10-μl reaction mixture volumes using 0.5 to 1 μg of pJFW051 DNA. Various amounts of CFE (from 0 to 20 μg total protein) were added to separate reaction mixtures that were incubated in NEBuffer 4 (20 mM Tris-acetate, pH 7.9, 50 mM potassium acetate, 10 mM magnesium acetate, and 1 mM dithiothreitol; New England BioLabs). Reaction mixtures were prepared on ice and incubated at 90°C for 15 min. Similar assays were performed to detect the type I or type III restriction endonucleases. These assays were performed as described above except with the inclusion of 1 mM ATP, 80 μM S-adenosylmethionine (SAM), or both.
PCR amplification and transformation of the wild-type pyrF gene.
PCR amplification of the wild-type pyrF gene with flanking regions ranging in length from 1,000 to 0 bp (Fig. 1A) was performed using the following primer sets: GL055-GL058, pyrF500bpF-pyrF500bpR, pyrF250bpF-pyrF250bpR, pyrF150bpF-pyrF150bpR, pyrF100bpF-pyrF100bpR, pyrF50bpF-pyrF50bpR, pyrF40bpF-pyrF40bpR, pyrF30bpF-pyrF30bpR, pyrF20bpF-pyrF20bpR, pyrF10bpF-pyrF10bpR, and pyrF0bpF-pyrF0bpR (see Table S1 in the supplemental material). These products were purified using a DNA Clean & Concentrator-25 column (Zymo Research) and transformed into the GLW101 strain. Three biological replicates were performed for each PCR product.
Fig 1.

(A) The wild-type pyrF region and PCR-amplified fragments (indicated as lines below the chromosomal region) with various lengths of flanking sequence used to transform GLW101, selecting uracil prototrophy. (B) Transformation efficiencies using PCR products. Amounts of 1 μg of DNA were used to transform ∼107 cells.
trpAB pop-out marker replacement strategy.
Sequence Manipulation Suite (32) was used to generate a random 40-bp sequence (5′ AAGTGAGCGTGTTACGCCGAGACCCGGTTTCGTCTCTCAT 3′) that was altered slightly at the 3′ end to prevent hairpin or self-annealing structures that could be problematic in PCR. This sequence was introduced into the pop-out PCR product using 5′ primer tails. Two primer sets (JF392-JF393 and JF394-JF395; see Table S1 in the supplemental material) were used to amplify 1-kb regions flanking trpAB. The Pgdh-pyrF marker cassette was amplified from pJFW017 (9) plasmid DNA using primers JF355.3 and JF356.3. The specific annealing regions of these primers were designed for melting temperatures at 55 ± 4°C (mean ± standard deviation). The overlap tails were 30 to 35 bases in length and designed so that the overlapping regions between PCR products would be 20 to 25 bases in length, melting at 62 ± 2°C. PCR was performed using PfuTurbo polymerase in a 50-μl reaction mixture volume according to the manufacturer's instructions (Stratagene). Thermal cycling included 30 cycles with annealing at 58°C and a 70-s extension at 72°C. Products were purified using a DNA Clean & Concentrator-25 column (Zymo Research). Three fragments were put together by two rounds of splicing by overlap extension (SOE) PCR (16). The trpAB upstream flanking region was joined to the Pgdh-pyrF marker cassette, and in a separate reaction, the Pgdh-pyrF marker cassette was also joined to the trpAB 3′ flanking region. SOE PCR was performed using ∼50 ng of each template DNA in a 50-μl reaction mixture. Prior to thermal cycling, the template was denatured without primers, allowed to anneal at 58°C, and extended for 10 min at 72°C. Thirty cycles of amplification were performed as described above, with the same end primers used to generate the template products but with the extension time increased to 120 s. These two products were purified and used as the template for another SOE PCR. In the second SOE PCR, the first annealing step was omitted and the two fragments were allowed to anneal and extend at 72°C for 10 min. Thirty cycles of amplification were performed as described above, with primers JF392 and JF395 and extension time increased to 180 s. The 3-kb PCR product was then purified and transformed into the GLW101 strain. Eight uracil prototrophs were picked into liquid defined medium without uracil and grown overnight at 90°C. Putative transformants were screened for the marker replacement by PCR with JF392 and JF395, using the conditions described to generate the pop-out PCR product (see Fig. 2). The final deletion mutant was confirmed by sequencing of the trpAB region, which contained the sequence as designed.
Fig 2.

Pop-out marker replacement strategy. Six primers are used to construct a pop-out PCR product that is used to direct marker replacement and subsequent excision of the selected marker. (A) One-kilobase flanking regions are amplified from genomic DNA, and Pgdh-pyrF is amplified from pJFW017. Overlap tails for SOE PCR introduce the “pop-out scar” sequence and are indicated in red. (B) SOE PCR generates two overlap products. (C) A second SOE PCR generates the final pop-out construct. (D) Transformation into P. furiosus allows for selection of the marker replacement event. (E) 5-FOA selection of the pop-out cassette generates a markerless deletion.
Construction of plasmids.
To construct pJFW051, a 4.4-kb fragment was amplified by PCR from pJFW018 using primers JF264 and JF269. The Pgdh-hmg cassette (21) was amplified from pGLW28 (20) using primers GL021 and GL022, treated with T4 polynucleotide kinase, and ligated into the 4.4-kb fragment. A cassette containing the wild-type trpAB alleles under the transcriptional control of the phosphoenolpyruvate (PEP) synthase (PF0043) promoter (Ppep-trpAB) was constructed by SOE PCR. A 126-bp portion of the intergenic region upstream of the PEP synthase gene was amplified from wild-type genomic DNA using primers GL158 and WN008. The trpAB genes (PF1705 and PF1706) were amplified using primers WN009 and WN010 and joined to the fragment containing the PEP regulatory region and a 12-bp terminator from the hpyA1 gene (PF1722) (30). The Ppep-trpAB cassette was treated with T4 polynucleotide kinase and ligated into the pJFW018 plasmid (9) that had been digested with EcoRV and treated with shrimp alkaline phosphatase. Escherichia coli strain DH5α cells were transformed by electroporation in a 2-mm-gap cuvette at 2.5 V. The plasmid constructions were confirmed by restriction analysis. Plasmid DNA was isolated from liquid cultures by using QIAprep spin miniprep columns (Qiagen, Inc.).
RESULTS AND DISCUSSION
Fewer than 40 bp of homologous DNA allows selection of marker replacements in P. furiosus.
To investigate the minimum homology required for recombination in P. furiosus GLW101 (COM1 ΔpyrF), PCR products containing the pyrF gene with flanking DNA regions ranging in length from 0 to 1,000 bp were used to restore the ΔpyrF locus in GLW101 to the wild type, selecting transformants for uracil prototrophy (Fig. 1A). The transformation efficiency was measured as the number of uracil prototrophic transformants per μg of DNA. The transformation efficiency increased exponentially with the increase in flanking region length, with up to 103 transformants per μg of DNA containing 1-kb flanking regions and a few transformants detectable for DNA with flanking regions as short as 20 nucleotides (Fig. 1B). It is important to note that the annotated pyrF open reading frame overlaps with the open reading frame of the downstream gene, so the constructed pyrF deletion retains the last 16 bases of pyrF to include the full-length downstream open reading frame. As a result, the 3′ flanking region contained 16 bases of additional homology to the pyrF gene (20). This may contribute little for larger fragments of homology but may well affect the interpretation of the data for the very short fragments. However, our results clearly show that recombination can occur between 40-base sequences.
In previous work, we routinely used high concentrations (2 to 10 μg/ml) of transforming DNA (20). To determine the relationship between transforming DNA concentration and transformation frequency, we transformed GLW101 with a range of DNA concentrations, using both a replicating shuttle vector, pJFW018 (9), and a linear wild-type pyrF-containing fragment with 1 kb of flanking homology. For both DNA types, transformants were detectable with DNA concentrations as low as 1 ng/ml. When the transforming DNA concentration is high (10 μg/ml), the transformation frequency of linear DNA is approximately 1 out of 100 to 500 viable cells. Transformation with linear DNA fragments requires both DNA uptake and integration into the genome. Given the long regions of homology and the overabundance of DNA, it is likely that DNA uptake is the more (though not necessarily the only) limiting factor. These results are consistent with a model of natural competence in which only a small subset of cells are competent but they are very efficiently transformed. Taken together, these data suggest that transformation is probably very efficient with ample regions of homology, even at very low DNA concentrations. Transformation with very short regions of homology may be possible, but the efficiency may be prohibitively low for practical applications.
Sequence homology within the PCR products used for selection of marker replacement allows pop-out of the selected marker.
Selection of marker replacements using the wild-type copy of pyrF results in strains that are uracil prototrophs not useful for further mutant construction using pyrF as a selectable marker. To overcome this, we adapted a strategy that had been used successfully in yeast (1) and T. kodakaraensis (28) for pop-out of the wild-type pyrF allele. An example of this strategy, targeting two genes involved in tryptophan biosynthesis, trpAB, is shown in Fig. 2. The transforming DNA fragment containing the deletion cassette contains pyrF under the control of the promoter for the gene encoding glutamate dehydrogenase (PF1602), Pgdh-pyrF (20), flanked by an additional 40-bp direct-repeat sequence with minimal homology to the P. furiosus chromosome and constructed using splicing by overlap extension (SOE) PCR (16). A direct-repeat sequence of 40 bp was sufficient to allow for pop-out of the Pgdh-pyrF marker, and these regions were introduced into the transformation construct using PCR primers shorter than 60 bases. Three separate PCRs were used to amplify the upstream and downstream flanking regions of trpAB, as well as the pyrF expression cassette (Fig. 2 and 3), and these products were then joined by two successive rounds of SOE PCR, using a total of six primers for the construction. A proofreading polymerase was used to minimize the potential for introducing changes during polymerization. It is interesting to note that, while larger regions of homology are necessary for efficient marker replacement, as little as 40 bp allows efficient pop-out of the selected marker, suggesting that recombination, apart from transformation, is also very efficient.
Fig 3.
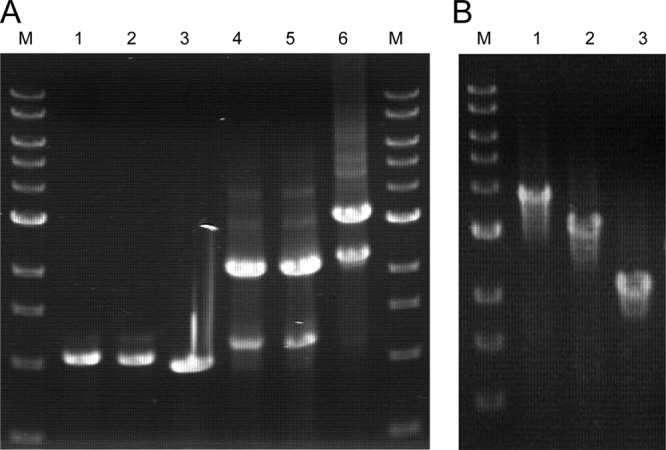
Construction of the trpAB pop-out markerless deletion. (A) Lanes: M, 1-kb DNA ladder; 1, 1-kb 5′ flanking amplicon; 2, 1-kb 3′ flanking amplicon; 3, Pgdh-pyrF marker cassette; 4, overlapped 5′ flanking region to Pgdh-pyrF marker cassette; 5, overlapped 3′ flanking region to Pgdh-pyrF marker cassette; 6, trpAB pop-out marker replacement cassette. (B) PCR amplification of the genomic regions surrounding the trpAB loci showing the marker replacement and subsequent pop-out (confirmed by DNA sequencing of the PCR products). Lanes: M, 1-kb DNA ladder; 1, trpAB locus in GLW101; 2, trpAB locus in JFW01; 3, trpAB locus in JFW02.
Transformation of the trpAB pop-out construction into the GLW101 strain resulted in hundreds of uracil prototrophic colonies, suggesting that marker replacement at this locus was also very efficient. Eight of these colonies were picked for PCR screening (see Fig. S1 in the supplemental material), and one was purified to homogeneity. This intermediate strain, JFW01, was a uracil prototroph and a tryptophan auxotroph. To select the pop-out event at the trpAB locus, JFW01 was grown in liquid medium containing uracil and then (1 ml of culture) was grown on solid medium containing both uracil and 5-fluoroorotic acid (5-FOA). All 5-FOA-resistant colonies (10 total) were screened for pop-out of the pyrF cassette, and the frequency of pop-out was approximately 10−7. One of these strains was designated JFW02. JFW02 is a tight tryptophan auxotroph, and excision of the Pgdh-pyrF marker restored 5-FOA resistance and uracil auxotrophy. JFW02 is therefore a double auxotroph and suitable for further genetic manipulation.
This strategy has several important advantages over conventional deletion construction. It does not require cloning, and only six primers are needed to provide specificity for the target gene (see Table S1 in the supplemental material). Primers of 60 bases are significantly less expensive and eliminate the need for primer purification, making pop-out construction of deletions amenable to a high-throughput system. Since both the integration and excision of the Pgdh-pyrF cassette are selected, this method may be used to target any nonessential gene. The pop-out constructs, as described here, leave a 40-bp “scar” sequence that remains in the genome after pop-out of the Pgdh-pyrF marker cassette. If a scarless deletion is desired, this strategy could be modified so that only one 40-bp pop-out sequence is included in the construct, which would recombine with the native sequence on the other side of the Pgdh-pyrF marker cassette to generate a scarless deletion of the target gene. Alternatively, the 40-bp scar sequence provides flexibility for modifying genomic targets by introducing specific sequences, such as signal peptides or affinity tags for protein purification. The utility of such tags has been demonstrated (6, 15).
For the trpAB deletion mutant, two rounds of purification, selecting uracil prototrophy, were required to resolve merodiploids generated by the initial marker replacement event. If the deletion is viable and produces a small or no growth defect, segregation and allelic fixation should be random. In the case of trpAB, which we expected to have a mild phenotype with tryptophan added to the medium, two rounds of colony purification were sufficient. The subsequent pop-out strain should have a neutral phenotype with both uracil and tryptophan added to the growth medium, and we found, in fact, that no additional purification was necessary after one round of selection on 5-FOA.
The ΔtrpAB strain is a tight tryptophan auxotroph but not resistant to 5-FAA.
As with uracil biosynthesis, tryptophan biosynthesis allows for selection for both prototrophy and auxotrophy because the wild-type allele is counterselectable. First demonstrated in Saccharomyces cerevisiae, deletion of various genes in the biosynthetic pathway results in a tryptophan auxotroph that is resistant to 5-fluoroanthranilic acid (5-FAA) (35). This anthranilic acid analog is converted to 5-fluorotryptophan by the tryptophan biosynthetic pathway, and incorporation of 5-fluorotryptophan into proteins is toxic. In addition, 5-fluorotryptophan inhibits anthranilate synthase, thereby reducing the synthesis of tryptophan and increasing 5-fluorotryptophan toxicity (22). We found that P. furiosus is sensitive to 5-FAA on defined medium at a concentration of approximately 2 g/liter. In P. furiosus, the tryptophan biosynthetic pathway is predicted to be in an operon consisting of seven genes (trpA to -G) (34). Our first targets for deletion mutagenesis were trpF (PF1707), trpE (PF1709), and trpD (PF1710). Mutants with deletions of trpE were not obtained, most likely for technical reasons, and mutants with deletions of trpF (PF1707) and trpD (PF1710) were leaky auxotrophs not resistant to 5-FAA. Deletion of the trpAB locus (PF1705 and PF1706) (Fig. 2) resulted in a tight tryptophan auxotroph but also did not confer resistance to 5-FAA, suggesting that there are other mechanisms of 5-FAA toxicity in P. furiosus.
Complementation of the ΔtrpAB strain by the wild-type trpAB alleles restores tryptophan prototrophy.
To test the utility of the trpAB deletion mutant for prototrophic selection, the wild-type trpAB alleles were cloned onto a replicating shuttle vector that also contained the pyrF expression cassette (9) to generate pJFW070 (Fig. 4). The COM1 ΔpyrF ΔtrpAB strain (JFW02) was readily transformed by this plasmid, selecting either uracil or tryptophan prototrophy. Since both markers are contained on the same plasmid, we were able to compare the transformation efficiencies of the two markers. The efficiencies (∼104 transformants per μg DNA) were similar to each other and comparable to that previously determined for pJFW018 (∼104 transformants per μg DNA [9]). JFW02 could also be transformed to tryptophan prototrophy using wild-type genomic DNA. This strain will be important for applications that require multiple simultaneous selections in the same strain, such as maintaining a replicating shuttle vector with one marker and using the other marker to perform chromosomal manipulations. Selecting tryptophan prototrophy also provides an additional alternative to uracil prototrophy or a requirement of agmatine for growth (15).
Fig 4.

Construction of pJFW070. (A) Primers GL158 and WN008 were used to amplify the PEP synthase promoter (Ppep). Primers WN009 and WN010 were used to amplify trpAB from wild-type genomic DNA. (B) These fragments were joined by SOE PCR to produce the Ppep-trpAB marker cassette, which was treated with T4 polynucleotide kinase (PNK) and ligated into the pJFW018 fragment produced by EcoRV digestion and shrimp alkaline phosphatase (SAP) treatment, producing pJFW070.
A deletion of radB has no obvious effect on recombination in the GLW101 genetic background.
To investigate the highly recombinant nature of GLW101 (COM1 ΔpyrF), we constructed a markerless deletion of radB (PF0021), a homologue of the eukaryotic rad51 gene (26). This protein has been implicated in recombination and repair in P. furiosus by its DNA binding affinity and interaction with other known recombination proteins (18). Its role as a recombination mediator rather than a true recombinase is supported by its weak ATPase and strand exchange activities in P. furiosus (18) and recombination and growth defects in deletion mutants of Haloferax volcanii (13, 14). The radB pop-out PCR product was constructed using the same approach used for trpAB. Hundreds of uracil prototrophic colonies were obtained, and of eight colonies screened, one was purified to homogeneity. This intermediate strain, designated JFW03, was grown in liquid medium containing uracil and plated on solid medium containing both uracil and 5-FOA. PCR amplification of the radB locus from the resulting colonies identified four that had pop-out of the pyrF allele, which was confirmed by DNA sequencing of the PCR products. One of these was designated JFW04. The fact that pop-out was readily selected in this radB marker replacement mutant suggests that deletion of radB had no significant effect on recombination.
To further investigate recombination in the radB deletion mutant, we transformed the JFW04 strain with several different DNA types: a replicating plasmid (pJFW018), a nonreplicating integrating plasmid (pGLW021) (20), integrating PCR products (trpAB::pyrF pop-out construction and amplified wild-type pyrF with 1-kb flanking regions), and wild-type (DSM3638) genomic DNA. All the DNA types transformed JFW04 to uracil prototrophy at equivalent frequencies (on the order of 10−2 to 10−3 transformants per viable cell count at a DNA concentration of 3 to 4 μg/ml, with plating efficiencies of approximately 1%).
Sensitivity to UV light was used to test recombination related to DNA repair. The survival of the radB mutant after exposure to UV doses in the range of 0 to 10 mJ on a plate surface was indistinguishable from the survival of the wild type and GLW101 at all intensities tested (Fig. 5A). There was also no difference in the growth rates of the two strains in defined medium (Fig. 5B) or under conditions of oxidative shock (Fig. 5C), as measured by sensitivity to hydrogen peroxide (33).
Fig 5.
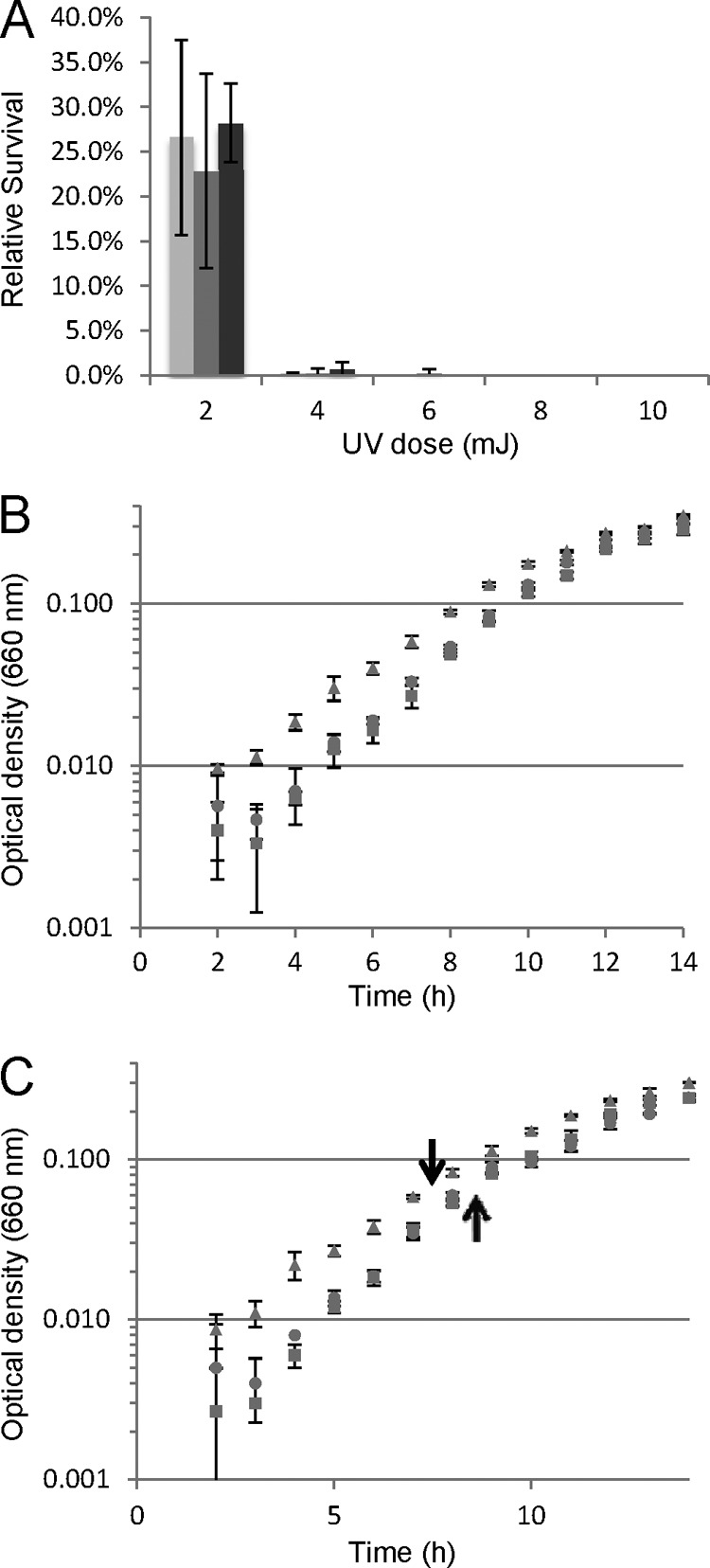
Characterization of JFW04. (A) Wild-type (light grey), GLW101 (grey), and JFW04 (dark grey) strains were exposed to UV radiation on a plate surface with doses ranging from 0 to 10 mJ. Relative survival was calculated as the proportion of colonies compared to the number for the unexposed control. (B) Growth curve of the JFW04 strain (circles) compared to the growth curves of GLW101 (squares) and the wild type (triangles). Culture growth was monitored by optical density at 660 nm. (C) Oxidative stress response of the JFW04 (circles) strain compared to the oxidative stress responses of GLW101 (squares) and the wild type (triangles). H2O2 was added at an OD660 of ∼0.07 (7.5 h for wild type, 8.5 h for GLW101 and JFW04). Times of H2O2 addition are indicated by arrows. Each point represents the average of samples from two or more independent cultures, and the error bars show standard deviations.
The apparent wild-type phenotype of JFW04 is somewhat surprising given the severe phenotype of radB mutants in the euryarchaeote Haloferax volcanii (13, 14; T. Allers, personal communication). On the other hand, radB is not present in any known member of the Crenarchaeota, including Sulfolobus species. The apparent wild-type phenotype of JFW04 may result from differences in radB functional divergence within the Euryarchaeota. It could also be the result of the GLW101 genetic background, but it is not possible to address this, as the wild-type strain, Pyrococcus furiosus DSM3638, has proven to be genetically intractable in our hands. Future work with radB in the closely related T. kodakaraensis could address this issue.
Attempts to construct a mutant with a deletion of radA were unsuccessful.
We also attempted to construct a mutant with a deletion of radA (PF1926), another rad51 homologue (27). In contrast to RadB, RadA is a true recombinase, with DNA binding, ATPase, and strand exchange activities in P. furiosus (18). Deletion of radA in Haloferax volcanii results in recombination and growth defects (37). Transformation of a radA pop-out construction into GLW101 also produced hundreds of uracil prototrophic colonies. Eight were screened for the marker replacement event at the radA locus, and all contained both radA and the Pgdh-pyrF marker replacement. Six rounds of colony purification, which were sufficient to purify other deletion mutants, failed to resolve these merodiploids, and repeated attempts to isolate a clean marker replacement strain were unsuccessful. In addition, the unresolved merodiploids showed a severe growth defect. Either in liquid medium or on plates, these strains took at least twice as long as the GLW101 parent to grow to a comparable cell density or colony size. These data suggest that a deletion of the radA gene results in a severe phenotype and that radA may, in fact, be essential for viability in P. furiosus, although further experiments would be required to prove this conclusively.
The exact nature of the mutation leading to the COM1 phenotype is unknown.
The nature of competence in GLW101 is of considerable biological interest. We hypothesized that the initial transformation event, integrating pGLW021, selected using simvastatin resistance, was a rare event, never seen again, but the deletion of pyrF in this transformant resulted in a strain that was starved for uracil, and uracil starvation resulted in competence as a mechanism to take up DNA as a source of uracil. Competence would then depend on uracil starvation, i.e., cell growth on plates without uracil, as in prototrophic selection using the wild-type pyrF allele. To test this, we constructed a shuttle vector, pJFW051, which is similar to pJFW018 but contains the 3-hydroxy-3-methylglutaryl coenzyme A gene for simvastatin resistance selection (see Fig. S2 in the supplemental material). We found that GLW101 was readily transformed by this plasmid on defined medium containing uracil and simvastatin. We performed the same experiment with GLW104, a GLW101-derived strain with a restored pyrF gene, and transformation of this strain was equally efficient. Since uracil starvation is not an issue with the restored pyrF deletion, this would indicate that competence does not result from uracil starvation.
We also hypothesized that competence might be the result of a mutation in a restriction system, as restriction of heterologous DNA is often a barrier to transformation. To test this, cell extracts were prepared from wild-type P. furiosus and GL101 cultures and incubated with pJFW051 plasmid DNA, using conditions suitable for other restriction enzymes from Pyrococcus species (17, 24), reported for their commercial use. Under all conditions tested, no restriction activity was detected for either the wild type or GLW101. In addition, there are no annotated restriction endonucleases in the P. furiosus genome. Interestingly, there are also no homologues of competence genes. We also emphasize that many “naturally competent” organisms do not exhibit competence under all conditions, and it is possible that wild-type P. furiosus will be competent if appropriate conditions are used.
Conclusions.
P. furiosus is an excellent model system for the study of DNA recombination, repair, and natural competence in the Archaea. The methods reported here will facilitate future studies by decreasing the time and expense required to generate marker replacement and deletion mutants. The trpAB deletion provides another selectable marker and will enable more sophisticated genetic analyses involving the maintenance of multiple selectable markers. In the GLW101 (COM1 ΔpyrF) background, we have found that the deletion of radB has no detectable phenotype but the deletion of radA has a severe, possibly lethal phenotype. It is not known if this is true for P. furiosus in general or is peculiar to the GLW101 strain, since the nature of competence in GLW101 is not yet understood. This is the subject of ongoing and future investigations, which will be further facilitated by the work presented here.
The application of this method in P. furiosus will be especially valuable in elucidating the function of the ∼600 genes which are unique to P. furiosus as compared to T. kodakaraensis (11). The increased transformation frequency associated with the GLW101 strain makes it particularly useful for the study of natural competence, CRISPR (clusters of regularly interspaced short palindromic repeats) function, and homologous recombination in general. In addition, the pop-out strategy can be adapted to generate tagged proteins in two steps in vivo and will have other uses that make strain construction rapid.
Supplementary Material
ACKNOWLEDGMENTS
We thank Daehwan Chung for technical advice throughout the course of the work and Jennifer Copeland for technical assistance.
This work was supported by grants from the Bio-Energy Science Center (grant DE-PS02-06ER64304), administered by Oak Ridge National Laboratory, and from the Office of Biological and Environmental Research (grant FG02-08ER64690) and the Chemical Sciences, Geosciences and Biosciences Division (grant DE-FG05-95ER20175), Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy. J.F. was supported by a predoctoral Graduate Training In Genetics grant (grant NIH 5T32GM007103-30) to the Genetics Department of the University of Georgia.
Footnotes
Published ahead of print 27 April 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Alani E, Cao L, Kleckner N. 1987. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics 116:541–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Allers T, Ngo HP, Mevarech M, Lloyd RG. 2004. Development of additional selectable markers for the halophilic archaeon Haloferax volcanii based on the leuB and trpA genes. Appl. Environ. Microbiol. 70:943–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bitan-Banin G, Ortenberg R, Mevarech M. 2003. Development of a gene knockout system for the halophilic archaeon Haloferax volcanii by use of the pyrE gene. J. Bacteriol. 185:772–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blumer-Schuette SE, Kataeva I, Westpheling J, Adams MWW, Kelly RM. 2008. Extremely thermophilic microorganisms for biomass conversion: status and prospects. Curr. Opin. Biotechnol. 19:210–217 [DOI] [PubMed] [Google Scholar]
- 5. Bridger SL, et al. 2011. Deletion strains reveal metabolic roles for key elemental sulfur-responsive proteins in Pyrococcus furiosus. J. Bacteriol. 193:6498–6504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chandrayan SK, et al. 2012. Engineering hyperthermophilic archaeon Pyrococcus furiosus to overproduce its cytoplasmic [NiFe]-hydrogenase. J. Biol. Chem. 287:3257–3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chung D, Huddleston JR, Farkas J, Westpheling J. 2011. Identification and characterization of CbeI, a novel thermostable restriction enzyme from Caldicellulosiruptor bescii DSM 6725 and a member of a new subfamily of HaeIII-like enzymes. J. Ind. Microbiol. Biotechnol. 38:1867–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deng L, Zhu H, Chen Z, Liang YX, She Q. 2009. Unmarked gene deletion and host-vector system for the hyperthermophilic crenarchaeon Sulfolobus islandicus. Extremophiles 13:735–746 [DOI] [PubMed] [Google Scholar]
- 9. Farkas J, Chung D, Debarry M, Adams MWW, Westpheling J. 2011. Defining components of the chromosomal origin of replication of the hyperthermophilic archaeon Pyrococcus furiosus needed for construction of a stable replicating shuttle vector. Appl. Environ. Microbiol. 77:6343–6349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fiala G, Stetter KO. 1986. Pyrococcus furiosus sp. nov. represents a novel genus of marine heterotrophic archaebacteria growing optimally at 100°C. Arch. Microbiol. 145:56–61 [Google Scholar]
- 11. Fukui T, et al. 2005. Complete genome sequence of the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1 and comparison with Pyrococcus genomes. Genome Res. 15:352–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grogan DW, Stengel KR. 2008. Recombination of synthetic oligonucleotides with prokaryotic chromosomes: substrate requirements of the Escherichia coli/λRed and Sulfolobus acidocaldarius recombination systems. Mol. Microbiol. 69:1255–1265 [DOI] [PubMed] [Google Scholar]
- 13. Haldenby S. 2007. Genetic analysis of RadB, a paralogue of the archaeal Rad51/RecA homologue, RadA. Ph.D. thesis. University of Nottingham, Nottingham, United Kingdom [Google Scholar]
- 14. Haldenby S, White MF, Allers T. 2009. RecA family proteins in archaea: RadA and its cousins. Biochem. Soc. Trans. 37:102–107 [DOI] [PubMed] [Google Scholar]
- 15. Hopkins RC, et al. 2011. Homologous expression of a subcomplex of Pyrococcus furiosus hydrogenase that interacts with pyruvate ferredoxin oxidoreductase. PLoS One 6:e26569 doi:10.1371/journal.pone.0026569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528–535 [PubMed] [Google Scholar]
- 17. Ishikawa K, et al. 2005. Discovery of a novel restriction endonuclease by genome comparison and application of a wheat-germ-based cell-free translation assay: PabI (5′-GTA/C) from the hyperthermophilic archaeon Pyrococcus abyssi. Nucleic Acids Res. 33:e112 doi:10.1093/nar/gni113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Komori K, et al. 2000. Both RadA and RadB are involved in homologous recombination in Pyrococcus furiosus. J. Biol. Chem. 275:33782–33790 [DOI] [PubMed] [Google Scholar]
- 19. Kurosawa N, Grogan DW. 2005. Homologous recombination of exogenous DNA with the Sulfolobus acidocaldarius genome: properties and uses. FEMS Microbiol. Lett. 253:141–149 [DOI] [PubMed] [Google Scholar]
- 20. Lipscomb GL, et al. 2011. Natural competence in the hyperthermophilic archaeon Pyrococcus furiosus facilitates genetic manipulation: construction of markerless deletions of genes encoding the two cytoplasmic hydrogenases. Appl. Environ. Microbiol. 77:2232–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matsumi R, Manabe K, Fukui T, Atomi H, Imanaka T. 2007. Disruption of a sugar transporter gene cluster in a hyperthermophilic archaeon using a host-marker system based on antibiotic resistance. J. Bacteriol. 189:2683–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miozzari G, Niederberger P, Hutter R. 1977. Action of tryptophan analogues in Saccharomyces cerevisiae. Arch. Microbiol. 115:307–316 [DOI] [PubMed] [Google Scholar]
- 23. Peck RF, DasSarma S, Krebs MP. 2000. Homologous gene knockout in the archaeon Halobacterium salinarum with ura3 as a counterselectable marker. Mol. Microbiol. 35:667–676 [DOI] [PubMed] [Google Scholar]
- 24. Pingoud V, et al. 2003. PspGI, a type II restriction endonuclease from the extreme thermophile Pyrococcus sp.: structural and functional studies to investigate an evolutionary relationship with several mesophilic restriction enzymes. J. Mol. Biol. 329:913–929 [DOI] [PubMed] [Google Scholar]
- 25. Pritchett MA, Zhang JK, Metcalf WW. 2004. Development of a markerless genetic exchange method for Methanosarcina acetivorans C2A and its use in construction of new genetic tools for methanogenic archaea. Appl. Environ. Microbiol. 70:1425–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sandler SJ, et al. 1999. Diversity of radA genes from cultured and uncultured Archaea: comparative analysis of putative RadA proteins and their use as a phylogenetic marker. J. Bacteriol. 181:907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sandler SJ, Satin LH, Samra HS, Clark AJ. 1996. recA-like genes from three archaean species with putative protein products similar to Rad51 and Dmc1 proteins of the yeast Saccharomyces cerevisiae. Nucleic Acids Res. 24:2125–2132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sato T, Fukui T, Atomi H, Imanaka T. 2005. Improved and versatile transformation system allowing multiple genetic manipulations of the hyperthermophilic archaeon Thermococcus kodakaraensis. Appl. Environ. Microbiol. 71:3889–3899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sato T, Fukui T, Atomi H, Imanaka T. 2003. Targeted gene disruption by homologous recombination in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Bacteriol. 185:210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Spitalny P, Thomm M. 2008. A polymerase III-like reinitiation mechanism is operating in regulation of histone expression in archaea. Mol. Microbiol. 67:958–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stetter KO. 1996. Hyperthermophilic procaryotes. FEMS Microbiol. Rev. 18:149–158 [Google Scholar]
- 32. Stothard P. 2000. The Sequence Manipulation Suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28:1102, 1104. [DOI] [PubMed] [Google Scholar]
- 33. Strand KR, et al. 2010. Oxidative stress protection and the repair response to hydrogen peroxide in the hyperthermophilic archaeon Pyrococcus furiosus and in related species. Arch. Microbiol. 192:447–459 [DOI] [PubMed] [Google Scholar]
- 34. Tang X, et al. 1999. The tryptophan biosynthesis gene cluster trpCDEGFBA from Pyrococcus kodakaraensis KOD1 is regulated at the transcriptional level and expressed as a single mRNA. Mol. Gen. Genet. 262:815–821 [DOI] [PubMed] [Google Scholar]
- 35. Toyn JH, Gunyuzlu PL, White WH, Thompson LA, Hollis GF. 2000. A counterselection for the tryptophan pathway in yeast: 5-fluoroanthranilic acid resistance. Yeast 16:553–560 [DOI] [PubMed] [Google Scholar]
- 36. Wang G, Kennedy SP, Fasiludeen S, Rensing C, DasSarma S. 2004. Arsenic resistance in Halobacterium sp. strain NRC-1 examined by using an improved gene knockout system. J. Bacteriol. 186:3187–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Woods WG, Dyall-Smith ML. 1997. Construction and analysis of a recombination-deficient (radA) mutant of Haloferax volcanii. Mol. Microbiol. 23:791–797 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.