

Abstract
In this paper, we have characterized two malic enzymes (ME), SCO2951 and SCO5261, from Streptomyces coelicolor and analyzed their role in antibiotic and triacylglycerol (TAG) production. Biochemical studies have demonstrated that Sco2951 and Sco5261 genes encode NAD+- and NADP+-dependent malic enzymes, respectively. Single or double mutants in the ME-encoding genes show no effect on growth rate compared to the parental M145 strain. However, the single Sco2951 and the double Sco2951 Sco5261 mutants display a strong reduction in the production of the polyketide antibiotic actinorhodin; additionally, the Sco2951 Sco5261 mutant shows a decrease in stored TAGs during exponential growth. The lower production of actinorhodin in the double mutant occurs as a consequence of a decrease in the expression of actII-ORF4, the transcriptional activator of the actinorhodin gene cluster. On the other hand, the reduced TAG accumulation is not due to reduced transcript levels of fatty acid biosynthetic genes nor to changes in the amount of the precursor acetyl coenzyme A (acetyl-CoA). This mutant accumulates intermediates of the tricarboxylic acid (TCA) cycle that could alter the regulation of the actinorhodin biosynthetic pathway, suggesting that MEs are important anaplerotic enzymes that redirect C4 intermediates from the TCA cycle to maintain secondary metabolism and TAG production in Streptomyces.
INTRODUCTION
Streptomyces species produce more than half of the natural products used in medicine and agriculture (9). An important group of natural compounds are polyketides, which are synthesized by a series of condensation-reduction reactions between small carboxylic acids by using a similar mechanism to that in fatty acid biosynthesis (6). Due to the importance of natural products in human health, substantial efforts have been made to understand the biosynthesis and regulation of these compounds in the genus Streptomyces (3, 18). However, much less attention has been devoted to the central metabolic network that supplies the building blocks for antibiotic biosynthesis, including anaplerotic enzymes, which are especially important to interconnect the main metabolic pathways in the cell. Previous work in other bacteria has demonstrated that anaplerotic enzymes involved in the phosphoenolpyruvate (PEP)-pyruvate (PYR)-oxaloacetate (OXA) node constitute an important switch point for carbon flux (30) (Fig. 1). This node gives rise to branch points between energy-producing, catabolic, and biosynthetic pathways. As an example, phosphoenolpyruvate carboxykinase and/or ME in combination with PEP synthetase direct intermediates from the tricarboxylic acid (TCA) cycle to yield PEP under gluconeogenic conditions (30). All the anaplerotic enzymes at the PEP-PYR-OXA node are distinctive among different bacteria, in contrast to the fairly widespread set of enzymes for the central metabolic pathways. Therefore, the identification and characterization of anaplerotic reactions in relation to their role in a particular bacterial metabolism are important starting points to manipulate growth and fermentation processes for biotechnological purposes (32).
Fig 1.

Metabolic pathways connected by the PEP-pyruvate-oxalacetate node. Abbreviations: PEPCK, PEP carboxykinase; ME-NAD(P), malic enzyme-NAD(P) dependent; MDH, malate dehydrogenase; PPS, PEP synthase.
Among the PEP-PYR-OXA node enzymes, MEs are interesting since they catalyze the oxidative decarboxylation of l-malate into PYR with the concomitant generation of NADH or NADPH as reductive equivalents. Depending on the cofactor utilized, MEs can be classified as NAD+ dependent (EC 1.1.1.38 and EC 1.1.1.39; the latter is also able to decarboxylate OXA) or NADP+ dependent (EC 1.1.1.40). Different roles have been assigned to MEs in plants and mammals (7, 16, 23); however, little is known about the role of these enzymes in prokaryotes. In mammals, the relevance of cytosolic and mitochondrial NADP+-dependent MEs is related to their ability to generate reduction equivalents for the biosynthesis of saturated fatty acids, while the human mitochondrial NAD+-dependent ME is believed to play an important role in glutamine metabolism in fast-growing tissues and tumors (16, 23). In plants, different MEs are involved in a wide range of metabolic pathways, such as the carbon flux of C3-C4 intermediates between different organelles and in C4 photosynthesis (7). In Escherichia coli, two MEs have been characterized, and their participation was demonstrated in directing the malate, produced by the glyoxylate cycle, to gluconeogenesis when bacteria grows on acetate as a sole carbon source (26). In Streptomyces spp., which represent one of most biotechnologically important genera, the role for malic enzymes has not been clearly established (17). This genus is an interesting system to study the role of this enzyme, since it not only produces important bioactive metabolites, such as those belonging to the polyketide family of compounds, but also accumulates triacylglycerols (TAGs), both of which require acetyl coenzyme A (acetyl-CoA) and NADPH for their biosynthesis (2). In this paper, we have characterized the two MEs present in S. coelicolor and studied their roles in growth, polyketide production, and TAG accumulation. In vitro and in vivo studies of these enzymes allowed the identification of SCO2951 and SCO5261 as an NAD+- and an NADP+-dependent ME, respectively. In addition, we have constructed single and double mutants in the ME-encoding genes and studied the impact of these enzymes in secondary metabolism and storage lipid accumulation.
MATERIALS AND METHODS
Strains and growth conditions.
Bacterial strains and plasmids used in this study are shown in Table 1. E. coli strains were grown either on solid or in liquid Luria-Bertani medium at 37°C and supplemented with the following antibiotics: 100 mg/liter ampicillin (Ap), 50 mg/liter kanamycin (Km), 20 mg chloramphenicol (Cm), or 100 mg/liter apramycin (Am) when needed. Streptomyces strains were grown at 30°C on MS agar, SMM medium (19), or SMM medium supplemented with 0.5% l-malate (SMM-Mal). For Streptomyces selection, the antibiotics Am, hygromycin (Hyg), and Km were supplemented to solid or liquid medium at final concentrations of 50, 50, and 200 mg/liter, respectively.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| Strains | ||
| S. coelicolor | ||
| M145 | Parental strain, SCP1− SCP2− | 19 |
| TR2951 | M145, Sco2951::Tn5062 (Amr) | This study |
| TR5261 | M145, Sco5261::Tn5062 (Amr) | This study |
| TRD | M145, Sco2951::Tn5062 (Amr), Sco5261::Tn 5066 (Hygr) | This study |
| TR2951c | TR2951 derivative carrying the integrative plasmid pTR2951 (Kmr) | This study |
| TR5261c | TR5261 derivative carrying the integrative plasmid pTR5261 (Kmr) | This study |
| TRDc2951 | TRD derivative carrying the integrative plasmid pTR2951 (Kmr) | This study |
| TRDc5261 | TRD derivative carrying the integrative plasmid pTR5261 (Kmr) | This study |
| E. coli | ||
| DH5α | E coli K12 F− lacU169(f80lacZDM15) endA1 recA1 hsdR17 deoR supE44 thi-1 l2 gyrA96 relA1 | 12 |
| BL21λ(DE3) | E. coli B F− ompt rB− mB− (DE3) | 33 |
| ET 12567 | supE44 hsdS20 ara-14 proA2 lacY galK2 rpsL20 xyl-5 mtl-1 dam− dcm− hsdM− (Cmr) | 29 |
| Plasmids | ||
| pET28a | Phagemid vector for expression of recombinant proteins under the control of strong T7 transcription and translation signals (Kmr) | Novagen |
| PCR-Blunt | Used for cloning PCR products (Apr) | Invitrogen |
| pTR285 | pRT802 derivative carrying the PermE*promoter | 2 |
| pTR252 | pET28a with an insert carrying the sco2951 His tag fusion gene under the control of strong T7 transcription and translation signals | This study |
| pTR5261 | pRT285 derivative plasmid carrying the sco5261-His tag gene under the control of PermE* | This study |
| pTR2951 | pRT285 derivative plasmid carrying the sco2951-His tag gene under the control of PermE* | This study |
Cloning of Sco2951 and Sco5261 genes.
Sco2951 and Sco5261 genes were amplified by PCR from genomic S. coelicolor DNA M145 using the following oligonucleotides: for sco2951, 5′-CATATGGCAACGGCGCCCAGC-3′ (upper) and 5′-GAATTCACTAGTACGCCACCACTCCGGC-3′ (lower), and for sco5261, 5′-CATATGGCAGCGGAGATCGTC-3′ (upper) and 5′-GAATTCACTAGTCACCGGCGCGCAACCCC-3′ (lower). The upper primers used were designed to have an NdeI site (in bold) overlapping the translational initiation codon, changing GTG start codons to ATG for the Sco5261 gene. The lower primers contained EcoRI and SpeI sites (in bold) downstream of the stop codon. The resulting PCR products were verified by DNA sequencing and cloned as NdeI-EcoRI fragments into the expression vector pET28a, which contains six His codons upstream of the NdeI site, to obtain pET28a::2951 (pTR252) and pET28a::5261 (pTR255). For complementation and overexpression in S. coelicolor, each XbaI-NotI fragment from the pET28a derivatives (pTR252 and pTR255) was cloned into the integrative vector pTR285 containing the PermE* promoter (2) to make pTR2951 (PermE*::sco2951) and pTR5261 (PermE*::sco5261), respectively.
Expression and purification of malic enzymes.
E. coli BL21(DE3) host strains (Stratagene) carrying each of the plasmids pRT252 and pTR255 were grown in Luria-Bertani medium at 37°C, induced with 0.5 mM isopropyl-d-thiogalactopyranoside (IPTG), and incubated for 20 h at 30°C. Cells were harvested by centrifugation at 4,000 × g for 20 min at 4°C, washed twice, and resuspended with buffer containing 50 mM Tris-HCl (pH 7.0), 150 mM NaCl, 10% glycerol, and 10 mM MgCl2 (buffer A). Cell disruption was carried out in a French pressure cell at 1,000 MPa in the presence of 1% (vol/vol) protease inhibitor cocktail (Sigma-Aldrich). The protein extract was cleared by centrifugation at 15,000 × g for 30 min at 4°C, and the supernatant was applied to a Ni2-nitrilotriacetic acid-agarose affinity column (Qiagen) equilibrated with the same buffer supplemented with 20 mM imidazole. The column was washed and the His-tagged proteins were eluted using buffer A containing 60 to 250 mM imidazole. Fractions were collected and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Fractions containing purified proteins were dialyzed overnight using buffer A at 4°C. Pure proteins were stored at −80°C.
Malic enzyme assay.
NAD+- and NADP+-ME activities were determined spectrophotometrically at 30°C by monitoring NADH and NADPH production at 340 nm, respectively. The reaction was assayed in a 0.5-ml reaction volume containing 50 mM Tris-HCl (pH 8.0), 0.5 mM NAD+ or NADP+, 20 mM l-malate, and 10 mM MgCl2. One unit of enzyme activity is defined as the amount of enzyme resulting in the production of 1 μmol of NADPH min−1 (5).
Isolation of Sco2951 and Sco5261 mutant strains of S. coelicolor.
To disrupt Sco2951 and Sco5261, two cosmids from the transposon insertion cosmid library of S. coelicolor (13) were used. Cosmids SCE59.1.F10 and 7g11-1.F12, carrying individual Tn5062 insertions in each gene, were introduced into S. coelicolor M145 by conjugation using E. coli ET12567/pUB307 as a donor (29). For each mutant, two independent Amr Kms exconjugants were isolated and checked by PCR using specific primers for each gene and for the transposon, verifying that allelic replacement had occurred. To isolate an Sco2951 Sco5261 double mutant, the Amr marker of Tn5062 in cosmid 7g11-1.B03 was replaced by Tn5066 Hygr using plasmid pQM5066 (P. Dyson, personal communication). This 7g11-1.F12 Hygr cosmid was introduced into the TR2951 strain to obtain the TRD strain.
Antibiotic determination.
For actinorhodin determination, 1 ml of whole broth was added to a KOH solution giving a final concentration of 1 M; the solution was mixed vigorously and centrifuged at 4,000 × g for 5 min. Supernatant absorbance at 640 nm was determined, and the actinorhodin concentration was calculated using a molar absorption coefficient at 640 nm of 25,320 (4). For undecylprodigiosin determination, 1 ml of broth was centrifuged at 5,000 × g for 10 min, and the cells were resuspended in 1 ml methanol. pH was taken to 1.5 with HCl 1 N, and the solution was mixed vigorously and centrifuged at 4,000 × g for 5 min. Supernatant absorbance at 530 nm was determined and the undecylprodigiosin concentration was calculated using a molar absorption coefficient at 530 nm of 100,500 (35).
Lipid analysis.
Total lipids were extracted twice from lyophilized cell material (1.5 to 3 mg) with chloroform and methanol (2:1, vol/vol). The combined extracts were evaporated and analyzed by thin-layer chromatography (TLC) on silica gel 60 F254 plates (0.2 mm; Merck), as described previously (2), using hexane-diethyl ether-acetic acid (80:20:1, vol/vol/vol) as developing solvent for TAG analysis. Lipid fractions were visualized by Cu-phosphoric staining.
Metabolite profiling.
Extraction, liquid partition, and derivation were performed as described by Lisec et al. (22). Four biological replicates per treatment with a second group of technical replicates were utilized (8 total data points used during analysis) for this determination. Gas chromatography coupled to mass spectrometry (GC-MS) analysis was performed using an autosystem XL gas chromatograph and a Turbo mass spectrometer (Perkin Elmer) at Facultad de Ciencias Bioquímicas y Farmacéuticas–UNR facilities. A 1-μl split injection (split ratio of 1:40) was injected at 280°C. The capillary column used was a VF-5ms (Varian, Darmstadt, Germany) with the following dimensions: 30-m by 0.25-mm inner diameter and a 0.25-μm film with helium as carrier gas at a constant flow of 1 ml/min. The temperature program was 5 min at 70°C, 5 min ramp to 310°C, and final heating for 2 min at 310°C. The transfer line to the MS was set to 280°C. Spectra were monitored in the mass range of m/z 70 to 600. Tuning and all other settings were according to manufacturer's recommendations.
Chromatograms were acquired with TurboMass 4.1 software (Perkin Elmer). The NIST98mass spectral search program (http://www.nist.gov/srd/mslist.htm; National Institute of Standards and Technology, Gaithersburg, MD) was the software platform. The MS and retention time index were compared with the collection of the Golm Metabolome Database (20). MS matching was manually supervised, and matches were accepted with thresholds of match of >650 (with maximum match equal to 1,000) and retention index deviation of <1.0%. Peak heights were normalized using the amount of the sample fresh weight and ribitol as the internal standard. Relative metabolite contents were determined, and statistical analyses were performed using analysis of variance (ANOVA) tests in Sigma Stat 3.1.
Acetyl-CoA determination.
Acetyl-CoA and other CoA thioesters were quantified as described by Wadler and Cronan (38). Briefly, metabolites were extracted with trichloroacetic acid using 50 ml of M145 and TRD cultures grown for 24 h in liquid SMM-Gluc (1%) medium. Recovery of metabolites was performed using a C18 cartridge previously equilibrated with 100% methanol and 1 mM HCl sequentially. CoA and CoA esters were then eluted with 0.1 M ammonium acetate in 65% ethanol. The eluate was evaporated under vacuum at room temperature until a final volume of 100 μl was reached. The sample was then treated with shrimp alkaline phosphatase to release the 3′-phosphates from CoA and its esters as inorganic phosphate. Following phosphatase removal, the dephosphorylated CoA metabolites were rephosphorylated by treatment with a dephospho-CoA kinase and [γ-33P]ATP. The resulting radioactive CoA metabolites were then separated by reverse-phase high-performance liquid chromatography and quantified by flowthrough scintillation counting.
RNA extraction and real-time qRT-PCR assay.
RNA was extracted from M145 and TRD cultures grown in liquid SMM-Gluc (1%) medium using the SV total RNA isolation system (Promega). Second-strand cDNA generated with random primers was used in quantitative PCR (qPCR) with SYBR green as the indicator dye. Primer pairs RTactIIORF4R (5′-TACACGAGCACCTTCTCACC-3′) and RTactIIORF4L (5′-TGGAATCGTATCGGAATCTC-3′), RTfabHup (5′-CAAGTTTCCTGCGATCACG-3′) and RTfabHdn (5′-GGGATGAAGACGTCCAGGT-3′), and RThrdBR (5′-GTTGATGACCTCGACCATGT-3′) and RThrdBL (5′-CAAGGGCTACAAGTTCTCCA-3′) were used to analyze transcript levels of actII-ORF4, fabH, and hrdB, respectively. The expression of each gene was quantified after normalization to hrdB mRNA levels. qPCR cycling conditions were as follows: 95°C for 2 min followed by 40 cycles at 94°C for 15 s, 58°C for 15 s, and 68°C for 20 s. qPCR data are presented as a fold difference of expression in M145 cells relative to that in TR50 cells using the Pfafll method (27), with hrdB used as a normalizing gene.
Phylogenetic analysis.
Sequence alignment was performed with CLUSTAL W (21) using default parameters. The alignment was edited, gaps were deleted, and the phylogenetic tree was constructed using MEGA 5 software (34) using 100 bootstrap replicas.
RESULTS
Biochemical characterization of Sco2951 and Sco5261, two malic enzymes from S. coelicolor.
A bioinformatic search of putative MEs in the S. coelicolor genome database (strepdb.streptomyces.org.uk) revealed the presence of two open reading frames, ORFs, SCO2951 and SCO5261, highly similar to previously characterized MEs in prokaryotes. Phylogenetic analysis of prokaryotic and eukaryotic MEs, including orthologs of SCO2951 and SCO5261 from several Streptomyces spp., showed that they are highly conserved throughout the genus and clearly divided in two discrete subclades (Fig. 2A). However, this analysis does not allow assigning cofactor selectivity to the enzymes found in each subclade. Recently, crystal structures and site-directed mutagenesis studies permitted the identification of the amino acid residues involved in cofactor specificity for eukaryotic MEs (15). Several residues have been demonstrated to participate in cofactor binding (Fig. 2B). Among those, amino acid residue 362 (the residue number corresponds to the mitochondrial NAD+-ME from humans, Fig. 2B) seems to be critical for cofactor selectivity. For example, human mitochondrial NADP+-ME contains a Lys in this position, while mitochondrial NAD+-ME contains a Gln. A single Glu-362-Lys substitution in the NAD-ME isoform shifts its cofactor preference from NAD+ to NADP+ (15). Analyzing the putative cofactor binding site of the S. coelicolor MEs by amino acid sequence alignment against eukaryotic MEs, we found that SCO5261 contains a Lys, whereas SCO2951 has an Ala residue at position 362 (Fig. 2B). This analysis suggests that SCO2951 and SCO5261 could be NAD+- and NADP+-dependent enzymes, respectively. In order to verify this hypothesis, the Sco2951 and Sco5261 genes were expressed in E. coli, and the corresponding proteins purified to homogeneity as His6-tagged fusion proteins. Malic enzyme activity was monitored spectrophotometrically following the formation of NADH or NADPH at 340 nm (5), and kinetic parameters were determined at pH 8.0 (Table 2). SCO2951 showed no enzyme activity in the presence of NADP+ as a substrate, while a specific activity of 36.97 U mg−1 was measured for SCO5261 (Table 2). When ME activity was assayed in the presence of NAD+, SCO2951 showed a specific activity of 2.35 U mg−1, while SCO5261 showed a lower specific activity of 0.0824 U mg−1. Even though SCO5261 utilizes both NAD+ and NADP+ as cofactors, analysis of cofactor specificity showed a clear preference for NADP [Vmax (NADP)/Vmax (NAD) = 448.66]. On the other hand, Km values for l-malate and the NADP+/NAD+ ratio are similar to those reported for malic enzymes from other bacteria (Table 2) (10). In addition, the Vmax d/Vmax r ratio (where d is the decarboxylation reaction of l-malate into pyruvate and r refers to the carboxylation of pyruvate into l-malate) of the NADP+-ME showed a marked preference for the direct reaction (Vmax d/Vmax r = 450-fold).
Fig 2.

Phylogenic analysis of malic enzymes. (A) Unrooted phylogenetic tree constituted by 37 ME members from various origins (see Table S1 in the supplemental material for details). Putative gene sequences encoding malic enzymes from Streptomyces spp. were retrieved using Sco2951 and Sco5261 as the query using BLAST (31). The analysis included other prokaryotic and eukaryotic malic enzymes for which cofactor preference is known. The evolutionary history was inferred by using the maximum likelihood method based on the Dayhoff matrix-based model (34). (B) Multiple amino acid sequence alignments of the cofactor binding region in the active sites of malic enzymes using ClustalW (21). Residue numbers correspond to the human mitochondrial NAD+-ME. Residues involved in the adenosine binding site of the cofactor are shown in a box.
Table 2.
Kinetic parameters of SCO2951 (NAD+-ME) and SCO5261 (NADP+-ME)a
| Protein | Reaction | Substrate | Vmax (U mg−1) | Km (mM) |
|---|---|---|---|---|
| SCO5261 | Direct | NADP | 36.97 ± 1.63 | 0.107 ± 0.015 |
| l-Malate | 6.4 ± 0.6 | |||
| Reverse | Pyruvate | 0.0824 ± 0.0114 | 18.1 ± 3.7 | |
| SCO2951 | Direct | NAD | 2.35 ± 0.21 | 4.3 ± 0.9 |
| l-Malate | 8.1 ± 1.4 |
Recombinant proteins were expressed and purified using affinity columns. NAD+- and NADP+-ME activities were determined spectrophotometrically at 30°C by monitoring NADH and NADPH production at 340 nm, respectively. Results are the means from three determinations ± standard errors.
Isolation of Sco2951 and Sco5261 mutant strains.
In order to study the effect of each ME mutation in the metabolism of S. coelicolor, Sco2951::Tn5062 (TR2951, Amr) and Sco5261::Tn5062 (TR5261; Amr) single mutant strains and a double sco2951::Tn5062 and sco5261::Tn5066 mutant strain (TRD; Amr and Hygr) were isolated as described previously (2). All mutants were grown under different conditions to analyze the effect of each mutation on growth, as well as on antibiotic and TAG production. All mutants grew similarly to the parent M145 strain in minimal medium SMM-Glc (Fig. 3). However, as it is shown in Fig. 4, the single TR2951 and the double TRD mutant strains showed a strong impairment in actinorhodin production compared with the wild-type M145, in both SMM-Glc (Fig. 4A) and SMM-Glc-Mal solid media (Fig. 4B). To quantify this effect, SMM-Glc liquid cultures of M145 and mutant strains were prepared, and antibiotic production was determined spectrophotometrically. As shown in Fig. 4C, actinorhodin production is reduced in the single TR2951 and in the double TRD mutant strains, with a stronger effect in the double mutant. On the contrary, a minor but still significant effect in antibiotic production was observed for the single TR5261 mutant (Fig. 4). Additionally, no difference in undecylprodigiosin levels was detected in any of the strains or media tested (Fig. 4D).
Fig 3.

Time growth curve of ME mutants and the M145 parent strain of S. coelicolor grown on minimal medium SMM-Glc. Results are the average of three independent experiments ± standard deviations (SD) with statistical significance (P < 0.05).
Fig 4.
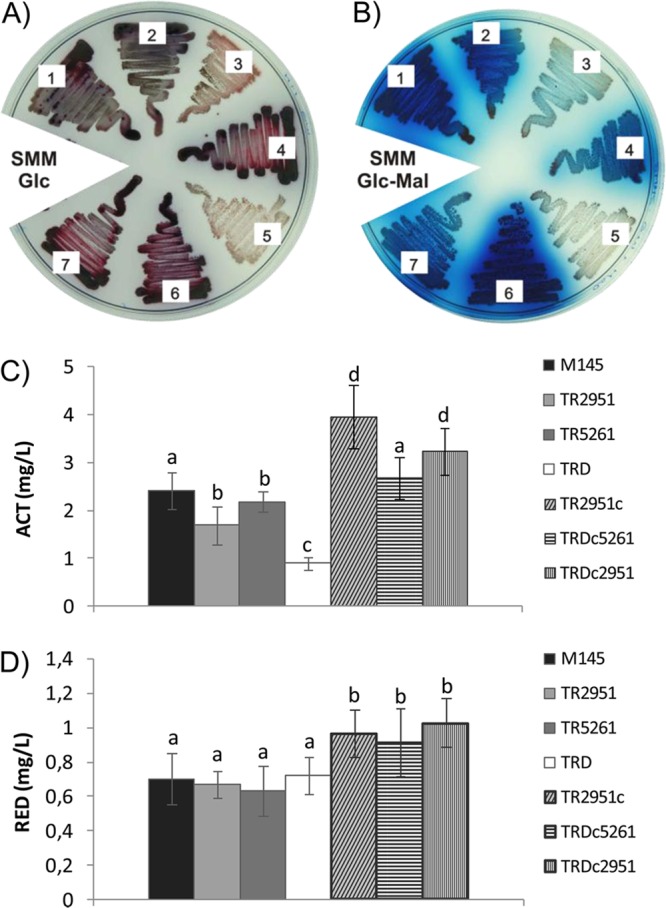
Antibiotic production in ME mutants and the M145 strain of S. coelicolor. Growth of mutant strains on SMM-agar plates supplemented with 1% glucose (A) or plus the addition of 0.5% l-malate (B); plates were incubated for 7 days at 30°C. Strains used are as follows: 1, S. coelicolor M145; 2, TR5261; 3, TR2951; 4, TR2951c; 5, TRD; 6, TRDc5261; and 7, TRDc2951. Determination of antibiotics of the aforementioned strains grown in liquid medium SMM-Gluc for 72 h to detect actinorhodin (C) and for 60 h to detect undecylprodigiosin (D). Results represent the average of three independent experiments ± SD. Different letters denote statistical differences applying an ANOVA test using Sigma Stat 3.1 (P < 0.05).
To validate that the phenotypes observed were due to ME gene inactivation, a wild-type copy of each gene under the strong and constitutive PermE* promoter was integrated into the ϕBT1 attachment site of each mutant strain using derivatives of pRT802 (11). As shown in Fig. 4C, the expression of a wild-type copy of the Sco2951 (NAD+-ME) gene in either the TR2951 or TRD mutant strains restored actinorhodin production to even higher levels than those found in the parental strain. It is interesting to note that overexpression of Sco5261 (NADP+-ME) in the TRD strain also restored actinorhodin production. This result suggests that restoration of the carbon flux is a requisite for activation of actinorhodin production independently of NADH or NADPH production.
In vivo malic enzyme activity assays.
In order to demonstrate that the two proteins under study were the only malic enzymes expressed in S. coelicolor, NAD- and NADP-dependent ME activities were assayed at different growth stages in M145 and in the single and double mutant strains. As shown in Fig. 5, cell extracts prepared from the TR2951 and TRD strains have undetectable levels of NADP+-dependent ME activity; however, the same protein extracts showed only a partial decrease in NAD reduction in the presence of malate. This enzyme activity, measured in the cell extracts of both strains, could be assigned to the malate dehydrogenase (MDH) activity, which is present in S. coelicolor and uses the same substrates as NAD+-ME (8). S. coelicolor MDH is highly NAD+ specific, being almost inactive with NADP. The reported Km value for NAD is 0.15 mM, while the Km for malate is 0.494 mM (9); both values are lower than those determined for the NAD+-ME in this study. Thus, it is not surprising that both TR2951 and TRD mutants can still reduce NAD to NADH in the presence of l-malate. Another possibility is that a different class or family of MEs could exist in Streptomyces, although we rule out this possibility based on the phenotypes observed for the mutant and complemented strains.
Fig 5.

Determination of NADP+-ME (A) and NAD+-ME (B) activity. ME activity was assayed in cell extracts of M145, TR2951, TR5261, TRD strains and in their corresponding complemented strains grown on SMM-Glc for 30 h. Values represent the average of experiments performed in triplicate ± SD. N.D., not detected. Different letters denote statistical differences applying an ANOVA test using Sigma Stat 3.1 (P < 0.05).
As shown in Fig. 5, introduction of a wild-type copy of each ME-encoding gene under the PermE* promoter into the mutant strains restored the corresponding ME activities to even higher levels than those found in the M145. This result could be expected since PermE* is a strong constitutive promoter.
Effect of malic enzyme activity on triacylglycerol accumulation.
The absence of ME activities could provoke a reduction on the levels of acetyl-CoA and/or NADPH and subsequently alter the levels of fatty acid biosynthesis and the ensuing accumulation of membrane lipids and TAG. In our experiments, however, we did not observe any effect on the growth of mutants compared with the M145 strain, suggesting that fatty acid biosynthesis and cell membrane integrity were not affected by the mutations introduced in the ME-encoding genes. Moreover, to analyze the potential impact of the absence of ME activity on TAG accumulation, liquid cultures of M145 and the mutant strains were prepared and their total lipid content was extracted with chloroform and methanol and analyzed as described previously (2). As shown in Fig. 6, only the double mutant strain showed a decrease in TAG accumulation at early stages of growth (20 to 30 h), but this effect declined at later stages. Complementation of this mutant with a wild-type copy of either Sco2951 (NAD+-ME) or Sco5261 (NADP+-ME) under the control of a constitutive strong promoter restored TAG accumulation (Fig. 6).
Fig 6.

Lipid analysis of ME mutants and M145 parent strain of S. coelicolor. Total lipids were extracted with chloroform and methanol, separated by TLC, and visualized by Cu2+-phosphoric staining. TAG content was determined by densitometry. Values represent TAG content relative to total lipids for the M145, ME mutant, and their complemented derivative strains at three time points (20, 24, and 30 h). Values represent the average of three independent experiments ± SD. Asterisks denote statistical differences applying Student's t test (P < 0.05).
Metabolite analysis of mutant strains.
In order to understand the effect of malic enzyme inactivation on S. coelicolor metabolism, we carried out metabolomic analysis of the wild-type M145 and the double mutant strain TRD using gas chromatography coupled to mass spectrometry (GC-MS) (22). In this manner, strains were grown in SMM-Glc liquid culture for 24 h, organic solvent extracts were prepared, and the metabolites were analyzed after a derivatization reaction by GC-MS (22). Quantification of changes measured for different metabolites was performed using ribitol as the internal standard. We identified 42 compounds by this methodology, and 19 of these had a statistically significant modification by inactivation of both MEs (P < 0.05). Interestingly, four intermediate metabolites of the TCA cycle were increased in the TRD mutant strain compared to in the M145 parent strain (succinic, malic, fumaric, and citric acids) (Fig. 7A). Among these metabolites, malic acid displayed the highest accumulation rate in the double mutant TRD compared to the M145 parent strain.
Fig 7.

Metabolic analysis of ME mutant and M145 parent strains of S. coelicolor. Relative content of TCA intermediates by GC-MS analysis (A) and acetyl-CoA quantification relative to total CoA thioesters content by high-performance liquid chromatography (HPLC) quantified by flowthrough scintillation counting (B). Values represent mean differences between the M145 control strain and the ME double mutant strain. Results represent the averages from three independent experiments ± SD. N.D., not detected. Asterisks denote statistical differences applying Student's t test (P < 0.05).
In addition, acetyl-CoA levels were determined as described previously (38). Consequently, acid stable metabolites were extracted from the same strains and separated using high-pressure liquid chromatography coupled to a radiochemical detector (38). As shown in Fig. 7B, no differences in the acetyl-CoA pool were found between M145 and TRD.
Transcript analysis of antibiotic and fatty acid biosynthesis gene expression.
To test if the decrease in actinorhodin production in the TRD double mutant was due to a downregulation of the transcriptional activator ActII-ORF4, total RNA was extracted from TRD and M145 cultures grown for 40 and 72 h in SMM-Gluc medium. Expression levels of actII-ORF4 mRNA were studied by qRT-PCR. The data obtained showed a significant decrease in the levels of actII-ORF4 mRNA at each time point in the TRD double mutant strain relative to the parent strain M145, indicating that the metabolic unbalance generated by the absence of ME activity has a negative effect on the transcriptional regulation of actinorhodin biosynthetic genes (Fig. 8).
Fig 8.

Transcriptional analysis of actII-ORF4 gene expression in the double ME mutant TRD and parent strain M145. The relative expression profile of actII-ORF4 was determined by qRT-PCR. The threshold cycle (CT) value of the target gene was normalized to the housekeeping gene hrdB. Values represent mean difference between M145 control and ME double mutant strains at two time points (40 and 72 h). Results are the averages from three independent experiments ± SD. Asterisks represent statistical differences applying Student's t test (P < 0.05).
On the other hand, to check if the decrease in TAG accumulation was due to a reduction in fatty acid biosynthesis, the expression levels of fabH (one of the fatty acid synthase condensing enzymes) mRNA were studied by qRT-PCR (1). The data obtained showed no significant changes in fabH mRNA levels at each time point in the TRD mutant relative to the parent strain M145 (data not shown).
DISCUSSION
Streptomyces spp. produce polyketide antibiotics and TAGs, the latter as reserve compounds. To synthesize these molecules, the bacterium uses the same precursors, NADPH and acetyl-CoA, a situation that represents an interesting metabolic challenge. Therefore, understanding the metabolic network in this genus is an important starting point for future biotechnological applications. Anaplerotic MEs have being demonstrated to have different roles in plants and mammals, interchanging C3-C4 intermediates between organelles and providing reducing equivalents to support fatty acid biosynthesis (7, 16). Thus, in this work, we have characterized two MEs from the polyketide producer S. coelicolor. The in vitro characterization of these two MEs demonstrated that Sco2951 encodes an NAD+-ME of 471 amino acids, while Sco5261 encodes an NADP+-ME of 409 amino acids. These two proteins have an overall amino acid sequence identity of 57%, with SCO2951 displaying an additional ACT domain at the amino terminal end of the protein. ACT domains are commonly involved in the specific binding of certain amino acids or other small ligands, leading to the regulation of enzyme activity. This fact is an indication that these enzymes have different roles and regulation in S. coelicolor metabolism (24). The two S. coelicolor MEs have a low amino acid sequence similarity with MEs from eukaryotes (about 25% identity compared with the human ME). However, our results indicate that the Streptomyces enzymes could contain determinants at the cofactor binding site similar to those demonstrated in the eukaryote enzymes. All known NADP+-dependent MEs from bacteria have the same Lys-362 residue at the cofactor binding site, similar to the human mitochondrial NADP+-ME (Fig. 2B) (14). On the contrary, NAD+-MEs have alternative amino acid residues (Ala, Glu, or His) at the 362 position, as it is found in mitochondrial NAD+-dependent MEs from eukaryotic sources.
MEs have not been shown to be essential enzymes for Streptomyces growth (Fig. 3). Phylogenetic analysis indicated that MEs are divided in two groups (Fig. 2A). Group I includes only prokaryotic enzymes, while group II is constituted by both eukaryotic and prokaryotic enzymes. This analysis also showed that both Sco2951 and Sco5261 are highly conserved in all of the Streptomyces sp. sequences currently known, and they are clearly divided in two discrete subclades within group I, suggesting that the function of these enzymes would be different (Fig. 2A). In addition, some actinobacteria have other MEs besides these two isoforms. For example, Steptomyces avermitilis, the producer of the potent anthelmintic and insecticidal avermectin, contains four MEs, two orthologues of SCO2951 and SCO5261, and the other two that cluster inside group II, each of which probably has specific roles in the metabolism of this microorganism (Fig. 2A).
Inactivation of Sco2951 and Sco5261 genes had no an impact on cell growth or morphogenesis compared with the parental M145 strain; however, Sco2951 and Sco2951 Sco5261 mutant strains showed a strong reduction in the production of the antibiotic actinorhodin (Fig. 4). Furthermore, the double mutant TRD also showed lower accumulation of TAGs at early time points (Fig. 6). The absence of at least NAD+-ME or even both MEs could provoke a decrease in the carbon flux from TCA intermediates to form acetyl-CoA and as a consequence reduced levels of secondary metabolites and fatty acid biosynthesis. Alternatively, changes in the pools of these intermediates could induce a metabolic imbalance and subsequent alterations in the regulation of the actinorhodin biosynthetic pathway. The absence of a phenotype for the NADP-ME mutant strain also suggests that this enzyme is not the main source of NADPH as a reducing equivalent, at least under the conditions tested.
Metabolic studies show that the double mutant strain accumulated different TCA cycle intermediates compared to the parent M145 strain. These results confirm the relevance of MEs, among all the anaplerotic enzymes, in the connection between the TCA cycle and other central metabolic pathways in Streptomyces. In this organism, there are two possible anaplerotic pathways using either PEP carboxykinase or ME in combination with PEP synthetase. Our results demonstrate that the last one seems to be the most important, especially during the exponential growth phase.
Previous studies for a citrate synthase (citA) mutant of S. coelicolor showed that medium acidification provoked defects in morphological differentiation and antibiotic biosynthesis. However, these defects were reverted by buffering the medium to maintain neutrality (37). It is worth mentioning that ME mutants did not show any defects in morphological differentiation and that impaired antibiotic production was still observed on SMM-Glc plates containing higher concentrations of TES buffer. Transcript analysis revealed that the impairment in actinorhodin production found in the double mutant strain is due to a lower expression of the transcriptional activator ActII-ORF4 and therefore to a downregulation of the actinorhodin gene cluster. One possible explanation for this observation is that accumulation of TCA intermediates may have a negative effect on the expression of the transcriptional activator of actinorhodin, although this hypothesis remains to be demonstrated. Among all pleiotropic regulatory proteins affecting secondary metabolism in S. coelicolor, only DasR has been demonstrated to sense an intracellular metabolite, N-acetylglucosamine, through which it regulates the expression of actII-ORF4 (28). Another transcriptional regulatory protein, AtrA, has been shown to bind to the promoter region of the actII-ORF4 gene, but no effectors have been identified for this regulator yet (25, 36).
Complementation of each mutant was achieved by overexpression of either NAD+- or NADP+-MEs; this result demonstrates that carbon flux reactivation of C4 intermediates from TCA into central metabolic pathways restores both actinorhodin production and TAG accumulation independently of NADH or NADPH production. TAG accumulation could be affected by reduced levels of acetyl-CoA and NADPH or by the downregulation of fatty acid biosynthesis (1). However, we were not able to detect any difference in transcript levels of the fatty acid biosynthetic gene fabH (data not shown) nor in acetyl-CoA levels in the double mutant strain compared to the parental M145 strain (1). In addition, complementation of TAG accumulation was achieved by overexpression of either NAD- or NADP-ME, independently of the production of NADH or NADPH. Thus, this result suggests that ME activity plays a crucial role in carbon flux homeostasis in Streptomyces by connecting TCA and other central carbon pathways rather than providing reduction power as NADPH for lipid biosynthesis.
In conclusion, this work demonstrates the relevance of studying key metabolic pathways by traditional biochemical and physiological methods and supports the importance of including the MEs from the polyketide producer Streptomyces spp. on the metabolic map of this biotechnologically important strain (Fig. 1). The mechanisms by which this bacterium senses the accumulation of tricarboxylic acid cycle intermediates and regulates the antibiotic and triacylglycerol production are under study.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by ANPCyT grants PICT 2006-01978 to E.R., PICT 2007-00711 to P.C., and PICT 2008-644 to H.G. E.R., P.C., and H.G. are members of the Research Career, and L.N. is a doctoral fellow of CONICET.
We thank Lorena Fernandez and Paul Dyson (Swansea University) for kindly providing the derivative cosmids carrying transposon insertions and Monica Hourcade (Universidad Nacional de Rosario) for technical assistance in the GC-MS analysis.
Footnotes
Published ahead of print 27 April 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Arabolaza A, D'Angelo M, Comba S, Gramajo H. 2010. FasR, a novel class of transcriptional regulator, governs the activation of fatty acid biosynthesis genes in Streptomyces coelicolor. Mol. Microbiol. 78:47–63 [DOI] [PubMed] [Google Scholar]
- 2. Arabolaza A, Rodriguez E, Altabe S, Alvarez H, Gramajo H. 2008. Multiple pathways for triacylglycerol biosynthesis in Streptomyces coelicolor. Appl. Environ. Microbiol. 74:2573–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bibb MJ. 2005. Regulation of secondary metabolism in streptomycetes. Curr. Opin. Microbiol. 8:208–215 [DOI] [PubMed] [Google Scholar]
- 4. Bystrykh LV, et al. 1996. Production of actinorhodin-related “blue pigments” by Streptomyces coelicolor A3(2). J. Bacteriol. 178:2238–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Casati P, Lara MV, Andreo CS. 2000. Induction of a C4-like mechanism of CO2 fixation in Egeria densa, a submersed aquatic species. Plant Physiol. 123:1611–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cronan JE, Thomas J. 2009. Bacterial fatty acid synthesis and its relationships with polyketide synthetic pathways. Methods Enzymol. 459:395–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drincovich MF, Casati P, Andreo CS. 2001. NADP-malic enzyme from plants: a ubiquitous enzyme involved in different metabolic pathways. FEBS Lett. 490:1–6 [DOI] [PubMed] [Google Scholar]
- 8. Ge YD, et al. 2010. Identification and biochemical characterization of a thermostable malate dehydrogenase from the mesophile Streptomyces coelicolor A3(2). Biosci. Biotechnol. Biochem. 74:2194–2201 [DOI] [PubMed] [Google Scholar]
- 9. Goodfellow M, Fiedler HP. 2010. A guide to successful bioprospecting: informed by actinobacterial systematics. Antonie Van Leeuwenhoek 98:119–142 [DOI] [PubMed] [Google Scholar]
- 10. Gourdon P, Baucher MF, Lindley ND, Guyonvarch A. 2000. Cloning of the malic enzyme gene from Corynebacterium glutamicum and role of the enzyme in lactate metabolism. Appl. Environ. Microbiol. 66:2981–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gregory MA, Till R, Smith MC. 2003. Integration site for Streptomyces phage phiBT1 and development of site-specific integrating vectors. J. Bacteriol. 185:5320–5323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580 [DOI] [PubMed] [Google Scholar]
- 13. Herron PR, Hughes G, Chandra G, Fielding S, Dyson PJ. 2004. Transposon Express, a software application to report the identity of insertions obtained by comprehensive transposon mutagenesis of sequenced genomes: analysis of the preference for in vitro Tn5 transposition into GC-rich DNA. Nucleic Acids Res. 32:e113 doi:10.1093/nar/gnh112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsieh JY, Chen MC, Hung HC. 2011. Determinants of nucleotide-binding selectivity of malic enzyme. PLoS One 6:e25312 doi:10.1371/journal.pone.0025312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hsieh JY, Liu GY, Chang GG, Hung HC. 2006. Determinants of the dual cofactor specificity and substrate cooperativity of the human mitochondrial NAD(P)+-dependent malic enzyme: functional roles of glutamine 362. J. Biol. Chem. 281:23237–23245 [DOI] [PubMed] [Google Scholar]
- 16. Hsu RY. 1982. Pigeon liver malic enzyme. Mol. Cell. Biochem. 43:3–26 [DOI] [PubMed] [Google Scholar]
- 17. Jechova V, Hostalek Z, Vanek Z. 1969. Regulation of biosynthesis of secondary metabolites. V. Decarboxylating malate dehydrogenase in Streptomyces aureofaciens. Folia Microbiol. (Praha) 14:128–134 [DOI] [PubMed] [Google Scholar]
- 18. Khosla C. 2009. Structures and mechanisms of polyketide synthases. J. Org. Chem. 74:6416–6420 [DOI] [PubMed] [Google Scholar]
- 19. Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics, 2000 ed. John Innes Foundation, Norwich, United Kingdom: ISBN 0-7084-0623-8 [Google Scholar]
- 20. Kopka J, et al. 2005. GMD@CSB.DB: the Golm Metabolome Database. Bioinformatics 21:1635–1638 [DOI] [PubMed] [Google Scholar]
- 21. Larkin MA, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 22. Lisec J, Schauer N, Kopka J, Willmitzer L, Fernie AR. 2006. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc. 1:387–396 [DOI] [PubMed] [Google Scholar]
- 23. Loeber G, Dworkin MB, Infante A, Ahorn H. 1994. Characterization of cytosolic malic enzyme in human tumor cells. FEBS Lett. 344:181–186 [DOI] [PubMed] [Google Scholar]
- 24. Marchler-Bauer A, et al. 2011. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 39:D225–D229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nothaft H, et al. 2010. The permease gene nagE2 is the key to N-acetylglucosamine sensing and utilization in Streptomyces coelicolor and is subject to multilevel control. Mol. Microbiol. 75:1133–1144 [DOI] [PubMed] [Google Scholar]
- 26. Oh MK, Rohlin L, Kao KC, Liao JC. 2002. Global expression profiling of acetate-grown Escherichia coli. J. Biol. Chem. 277:13175–13183 [DOI] [PubMed] [Google Scholar]
- 27. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45 doi:10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rigali S, et al. 2008. Feast or famine: the global regulator DasR links nutrient stress to antibiotic production by Streptomyces. EMBO Rep. 9:670–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rodriguez E, et al. 2003. Rapid engineering of polyketide overproduction by gene transfer to industrially optimized strains. J. Ind. Microbiol. Biotechnol. 30:480–488 [DOI] [PubMed] [Google Scholar]
- 30. Sauer U, Eikmanns BJ. 2005. The PEP-pyruvate-oxaloacetate node as the switch point for carbon flux distribution in bacteria. FEMS Microbiol. Rev. 29:765–794 [DOI] [PubMed] [Google Scholar]
- 31. Schaffer AA, et al. 2001. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 29:2994–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stols L, Donnelly MI. 1997. Production of succinic acid through overexpression of NAD+-dependent malic enzyme in an Escherichia coli mutant. Appl. Environ. Microbiol. 63:2695–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113–130 [DOI] [PubMed] [Google Scholar]
- 34. Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsao SW, Rudd BA, He XG, Chang CJ, Floss HG. 1985. Identification of a red pigment from Streptomyces coelicolor A3(2) as a mixture of prodigiosin derivatives. J. Antibiot. (Tokyo) 38:128–131 [DOI] [PubMed] [Google Scholar]
- 36. Uguru GC, et al. 2005. Transcriptional activation of the pathway-specific regulator of the actinorhodin biosynthetic genes in Streptomyces coelicolor. Mol. Microbiol. 58:131–150 [DOI] [PubMed] [Google Scholar]
- 37. Viollier PH, Minas W, Dale GE, Folcher M, Thompson CJ. 2001. Role of acid metabolism in Streptomyces coelicolor morphological differentiation and antibiotic biosynthesis. J. Bacteriol. 183:3184–3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wadler C, Cronan JE. 2007. Dephospho-CoA kinase provides a rapid and sensitive radiochemical assay for coenzyme A and its thioesters. Anal. Biochem. 368:17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.