

Abstract
Documenting the role of novel mutation versus homologous recombination in bacterial evolution, and especially in the invasion of new hosts, is central to understanding the long-term dynamics of pathogenic bacteria. We used multilocus sequence typing (MLST) to study this issue in Xylella fastidiosa subsp. pauca from Brazil, a bacterium causing citrus variegated chlorosis (CVC) and coffee leaf scorch (CLS). All 55 citrus isolates typed (plus one coffee isolate) defined three similar sequence types (STs) dominated by ST11 (85%), while the remaining 22 coffee isolates defined two STs, mainly ST16 (74%). This low level of variation masked unusually large allelic differences (>1% divergence with no intermediates) at five loci (leuA, petC, malF, cysG, and holC). We developed an introgression test to detect whether these large differences were due to introgression via homologous recombination from another X. fastidiosa subspecies. Using additional sequencing around these loci, we established that the seven randomly chosen MLST targets contained seven regions of introgression totaling 2,172 bp of 4,161 bp (52%), only 409 bp (10%) of which were detected by other recombination tests. This high level of introgression suggests the hypothesis that X. fastidiosa subsp. pauca became pathogenic on citrus and coffee (crops cultivated in Brazil for several hundred years) only recently after it gained genetic variation via intersubspecific recombination, facilitating a switch from native hosts. A candidate donor is the subspecies infecting plum in the region since 1935 (possibly X. fastidiosa subsp. multiplex). This hypothesis predicts that nonrecombinant native X. fastidiosa subsp. pauca (not yet isolated) does not cause disease in citrus or coffee.
INTRODUCTION
Xylella fastidiosa subsp. pauca is a bacterial plant pathogen causing citrus variegated chlorosis (CVC), a disease thus far only recorded in South America, primarily Brazil. It is considered to pose a serious potential threat to the citrus industry in the United States, and for this reason it is listed as a plant protection and quarantine select agent by the U.S. Department of Agriculture (USDA; see http://www.selectagents.gov/). It has caused significant economic losses within Brazilian agriculture since the first report of CVC in 1987 (5). The high economic impact of CVC promoted the sequencing of an X. fastidiosa subsp. pauca CVC strain (9a5c), the first plant pathogenic bacterium to have its genome completely sequenced and annotated (44). It also infects coffee, another economically important plant in Brazil: coffee leaf scorch (CLS) was first documented in Brazil in 1995 (3). X. fastidiosa subsp. pauca is presumed to be native to South America, and both citrus and coffee have been grown in Brazil and other South American countries since 1530/1540 and 1727, respectively (45, 35). This probable historical sympatry of host and pathogen raises the important question of why the diseases of CVC and CLS did not appear much earlier. We examine here the patterns of genetic variation seen within X. fastidiosa subsp. pauca to gain insight into this apparent paradox.
X. fastidiosa is a Gram-negative gammaproteobacterium limited to the xylem system of plants (46) and transmitted by xylem-feeding insects (38). X. fastidiosa has been divided into four subspecies (42, 43), and of these only X. fastidiosa subsp. pauca is absent from the United States. However, all four are restricted to the Americas.
This species is known to cause disease in a wide range of economically important plants in the United States (16); however, the three U.S. subspecies typically infect a limited range of different hosts, and none infect citrus. The X. fastidiosa subsp. fastidiosa causes Pierce's disease of grapevines and almond leaf scorch, the X. fastidiosa subsp. sandyi causes oleander leaf scorch, and the X. fastidiosa subsp. multiplex is associated with scorch disease in a range of trees, including almond, peach, and oak trees.
X. fastidiosa subsp. pauca isolated from citrus and coffee is generally reciprocally host specific (1, 20), although infection of coffee plants by citrus isolates has sometimes been observed (19, 37). These two forms of X. fastidiosa subsp. pauca are also found to be genetically distinct (1, 6, 39, 47; see also reference 28). Almeida et al. (1) used multilocus sequence typing (MLST) (21) to study the sequence diversity of X. fastidiosa subsp. pauca based on the scheme introduced by Scally et al. (41) for the North American X. fastidiosa. In its original form, this MLST system was not optimized for use on X. fastidiosa subsp. pauca, and only four of the genes could be amplified, a problem that has now been corrected (49). Based on these four genes (leuA, cysG, malF, and petC) plus one other gene (rfbD) included in the Scally et al. (41) study, Almeida et al. (1) showed that the coffee (CLS-causing) and citrus (CVC-causing) strains are genetically distinct and that the phylogenetic trees of different genes using the same the X. fastidiosa subsp. pauca isolates were heterogeneous (i.e., they were not consistent with clonal evolution), providing strong evidence of recombination within the subspecies. This last result was consistent with a previous analysis of North American X. fastidiosa, which had indicated that recombination contributed about three times more to genetic diversity (measured at the nucleotide level) than point mutation (41).
The high level of recombination-related diversity found in the North American X. fastidiosa was in part due to intersubspecific homologous recombination, a phenomenon analyzed in X. fastidiosa subsp. fastidiosa (31, 49), where X. fastidiosa subsp. multiplex was the donor. Almeida et al. (1) suggested that the same process may have occurred in X. fastidiosa subsp. pauca, with X. fastidiosa subsp. fastidiosa (or perhaps some other North American subspecies) as the possible donor. These researchers found patterns consistent with intersubspecific recombination at two loci (leuA and rfbD); however, the data were not analyzed in any detail or subjected to statistical testing.
Intersubspecific recombination obviously requires the sympatry of two subspecies. X. fastidiosa subsp. fastidiosa and X. fastidiosa subsp. multiplex have been sympatric in the United States for more than 100 years (32), providing ample opportunity for genetic exchange. There appears to have been a similar opportunity in South America between X. fastidiosa subsp. pauca and an introduced North American form. Xylella causing plum leaf scald was first detected in 1935 in Argentina and then in Paraguay and Brazil (13, 18). Four genetic analyses of one plum isolate showed that it is more closely related to the U.S. subspecies than to X. fastidiosa subsp. pauca (6, 17, 27, 30).
The present work was designed to analyze in detail the hypothesis that X. fastidiosa subsp. pauca has been involved in intersubspecific recombination and, if so, to determine whether it was likely to be a major source of genetic variability. Our reason for investigating this question was to gain further insight into the paradox mentioned above: that although citrus and coffee have been grown in South America for about 250 years or more, the diseases of CVC and CLS only appeared around 25 years ago. A host shift following intersubspecific recombination might explain the time delay.
Our approach was first to type the available X. fastidiosa subsp. pauca isolates using the seven-locus MLST scheme already used to type X. fastidiosa (49), thus enabling a direct comparison among the subspecies using standardized data. Second, the MLST data were used to determine whether the hypothesis of intersubspecific homologous recombination could be rigorously verified using a new test specifically designed to detect genetic introgression and, if so, whether introgressed sequence was a significant source of genetic diversity. Third, we sought to determine whether our introgression test was substantially more effective at detecting regions of introgression arising from homologous recombination than some commonly used recombination tests.
MATERIALS AND METHODS
MLST analysis.
The MLST analysis was based on 55 isolates of X. fastidiosa subsp. pauca from citrus and 23 from coffee, including the 26 citrus and 20 coffee samples previously analyzed by Almeida et al. (1). These isolates were sampled from symptomatic plants in different regions of Brazil, mostly from São Paulo state (79%) (see Table S1 in the supplemental material). The seven MLST genes (plus one cell surface protein coding gene, pilU) were sequenced using previously described procedures (49). According to the MLST protocol, each allele of a particular MLST gene region was given a different number, building on the preexisting database of known X. fastidiosa variation (maintained at www.pubmlst.org/xfastidiosa). Thus, each isolate was characterized by its allelic profile, consisting of the seven numbers defining the alleles at each of the seven loci. Each unique allelic profile was assigned a sequence type (ST) number. The STs were grouped into clonal complexes using the grouping criterion that within each complex the STs must share five or more alleles with at least one other member of the clonal complex (41, 49).
Introgression test.
To detect regions of intersubspecific recombination, we developed an introgression test. The test was designed to detect short regions of DNA that have introgressed into a native population (in this case, X. fastidiosa subsp. pauca) from some donor group via homologous recombination by comparing native sequence variation to reference sequence from the probable donor. It is not essential that the reference group is the actual donor; the test can still be effective provided the reference is an outgroup to both the native and the donor taxa. In the present case, the reference group consisted of the North American subspecies.
The test is based on the null expectation that within any given short region of the genome there will be a constant ratio of the number of fixed differences (F) between the native and reference populations compared to the number of polymorphic sites (P) in the native population that share at least one variant base with the reference population. In the absence of homologous recombination, bases are shared between the native and reference populations due to common ancestry or, more rarely, due to homoplasy, and the ratio (F:P) is dependent upon mutation, selection, and genetic drift. Neighboring regions a few hundred bases long are expected to be affected equally by all three processes. The introgression test detects local heterogeneity in the F:P ratio by comparing the ratio of adjacent regions. Heterogeneity arises when some genomes of the native taxon carry a short stretch of introgressed donor sequence (and some carry the ancestral sequence) next to a region with no history of introgression. The region with introgression will have decreased F sites (fixed differences) and increased P sites relative to the region with only ancestral native sequence. This pattern will continue to apply even if homologous recombination within the native taxon has mixed the ancestral and introgressed sequence.
Potential “native” regions (i.e., no introgression) and “introgressed” regions (i.e., those polymorphic for introgressed and native sequence) are identified from the data. The criteria used to define the breakpoints between regions were as follows: (i) a reversal of site type (F to P or P to F) of three or more consecutive sites; (ii) however, if this point differed depending on direction (evaluating 5′ to 3′ or 3′ to 5′), then to be conservative the partition maximizing the length of the nonrecombinant region was chosen; (iii) an informative P site was weighted as two sites in the application of criterion i; and (iv) if there was a potential breakpoint to a nonrecombinant region very close to the end of the sequence (typically involving two or three F sites), then (to be conservative) the ambiguous region was eliminated from the analysis, i.e., not included as part of the adjoining recombinant region.
Consider the case where the sequence data begins with a high F (native) region and ends with a lower F (possibly introgressed) region. (The argument is reversed if the first segment is high P and the second segment is high F.) We choose a dividing line as described above, with the first region containing L1 (= F1 + P1) sites of interest and the second containing L2 (= F2 + P2) sites, giving a total length of L12 (= L1 + L2) sites. We wanted to know the probability of randomly getting a result at least as extreme as that observed in the separation of F and P sites among these two regions, sampling from F12 (= F1 + F2) fixed and P12 (= P1 + P2) polymorphic sites. This is defined by the following equation:
| (1) |
where C equals the smaller of P1 and F2. The numerator is the number of combinations that are at least as extreme as the observed sequence (i.e., having at least F1 fixed sites in the 5′ region of L1 sites and at least P2 polymorphic sites in the 3′ region), i.e., the sum of the number of combinations given a partition as extreme (i = 0) or more extreme (i > 0) than the observed one. The total number of different combinations is L12!/(F12!P12!), but only half are relevant since half of these will have a higher F:P ratio at the 5′ end, and half will have a higher F:P ratio at the 3′ end. This defines the denominator.
The probability (equation 1) assumes that the two regions encompass the whole sequence; however, for any region internal to the DNA sequence one F (or P) must be ignored. This corrects the built-in bias due to the nonrandom choice of the beginning or ending base. For example, a 5′ internal high F region will be chosen to start with an F site, and a 3′ internal high P region will be chosen to end with a P site. This nonrandom base must be ignored when using equation 1. Consider the following specific case: if an internal high F segment (region 1, with 7 F and 2 P sites) is compared to a 3′ high P segment (region 2 with 3 F and 11 P sites) that runs all of the way to the end of the sequence, then the first F site is ignored in defining F1 (= 7 − 1), whereas, since the end of region 2 is objective (it corresponds to the end of the available sequence), all of its sites are counted. Thus, F1 = 6, P1 = 2, F2 = 3, P2 = 11, and C = 2. The probability, p, of a partition as extreme or more extreme is thus [10,192 + 728 + 14]/248,710 = 0.044, i.e., the partition is significant at the 5% level. A program calculating the probability is available on request.
The performance of the introgression test was compared to that of some published tests designed to detect recombination. We used the RDP3.44 package to implement RDP (22), GeneConv (34), MaxChi (23), Chimaera (36), and 3Seq (4).
Identifying regions of intersubspecific recombination.
X. fastidiosa subsp. pauca alleles from five of the seven MLST genes (49) and those from the rfbD sequence analyzed by Almeida et al. (1) showed unusual levels of divergence, so the sequence at these sites was extended at the 5′ and/or 3′ end of the MLST sequence using a representative isolate of each of the X. fastidiosa subsp. pauca STs: 1,465 bp 5′ of leuA (giving a total sequence length of 2173 bp), 695 bp 5′ and 700 bp 3′ of petC (total, 1,928 bp), 524 bp 5′ and 716 bp 3′ of cysG (total, 1,840 bp), 612 bp 5′ of malF (total, 1,342 bp), 544 bp 5′ of holC (total, 923 bp), and 699 bp 5′ and 559 bp 3′ of rfbD (total, 1,705 bp). The primer information is given in Table S2 in the supplemental material.
Our introgression test was applied across these regions by comparing the allelic variation in X. fastidiosa subsp. pauca to the intersubspecific variation represented by the corresponding sequence from the three other subspecies, using M12 (ALS0299, X. fastidiosa subsp. multiplex ST7), Temecula1 (PD0001, X. fastidiosa subsp. fastidiosa ST1), and Ann-1 (OLS0002, X. fastidiosa subsp. sandyi ST5).
Analysis of genetic distance and divergence time estimation.
Given evidence of recombination, the genetic differences among the STs are best documented by genetic distance rather than using a phylogenetic model (which assumes clonality). We compared the concatenation of the seven MLST sequences (4,161 bp) of all of the X. fastidiosa subsp. pauca STs and compared these STs to the other X. fastidiosa subspecies, represented by ST5 (the only X. fastidiosa subsp. sandyi ST), ST7 (representing X. fastidiosa subsp. multiplex), plus ST1 and ST18 representing X. fastidiosa subsp. fastidiosa from the United States and Costa Rica, respectively (29, 32). The distance tree with bootstrap values (from 500 replicates) was created using the programs Seqboot, Dnadist, Fitch, and Consense from Phylip 3.69 (11, 12).
To estimate a diversification date for X. fastidiosa subsp. pauca, we used sequence data from the seven MLST loci, including the supplementary sequencing that was obtained for the recombination analysis, plus the sequences from rfbD and pilU. However, since intersubspecific recombination would bias downward the estimate of X. fastidiosa subsp. pauca divergence from the other subspecies, we excluded all regions identified as recombinant by our Introgression Test. Noncoding intergenic regions were also excluded. The remaining sequence was concatenated in frame using data from the following strains: one isolate representing each of the X. fastidiosa subsp. pauca STs, plus reference sequences for the other subspecies, again using M12, Temecula1, and Ann-1.
These data were used to define a phylogeny based on changes at synonymous sites using PAML 4.4 (48), allowing the dN/dS ratio to vary among branches (model 1 in the program). Assuming the neutrality of synonymous substitutions allows the branch lengths to be used to estimate a time scale (43). Specifically, this is calculated using the following equation:
| (2) |
where T is the time in years, K is the estimated number of synonymous substitutions per synonymous site, μ is the per division mutation rate (using 5.4 × 10−10 changes per site per generation), and G is the division rate (using 1,000 generations per year). Measurement of the growth rate of X. fastidiosa under ideal culture conditions (10) showed a maximum rate (at 28°C) of G = 1,700, while G = 1,000 corresponds to growth at 22°C. We consider that the number of divisions in the field, under conditions of fluctuating temperature and low levels of nutrition, will be substantially lower than this value. Thus, using a value of G = 1,000 is expected to lead to estimated divergence times considerably shorter than the actual times.
To incorporate a measure of random variation and potential variation in the rate of evolution across the genome, we applied a jackknife procedure (see reference 43). Specifically, we divided the concatenated sequence into 10 regions, each containing 10% of the synonymous SNPs. Eliminating each of these 10 regions in turn, we created 10 sets of results from which we used the jackknife pseudovalues to estimate the mean and variance of each divergence time.
Nucleotide sequence accession numbers.
The MLST data are available at the MLST website (http://pubmlst.org), and the previously published gene sequences are available at GenBank (32, 49). Newly determined from this study are holC allele 13 (JQ290485), nuoL allele 8 (JQ290486), cysG allele 9 (JQ290487), and the longer sequences from CVC0145, CVC0251, COF0239, and COF0238 (JQ290488 to JQ290511). The other longer sequences were from published genomes.
RESULTS
MLST-based genetic relationships.
MLST of 78 isolates of X. fastidiosa subsp. pauca revealed only five STs that were divided into three clonal complexes (CCs) (Table 1). MLST of the 55 isolates from citrus defined three STs that grouped as a single clonal complex (CC1), and 86% had the same sequence type (ST11). The second most abundant citrus ST (9%) was ST13, which included the sequenced strain 9a5c (CVC0018) (44). ST12 was the final citrus type (5%). This complex also included a single coffee isolate (COF0237) of the common ST11. The other coffee isolates defined two clonal complexes (CC2 and CC3), CC2 consisting of ST16 and CC3 consisting of ST14. These two CCs represented 74 and 22% of the 23 coffee isolates (Table 1). The level of site polymorphism seen in citrus isolates was 0.31%, which is half of the variation (0.77%) seen among the coffee isolates (after excluding the one “citrus type” isolate, COF0237, which would further inflate the variation of the coffee isolates).
Table 1.
MLST of 55 CVC and 23 CLS isolates of X. fastidiosa subsp. pauca showing all STs founda
| ST | Allele type at each MLST locus |
Plant host | No. of isolates | Almeida et al.b |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| leuA | petC | malF | cysG | holC | nuoL | gltT | Type | No. of isolates | |||
| Clonal complex 1 | |||||||||||
| ST11 | 7 | 7 | 7 | 9 | 10 | 8 | 8 | Citrus | 47 | 1 | 22 |
| Coffee | 1 | 0 | |||||||||
| ST12 | 7 | 7 | 7 | 9 | 13 | 8 | 8 | Citrus | 3 | 0 | |
| ST13 | 7 | 6 | 7 | 9 | 10 | 7 | 8 | Citrus | 5 | 2 | 4 |
| Clonal complex 2 | |||||||||||
| ST16 | 7 | 6 | 8 | 10 | 11 | 8 | 8 | Coffee | 17 | 3 | 2* |
| 4 | 13 | ||||||||||
| 5 | 1* | ||||||||||
| Clonal complex 3 | |||||||||||
| ST14 | 8 | 8 | 8 | 11 | 12 | 9 | 9 | Coffee | 5 | 6 | 1* |
| 7 | 3 | ||||||||||
Each ST represents a unique allelic profile defined across the seven MLST loci, and each member of a clonal complex shares at least five alleles with another member. Data from Almeida et al. (1) are included in the last two columns of the table for comparison. The numbering of the alleles within each locus is arbitrary. It is not determined by genetic similarity.
All isolates used by Almeida et al. (1) were included in the MLST analysis. *, Subdivision of ST due to alleles at rfbD, a gene not included in the set of seven MLST genes.
The genetic distances among the concatenated sequences of the five STs of X. fastidiosa subsp. pauca and their similarity to representative STs of the other three subspecies are shown in Fig. 1. A maximum-likelihood tree with the same subspecific topology but including more STs from X. fastidiosa subsp. multiplex and X. fastidiosa subsp. fastidiosa was given in Nunney et al. (32). This topology is consistent with prior analyses (see, for example, references 1, 15, 43, and 49).
Fig 1.

Genetic distance tree relating the five X. fastidiosa subsp. pauca MLST sequence types (STs) identified from 78 Brazilian isolates to each other and to the other X. fastidiosa subspecies. The tree is based on a concatenation of the seven MLST sequences. The other subspecies are defined by a single representative sequence type, except subsp. fastidiosa, which is represented by one U.S. ST and one ST from Costa Rica. The numbers at each node indicate the bootstrap percentages.
Intersubspecific recombination.
Although the number of STs was found to be very low, the pattern of variability among the alleles making up these STs is complex. Specifically, it is bimodal, with some allele pairs differing at one to four sites, while others showed unexpectedly large single-nucleotide polymorphism (SNP) differences, with no intermediate alleles bridging the difference. Using the allele numbers given in Table 1, the allelic differences were as follows: three allelic pairs differed by one SNP (petC, 6 versus 8; cysG, 9 versus 10; and gltT, 8 versus 9), while pairwise comparisons of two trios of alleles differed by 1, 2, and 3 bp (nuoL, 7, 8, and 9) and 1, 3, and 4 bp (holC, 10, 11, and 13); however, collapsing these three pairs and two trios leaves five comparisons that show minimum differences of 8 to 14 sites: leuA, 7 versus 8 = 8 SNPs (1.1% divergence); cysG, 9 versus 11 = 9 SNPs (1.5% divergence); petC, 6 versus 7 = 10 SNPs (1.9% divergence); malF, 7 versus 8 = 14 SNPs (1.9% divergence); and holC, 10 versus 12 = 10 SNPs (2.6% divergence). Such large allelic differences, reaching the level of subspecific divergence (43), can arise (i) via point mutation over a very long period of clonal diversification in the absence of recombination or (ii) via the homologous recombination of one allele with some genetically distinct taxon. The first possibility is untenable given the strong evidence of homologous recombination in this species (49) and, specifically, in subspecies pauca (1).
Given the second possibility, the introgression of genetic variation from some distinct taxon, these divergent pairs were investigated further for evidence of recombination with the other X. fastidiosa subspecies. To increase the power of our analysis of these MLST data, we sequenced further upstream, downstream, or both depending on the pattern observed. These extended sequences were investigated using our new introgression test (see Materials and Methods). Regions of possible introgression via intersubspecific recombination were identified as segments of sequence with very few “fixed” (F) sites (fixed differences between X. fastidiosa subsp. pauca and all North American alleles) but with a concentration of “polymorphic” (P) sites (X. fastidiosa subsp. pauca sites sharing at least one variant with a North American allele). By itself, an excess of P sites is not strong evidence of recombination; however, a sharp reversal of this pattern in the neighboring region of sequence does provide strong evidence of a recombinational breakpoint.
We first tested leuA, one of the two genes that Almeida et al. (1) suggested might be involved in intersubspecific recombination based on the observation that some fragments of the alleles were not consistent with the expected phylogeny. We sequenced 2,173 bp (including the 1,256 bp analyzed in reference 1) and despite the increased sequence length we found no evidence of such recombination using five different preexisting tests for recombination (see Fig. 2). However, application of our introgression test revealed a different picture. We found strong evidence (P < 0.001) of a breakpoint between a nonrecombinant 5′ portion and a recombinant region 3′ portion (Fig. 2), with nine fixed differences and no shared polymorphism 5′ of the breakpoint (9 F sites to 0 P sites), compared to 6 F sites to 22 P sites 3′ of the breakpoint. The last nonrecombinant F site was 439 bp into the sequence and the first recombinant P site was at position 451. Between these limits, the precise location of the recombinational break is unknowable; however, we used the midpoint between the limits to provide a rough estimate. In this case, the midpoint defined a break at position 445 and hence a region of recombination >1,728, since the 3′ end extended beyond the limits of our sequencing. This region of recombination included all 708 bp of the leuA sequence used for MLST. The details of the sequence differences (Table 2) show that within the recombinant region ST14 is highly differentiated from the other X. fastidiosa subsp. pauca STs (at 12 sites), but there is no clear pattern in their relationship to the sequence of the other subspecies.
Fig 2.

Regions of intersubspecific recombination were identified in five out of seven MLST gene regions plus rfbD using the introgression test. Recombinant introgression was identified by a significant increase in the ratio of polymorphic (Poly) to fixed (Fixed) sites (as defined in the text). The Fixed/Poly pairs progress from left to right along the sequence from 5′ to 3′. The data from recombinant portions are in boldface, together with the number of informative sites (in parentheses) and an estimate of length of the event (Rec. length). The “break position” (measured from the start position) is the midpoint between sites defining where the regions start and end. Our detection of intersubspecific recombination was compared to the patterns revealed using five other recombination tests (RDP, GeneConv [GC], MaxChi [MC], Chimaera [CH], and 3Seq [3S]; see the text). Each introgression detected is defined by the subspecific donor, the region detected, and the significance. The sequence data available were extended beyond that available from MLST using one representative of each ST (see Table 2).
Table 2.
X. fastidiosa subsp. pauca sequence variability around the leuA gene showing a region of apparent intersubspecific recombination beginning around position 445 (indicated by “-” in the sequence), as detected using the introgression testa
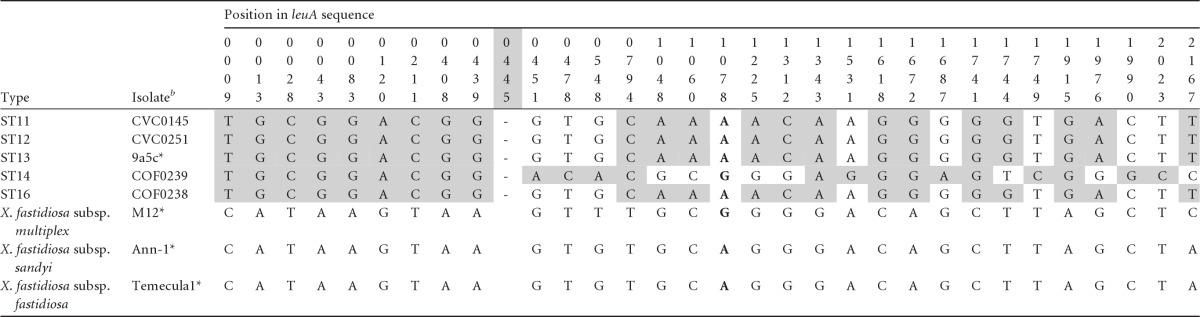
In comparison to sequences from the other three subspecies (using the isolates shown). Sites in white indicate bases found in those three subspecies, with informative sites indicated in boldface (in this case, a single site at position 1078).
*, Sequenced genomes with aliases CVC0018, ALS0299, OLS0002, and PD0001, respectively.
The second sequence that Almeida et al. (1) suggested showed some evidence of intersubspecific recombination was in a 437-bp region of rfpD, a gene that Scally et al. (41) had considered in developing MLST for X. fastidiosa but was not included in the final scheme. The five standard recombination tests applied to the original rfbD sequence failed to show recombination; however, when applied to our extended data, all of the tests detected a 3′ region of introgression of about 777 bp (Fig. 2). This region is shown in Table 3, 3′ of the switch point (also shown) that is discussed below. Two of them (GeneConv and 3Seq) identified another short region of recombination (124 bp) just 5′ of the switch point. All of these tests identified X. fastidiosa subsp. fastidiosa as the donor.
Table 3.
X. fastidiosa subsp. pauca sequence variability around the rfbD gene in a region of apparent intersubspecific recombination identified by the introgression testa

The sequence shown begins after a recombination breakpoint around position 262 and ends at position 1351 (an additional 3′ 354 bp of recombinant sequence are not shown). There is a “switch point” at around positions 676 to 680 (shaded numbers) so that ST13 and ST14 are very similar to the other three subspecies before this point but very different from them afterward, whereas ST16 shows exactly the opposite pattern. Five other recombination tests detected as recombinant the region in ST16 that is 3′ of the switch point (with slightly different start points, 772, as shown by the boldface letters, or 944). Two of these tests also identified a second region 5′ of the switch point (boldface italic letters). Three informative sites in this range were omitted. For further table details, see footnotes to Table 2.
Our introgression test indicated a rather different pattern consisting of a single region of introgression of >1,443 bp containing four informative P sites extending over all but the first 262 bp of the region (P < 0.001; Fig. 2). This sequence includes a short region of two fixed sites 4 bp apart (positions 676 and 680) that marks a switch in the pattern of recombination across the X. fastidiosa subsp. pauca STs (Table 3). In the 414 bp before this point there are 18 polymorphic sites. Of the polymorphic sites, 94% in ST13 and 78% in ST14 are identical to the North American subspecies, whereas for ST16 the figure is just 22%. This suggests that in this region ST16 carries a largely unrecombined X. fastidiosa subsp. pauca sequence. However, after the 676 to 680 boundary, there were 46 noninformative polymorphic sites and, of these, ST16 shared 83% with the North American subspecies, a highly significant increase (χ2 = 20.9, P < 0.001), whereas the other STs showed the opposite effect, a highly significant decrease (e.g., ST14: 17%, χ2 = 17.3, P < 0.001). This switch may reflect two separate recombination events or after introgression recombination within X. fastidiosa subsp. pauca.
Using the introgression test, we also detected a clear pattern of introgression in malF, the second MLST region with highly divergent alleles. We added 612 bp of 5′ sequence to the portion of the gene used for MLST, for a total sequence length of 1,341 bp. Our analysis revealed a recombinant region of 633 bp (both breakpoints P < 0.001; Fig. 2), which included 409 bp of the 5′ end of the MLST target sequence. The pattern of sequence divergence within the recombinant region (Table 4) shows that the coffee STs (ST14 and ST16) closely resemble the other three subspecies, uniquely sharing 21 SNPs with them, whereas the citrus STs (ST11 to ST13) are strongly differentiated from them, uniquely sharing only one SNP (at position 444). Four of the five other recombination tests detected intersubspecific introgression into both ST 14 and ST16, identifying the donor either as X. fastidiosa subsp. sandyi or as X. fastidiosa subsp. multiplex (Fig. 2). In the case of MaxChi, the recombinant region of ST16 identified was the same as that found with the introgression test (Table 4).
Table 4.
X. fastidiosa subsp. pauca sequence variability around the malF gene showing a region of apparent intersubspecific recombination beginning around position 387 and ending around position 1020 identified using the introgression testa
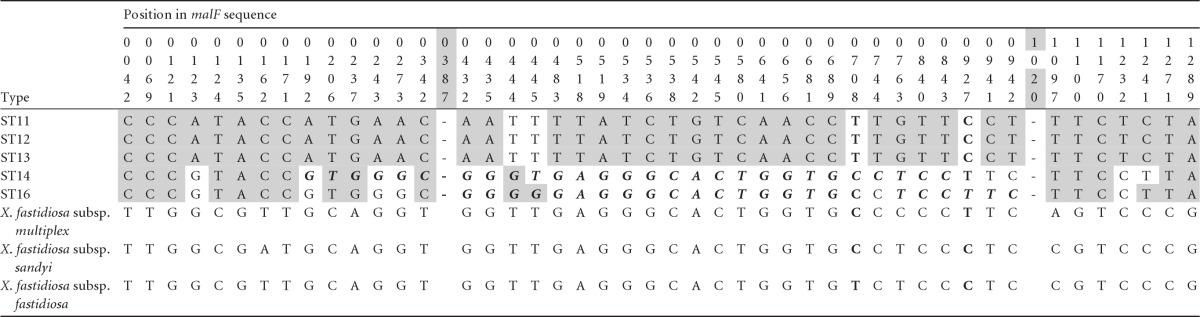
All five of the other recombination tests identified a recombination region in both ST14 and ST16 roughly corresponding to the same region. Precise start and endpoints differed, and as an example the results for the MaxChi test are indicated in italic and boldface font. For further table details, see footnotes to Table 2.
The third MLST locus with divergent alleles was petC. The petC MLST sequence data was increased to encompass the whole gene plus 150 bp downstream. This 1,928-bp sequence showed a region of recombination of about 698 bp (two breakpoints with P ≤ 0.003; Fig. 2) encompassing all of the gene except for roughly the first 75 bp, including all of the region used in MLST (533 bp). As in the case of malF, within the recombinant region some STs were very similar to their subspecific relatives; however, in this case they were not the coffee-type STs, but ST11 and ST12, both from citrus. These STs uniquely shared eight SNPs with the other subspecies, whereas ST13, ST14, and ST16 uniquely shared only two SNPs (Table 5). In addition, there were two informative sites involving all five STs. Although the petC recombinant region is easily detectable by eye (see Table 5), none of the five standard recombination tests detected it.
Table 5.
X. fastidiosa subsp. pauca sequence variability around the petC gene showing a region of apparent intersubspecific recombination beginning around position 568 and ending around position 1266 detected by the introgression testa

No recombination was detected by any of five other recombination tests applied (see the text). For further table details, see footnotes to Table 2.
The fourth MLST region with highly divergent alleles was cysG. We extended the sequencing both upstream (524 bp) and downstream (717 bp) of the MLST region to encompass all but 75 bp of the gene plus just over 500 bp downstream. None of the five standard recombination tests detected any introgression within the resulting sequence of 1,840 bp. In contrast, applying the introgression test indicated that the sequence contained two regions (201 and 649 bp) consistent with intersubspecific recombination (Fig. 2). The breakpoints were highly significant (P ≤ 0.002). We excluded 234 bp of sequence at the 3′ end, where there was a possible breakpoint defined by a region containing 3 F sites that was too short to provide a reliable test (although based on the limited data, the significance of the potential break was P = 0.052). This decision did not affect estimates of the recombinant fraction within the centrally located MLST sequence, which included 180 and 167 bp of the two recombinant regions at its 3′ and 5′ ends, respectively. In this sequence, it was X. fastidiosa subsp. pauca ST14 that showed the most similarity to the other subspecies (13 unique SNPs shared versus 5 for the other STs; data not shown).
The final MLST gene was holC. An additional 526 bp were sequenced 5′ of the MLST region; however, even with the extra data, none of the five recombination tests detected sequence heterogeneity. In contrast, the introgression test revealed evidence of two regions of intersubspecific recombination, one at each end of the sequence (>543 bp at the 5′ end and 158 bp at the 3′ end), with only a short region of about 18 bp between them where the variation was consistent with the South America versus a Central and North America split (breakpoints, P < 0.001 and 0.014, respectively; Fig. 2). The three most 3′ sites (2 F sites and 1 P site) were excluded from the analysis since they were suggestive of a nonrecombinant region too short to test.
The recombination history of this region is complex, since in the allelic phylogeny both ST16 and the X. fastidiosa subsp. fastidiosa group in the “wrong” place (Fig. 3). The details of these events will be considered elsewhere. In brief, the 5′ recombinant region (543 bp including 17 bp of the MLST sequence), included 93 polymorphic sites, of which 56 were informative. This number of informative sites was much greater than that seen in the other comparisons (60% versus 0 to 15% in other polymorphic regions: see Fig. 2), which is consistent with bidirectional introgression involving the transfer of X. fastidiosa subsp. pauca sequence to ST01 (X. fastidiosa subsp. fastidiosa), as well as a reverse transfer to X. fastidiosa subsp. pauca from the other subspecies. In the 358-bp 3′ region (all within the MLST site), the data suggest the one-way introgression from the other subspecies into X. fastidiosa subsp. pauca seen in the other examples, in this case into ST16.
Fig 3.

Genetic distance tree based on sequence data from the expanded holC MLST region showing representatives of the five X. fastidiosa subsp. pauca MLST sequence types (STs) and the other X. fastidiosa subspecies. The tree is consistent with two intersubspecific recombination events, one causing the similarity of ST16 to X. fastidiosa subsp. multiplex and X. fastidiosa subsp. sandyi and another causing the similarity of ST01 (X. fastidiosa subsp. fastidiosa) to X. fastidiosa subsp. pauca. The numbers at each node indicate the bootstrap percentages. For the isolates used, see Table 2.
To attempt to determine which subspecies was the donor for intersubspecific recombination, we examined sites within the recombinant regions where X. fastidiosa subsp. pauca was invariant, but the reference sequences of the other three subspecies differed. The donor DNA was indicated by whichever of the variants was present in X. fastidiosa subsp. pauca, so a site was scored as positive if the target subspecies and X. fastidiosa subsp. pauca have the same base. Based on the recombinant regions of leuA, petC, malF, cysG, and rfbD, there were 73 such sites, and the scores of the three subspecies were indistinguishable: X. fastidiosa subsp. multiplex 59%; X. fastidiosa subsp. sandyi, 58%; and X. fastidiosa subsp. fastidiosa, 53%.
We also considered the question of whether citrus and coffee STs differed in their levels of intersubspecific recombination. To this end, we summed the number of P sites within recombinant regions where a given ST shared a base with the three other subspecies (the unshaded sites in Tables 2 to 5). Summing across recombinant regions of leuA, petC, malF, cysG, and rfbD showed that ST16 and ST14 (coffee types) had 81 and 70 such shared sites, while the three citrus types (ST11 to ST13) all shared 47. This substantial difference indicates that, at least based on our limited sample of genes, the coffee forms have undergone more intersubspecific recombination than the citrus forms.
The MLST gene regions represent a small (4,161-bp) random sample of the sequence of housekeeping genes. Within this sample, we identified two regions of introgression within each of cysG (180 + 167 bp) and holC (17 + 158 bp), one region within malF (409 bp), and all of leuA (708 bp) and petC (533 bp). There were no indications of introgression within nuoL or gltT. Thus, seven regions of introgression were identified totaling 2,172 bp out of a total of 4,161 bp (52%). The other recombination tests detected a total of only 409 bp, all in malF (just 10% of the MLST data).
Estimated divergence time of X. fastidiosa subsp. pauca.
Documenting the time scale of the separation of X. fastidiosa subsp. pauca from the other X. fastidiosa subspecies is complicated by the evidence of extensive intersubspecific homologous recombination. We minimized this problem by eliminating the regions recognized as probable recombination sites from the data, since the remaining nonrecombinant regions could be expected to give an accurate phylogenetic picture. The data used for this analysis included our additional sequence as well as the MLST regions. A concatenation of the nonrecombinant coding regions consisted of 5,271 bp, and the resulting tree (based on synonymous substitutions) showed an early split of X. fastidiosa subsp. pauca from the other three subspecies (Fig. 4), consistent with the only other tree based on synonymous substitutions (43). If we assume that neutral evolution in X. fastidiosa subsp. fastidiosa, X. fastidiosa subsp. sandyi, and X. fastidiosa subsp. pauca has been occurring at similar rates (X. fastidiosa subsp. multiplex is consistently slower for reasons considered elsewhere [see reference 32]), then the time of the split between X. fastidiosa subsp. pauca and the other subspecies can be estimated at about 60,000 years ago. Our jackknife estimate of the variance suggests that the standard error of this estimate is roughly 14,000 years, i.e., roughly the square root of (3.4)2 + (54.2/84.2)(17.0)2 (Fig. 4). The time estimates shown in Fig. 4 are dependent upon specific parameter values (see Materials and Methods); however, the relative branch lengths are independent of these values.
Fig 4.

Estimates of the time of divergence of the S. American X. fastidiosa subsp. pauca from the Central and North American subspecies, based on the rate of synonymous substitution in the nonrecombinant gene regions. The time estimate of the long branch separating X. fastidiosa subsp. pauca from the other subspecies (84.2 thousand years [Kyrs]) is split into 54.2 and 30.0 Kyrs to approximately equalize the two halves of the tree; however, the tree is unrooted, and this split is for illustration only. For details of the estimation of divergence times and their jackknife standard errors, see Materials and Methods. Time estimates less than 5,000 years were omitted. For the isolates used, see Table 2.
DISCUSSION
The value of MLST schemes in documenting the genetic variation of bacterial species is well established, both in general (see reference 21) and specifically for the case of X. fastidiosa (32). The MLST scheme previously proposed for this species (41, 49) is based on seven randomly chosen housekeeping genes subject to the typical levels of selective constraint (as measured by a range of 0.08 to 0.32 in the ratio of nonsynonymous to synonymous substitution rates [dN/dS]; see reference 41). We typed 78 X. fastidiosa subsp. pauca isolates from Brazil, which included completing the typing of 46 isolates studied previously (1). Given this overlap with the earlier study, we focused on examining novel features of the sequence data. However, the basic genetic patterns of limited genetic variability (we found only five STs) and the genetic separation of coffee and citrus isolates (see Fig. 1) were similar to the patterns observed using SNPs (47), fluorescent amplified fragment length analysis (17), and partial MLST (1).
The lack of ST variation is consistent with strong host-plant selection, enabling only a specific limited range of genotypes to be pathogenic on a given plant host. Supporting this view is the finding that the coffee and citrus forms are generally host specific (1, 20), while the possibility of rare exceptions to this rule (19, 37) was supported by our finding of just one coffee isolate with a citrus genotype (ST11) among the 23 coffee isolates typed.
Contained within this relatively simple pattern of low variability was an unusual feature that has not previously been noted: high levels of sequence divergence (1 to 3%) between some alleles, levels that are typical of subspecific differences (see reference 43). For example, while the three STs observed on citrus (ST11 to ST13) were identical across four of the seven MLST genes and differed by one and three SNPs at two more of them, at the seventh locus 1 pair of alleles differed by 10 bp, creating the clear genetic separation of ST13 from the other two citrus STs (Fig. 1). Similarly, the two coffee STs (ST14 and ST16) were very similar across four of the MLST genes (≤1-bp difference); however, they were differentiated by widely divergent alleles at the other three loci (differing by 8, 10, and 13 bp). It is extremely difficult to construct any scenario in which such large allelic differences could have accumulated within X. fastidiosa subsp. pauca via point mutation. Two factors that would promote diverse alleles are clonality and a strong geographical genetic structuring of the population. However, this species (including this subspecies) is subject to significant levels of (intrasubspecific) homologous recombination (1, 41), and there is no evidence of genetic structure in this subspecies, at least among the different states within Brazil (1).
The most parsimonious explanation for these large allelic differences is that variation has been acquired by X. fastidiosa subsp. pauca through intersubspecific recombination. Such recombination has been observed at low levels in X. fastidiosa subsp. fastidiosa in North America (31, 32, 49), and very high levels of recombination between X. fastidiosa subsp. fastidiosa and X. fastidiosa subsp. multiplex have been implicated in the genesis of the strain that infects mulberry (31). We tested for the occurrence of intersubspecific recombination in X. fastidiosa subsp. pauca after combining MLST data with data from additional locally targeted sequencing. We showed that these large allelic differences were entirely consistent with intersubspecific recombination, confirming the previous tentative suggestion of its occurrence in X. fastidiosa subsp. pauca (1). More importantly, we found that the degree of intersubspecific recombination was extremely high. Based on the small but random sample of housekeeping genes used in the MLST of this species, our estimate was that there was evidence of subspecific introgression across 52% of the 4,161 bp that are used in typing. We also found that the coffee STs had undergone roughly 50 to 70% more intersubspecific recombination than the citrus STs, based on a count across all of our sequence data of the polymorphic sites (within X. fastidiosa subsp. pauca) where STs shared a base with the three potential donor subspecies.
We detected intersubspecific recombination using our introgression test. We were motivated to develop a new test because Scally et al. (41) found that, given the patterns of polymorphism found in X. fastidiosa, the available recombination tests were often ineffective at isolating regions of recombination, even those that were easily detected by eye. We compared the introgression test to five other tests (RDP (22), GeneConv (34), MaxChi (23), Chimaera (36), and 3Seq (4) (see Materials and Methods), and the introgression test was clearly superior. The introgression test identified eight regions of recombination (totaling 6,053 bp) in six loci, whereas the best-performing tests (3Seq, MaxChi, and Chimaera) identified just two of these regions (Fig. 2). 3Seq identified the most recombinant bases (1,793 bp across these two regions, subdivided into three pieces), just 30% of the total found with the introgression test. Examples of recombination breakpoints only detected by the introgression test are shown in Tables 2 and 5.
The effectiveness of the introgression test does not depend on including the true non-native donor in the analysis. It requires either that the reference taxa include the true donor or that they are an outgroup relative to the donor and native pair. Obviously, the power of the test declines as the donor and native taxa become more closely related relative to the reference taxa.
The genetic introgression into X. fastidiosa subsp. pauca acts to reduce its apparent phylogenetic distance from the other subspecies. Identifying recombinant and nonrecombinant regions in the sequence data allowed us to correct for this bias and to estimate the time of separation of X. fastidiosa subsp. pauca from the other three subspecies. Schuenzel et al. (41) introduced a method for estimating the age of bacterial taxa using a central result of neutral evolutionary theory: the rate of substitution at neutral sites is equal to the neutral mutation rate. Applying this result to synonymous substitutions, they estimated that the X. fastidiosa subsp. fastidiosa/X. fastidiosa subsp. sandyi clade split from X. fastidiosa subsp. multiplex about 30,000 years ago. X. fastidiosa subsp. pauca is an outgroup to these subspecies, and here we carried out a similar analysis to estimate when X. fastidiosa subsp. pauca diverged from the other subspecies. After removing all regions of suspected intersubspecific recombination from the sequence data, analysis of the remaining (∼5 kb) sequence from nine different genomic locations (the seven MLST loci, plus pilU and rfbD) indicated that X. fastidiosa subsp. pauca diverged from the other subspecies of X. fastidiosa about 60,000 years ago. The jackknife-estimated standard deviation of about 14,000 years showed that this age estimate is fairly consistent across the regions of the genome sampled.
Given such a long evolutionary history, we would expect that X. fastidiosa subsp. pauca would be a genetically diverse group through the accumulation of both beneficial and neutral mutations in different lineages. In contrast, it appears that most of the genetic variation detected has its origins in intersubspecific recombination. To resolve this paradox, we propose the following hypothesis: that intersubspecific recombination was instrumental in facilitating the recent invasion of citrus and coffee, that colonization of these new hosts created a genetic bottleneck, and that native nonrecombinant X. fastidiosa subsp. pauca (with its expected genetic variability) has yet to be found.
The natural plant hosts of X. fastidiosa subsp. pauca are unknown, and yet they must exist. We know of no studies in the native environment attempting to identify such hosts. To date, the only study examining alternate plant hosts was a study of weed infection within orange groves, where 10 of 23 weed species were found to be infected (20); however, within a grove a weed may test positive simply because of repeated feeding by leafhoppers carrying X. fastidiosa subsp. pauca from citrus. There is no evidence that any of these weed species support X. fastidiosa subsp. pauca infection at locations remote from citrus.
The known record of Xylella-related disease in South America goes back less than 50 years, even though the phylogenetic data indicate that X. fastidiosa is a long-term native of South America and the two affected crops, citrus and coffee, have been commercially grown for several hundred years. Citrus variegated chlorosis (CVC) was described in Minas Gerais, Brazil, in 1987, and in Sao Paulo soon after (5), and coffee leaf scorch (CLS) was first noted in Sao Paulo, Brazil, in 1995 (7), although Li et al. (19) suggested that that CLS probably appeared earlier than this, but only by about 30 years.
Why did these diseases appear only very recently? One possibility is that X. fastidiosa subsp. pauca invaded Brazil and Argentina from some other region of South America. Arguing against this possibility is the fact that there are relatively few natural geographical barriers in South America (excepting the extreme west), that the continent has abundant populations of potential vectors (40), and that both citrus and coffee are grown in other parts of the continent, and yet there are no reports of Xylella-related disease in those areas (i.e., areas other than Argentina, Paraguay, and Brazil).
The alternative possibility that we propose is that X. fastidiosa subsp. pauca was originally unable to infect coffee and citrus but that adaptation to these host plants only became possible following the introduction of novel genetic variation resulting from intersubspecific recombination. Obviously, such an event requires the proximity of donor DNA, and it appears likely that this condition prevails. Current evidence suggests that only one subspecies of X. fastidiosa evolved in South America; however, an apparently distinct form of X. fastidiosa causes plum leaf scald in Argentina, Brazil, and Paraguay (13, 18). Four genetic analyses of the Brazilian plum isolate PL9746 suggest that it is genetically distinct from X. fastidiosa subsp. pauca and is probably an example of X. fastidiosa subsp. multiplex. Array-based hybridization of genomic DNA against the X. fastidiosa subsp. pauca strain 9a5c showed that PL9746 has clear regions of similarity to North American X. fastidiosa (based on gene absences relative to 9a5c), but no subspecific comparisons were made (30). Da Costa et al. (6), using arbitrarily primed PCR, found that PL9746 was roughly equidistant from X. fastidiosa subsp. pauca isolates, a North American plum isolate, and a North American grape isolate. However, more recently, two genomic fingerprinting studies (17, 27) both showed that while PL9746 was distinct from both coffee and citrus isolates of X. fastidiosa subsp. pauca, it had a very close relationship with PLS0135 (ATCC 35871), an isolate from plum that Schaad et al. (42) named as the type of the subspecies multiplex (ST41 [see reference 33]), and it had a clear separation from grape isolates (X. fastidiosa subsp. fastidiosa). The conclusion that PL9746 is a representative of subspecies multiplex is consistent with its plum host. As noted above, the type for subspecies multiplex was isolated from plum, and furthermore two different plum isolates (PLS0026 and PLP0070) have been used to represent this subspecies in phylogenetic analyses (one each in references 43 and 49). We have typed an additional 16 plum isolates, and none of them were X. fastidiosa subsp. fastidiosa or X. fastidiosa subsp. sandyi (unpublished data).
Notwithstanding these data, at present we cannot exclude the possibility that PL9746 represents another, as-yet-undescribed, South American subspecies of X. fastidiosa that evolved in geographical isolation from X. fastidiosa subsp. pauca. This view gains some weak support from our finding that the sequence data failed to identify which of the known subspecies is the source that has recombined into X. fastidiosa subsp. pauca. Within the recombinant regions, the sites that are polymorphic within X. fastidiosa subsp. pauca are almost always monomorphic across the potential donor subspecies or they are “informative,” with the exact same polymorphism as X. fastidiosa subsp. pauca (see Tables 2 to 5). Additional information was gleaned from sites within recombinant regions where the potential donor sequences varied but X. fastidiosa subsp. pauca did not. If variants present in only one particular subspecies are also present in the X. fastidiosa subsp. pauca sequences then this would strongly support that subspecies as the donor. However, although X. fastidiosa subsp. multiplex does have the highest percentage of such sites (59%), the values for the other two subspecies (58 and 53%) are indistinguishable.
Over the last 20 years it has become increasingly apparent that homologous recombination is almost ubiquitous among bacteria, although the degree to which it occurs apparently varies widely among species (9, 14, 26). It typically involves short pieces of DNA (often <1 kb [8, 24, 26]), which is consistent with the sizes of the recombinant fragments that we have identified in X. fastidiosa subsp. pauca, which averaged 757 bp (an underestimate, since the eight fragments included three with only one end identified [see Fig. 2]).
It is generally assumed that homologous recombination is beneficial by enabling bacteria to avoid host defenses (9, 25), but it has also been speculated that it may promote adaptation to novel hosts (2). Moreover, there is another example in X. fastidiosa where massive intersubspecific recombination has resulted in the colonization of a new host. Isolates from mulberry are a mix of X. fastidiosa subsp. fastidiosa and X. fastidiosa subsp. multiplex genomes, and mulberry is a plant host that neither parent subspecies is capable of infecting (31). In the present study, we have established a strong circumstantial case for the involvement of the same kind of intersubspecific homologous recombination in the shift of X. fastidiosa subsp. pauca from its unknown native hosts to citrus and coffee. The next step is to determine whether a nonrecombinant native form of X. fastidiosa subsp. pauca can be found and tested for its host specificity. Our prediction is that it will infect citrus and coffee poorly or not at all.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a USDA-CSREES-NRI grant 2007-55605-17834 to L.N. and R.S.
We thank Rodrigo Almeida for providing DNA samples and Stephanie Russell for sequence curation. We thank Laramy Enders, Josh Wang, Elizabeth Mah, Qui Luong, and Mariel Garcia for help in sequencing genes from many of the isolates used in this study. We also thank Adam Retchless and two anonymous reviewers for their valuable comments.
Footnotes
Published ahead of print 27 April 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Almeida RPP, et al. 2008. Genetic structure and biology of Xylella fastidiosa strains causing disease in citrus and coffee in Brazil. Appl. Environ. Microbiol. 74:3690–3701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baldo L, Bordenstein S, Wernegreen JJ, Werren JH. 2005. Widespread recombination throughout Wolbachia genomes. Mol. Biol. Evol. 23:437–449 [DOI] [PubMed] [Google Scholar]
- 3. Beretta MJG, Harakava R, Chagas CM. 1996. First report of Xylella fastidiosa in coffee. Plant Dis. 80:821 [Google Scholar]
- 4. Boni MF, Posada D, Feldman MW. 2007. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 176:1035–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chang CJ, Garnier RM, Zreik L, Rossetti V, Bove JM. 1993. Culture and serological detection of the xylem-limited bacterium causing citrus variegated chlorosis and its identification as a strain of Xylella fastidiosa. Curr. Microbiol. 27:137–142 [DOI] [PubMed] [Google Scholar]
- 6. da Costa PI, Franco CF, Miranda VS, Teixeira DC, Hartung JS. 2000. Strains of Xylella fastidiosa rapidly distinguished by arbitrarily primed-PCR. Curr. Microbiol. 40:279–282 [DOI] [PubMed] [Google Scholar]
- 7. de Lima JEO, et al. 1998. Coffee leaf scorch bacterium: axenic culture, pathogenicity, and comparison with Xylella fastidiosa of citrus. Plant Dis. 82:94–97 [DOI] [PubMed] [Google Scholar]
- 8. Falush D, et al. 2001. Recombination and mutation during long-term gastric colonization by Helicobacter pylori: estimates of clock rates, recombination size, and minimal age. Proc. Natl. Acad. Sci. U. S. A. 98:15056–15061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feil EJ, Spratt BG. 2001. Recombination and the population structures of bacterial pathogens. Annu. Rev. Microbiol. 55:561–590 [DOI] [PubMed] [Google Scholar]
- 10. Feil H, Purcell AH. 2001. Temperature-dependent growth and survival of Xylella fastidiosa in vitro and in potted grapevines. Plant Dis. 85:1230–1234 [DOI] [PubMed] [Google Scholar]
- 11. Felsenstein J. 1989. PHYLIP: phylogeny inference package (version 3.2). Cladistics 5:164–166. [Google Scholar]
- 12. Felsenstein J. 2005. PHYLIP (phylogeny inference package) version 3.6 Department of Genome Sciences, University of Washington, Seattle [Google Scholar]
- 13. French WJ, Kitajima EW. 1978. Occurrence of plum leaf scald in Brazil and Paraguay. Plant Dis. Reporter 62:1035–1038 [Google Scholar]
- 14. Hanage WP, Fraser C, Spratt BG. 2006. Sequences, sequence clusters and bacterial species. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361:1917–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hernandez-Martinez R, de la Cerda KA, Costa HS, Cooksey DA, Wong FP. 2007. Phylogenetic relationships of Xylella fastidiosa strains isolated from ornamentals in southern California. Phytopathology 97:857–864 [DOI] [PubMed] [Google Scholar]
- 16. Hopkins DL. 1989. Xylella fastidiosa: xylem-limited bacterial pathogen of plants. Annu. Rev. Photopathol. 27:271–290 [Google Scholar]
- 17. Kishi L, et al. 2008. Evaluation of Xylella fastidiosa genetic diversity by fAFLP markers. Rev. Bras. Frutic. 30:202–208 [Google Scholar]
- 18. Kitajima EW, Bakarcic M, Fernandez-Valiela MV. 2003. Association of Rickettsia-like bacteria with plum leaf scald disease. Phytopathology 65:476–479 [Google Scholar]
- 19. Li WB, et al. 2001. Coffee leaf scorch caused by a strain of Xylella fastidiosa from citrus. Plant Dis. 85:501–505 [DOI] [PubMed] [Google Scholar]
- 20. Lopes SA, et al. 2003. Weeds as alternative hosts of the citrus, coffee, and plum strains of Xylella fastidiosa in Brazil. Plant Dis. 87:544–549 [DOI] [PubMed] [Google Scholar]
- 21. Maiden MCJ. 2006. Multilocus sequence typing of bacteria. Annu. Rev. Microbiol. 60:561–588 [DOI] [PubMed] [Google Scholar]
- 22. Martin D, Rybicki E. 2000. RDP: detection of recombination amongst aligned sequences. Bioinformatics 16:562–563 [DOI] [PubMed] [Google Scholar]
- 23. Maynard Smith J. 1992. Analyzing the mosaic structure of genes. J. Mol. Evol. 34:126–129 [DOI] [PubMed] [Google Scholar]
- 24. Maynard Smith J, Dowson CG, Spratt BG. 1991. Localized sex in bacteria. Nature 349:29–31 [DOI] [PubMed] [Google Scholar]
- 25. Maynard Smith J, Smith NH, O'Rourke M, Spratt BG. 1993. How clonal are bacteria? Proc. Natl. Acad. Sci. U. S. A. 90:4384–4388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mau B, Glasner JD, Darling AE, Perna NT. 2006. Genome-wide detection and analysis of homologous recombination among sequenced strains of Escherichia coli. Genome Biol. 7:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mehta A, Leite RP, Rosato YB. 2001. Assessment of the genetic diversity of Xylella fastidiosa isolated from citrus in Brazil by PCR-RFLP of the 16S rDNA and 16S-23S intergenic spacer and rep-PCR fingerprinting. Antonie Van Leeuwenhoek 79:53–59 [DOI] [PubMed] [Google Scholar]
- 28. Miranda VS, Farias PRS, Roberto SR, Lacava PM. 2007. Genetic characterization of Xylella fastidiosa isolated from citrus and coffee plants. Sci. Agric. 64:482–485 [Google Scholar]
- 29. Montero-Astua M, et al. 2007. Genetic diversity of Xylella fastidiosa strains from Costa Rica, São Paulo, Brazil, and the United States. Phytopathology 97:1338–1346. [DOI] [PubMed] [Google Scholar]
- 30. Nunes LR, et al. 2003. Microarray analyses of Xylella fastidiosa provide evidence of coordinated transcription control of laterally transferred elements. Genome Res. 13:570–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nunney L. 2011. Homologous recombination and the invasion of a new plant host by the pathogenic bacterium, Xylella fastidiosa. Phytopathology 101:S130 [Google Scholar]
- 32. Nunney L, et al. 2010. Population genomic analysis of a bacterial plant pathogen: novel insight into the origins of Pierce's disease of grapevine in the U.S. PLoS One 5:e15488 doi:10.1371/journal.pone.0015488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nunney L, Elfekih S, Stouthamer R. 2012. The importance of multilocus sequence typing: cautionary tales from the bacterium Xylella fastidiosa. Phytopathology 102:456–460 doi:10.1094/PHYTO-10-11-0298 [DOI] [PubMed] [Google Scholar]
- 34. Padidam M, Sawyer S, Fauquet CM. 1999. Possible emergence of new geminiviruses by frequent recombination. Virology 265:218–225 [DOI] [PubMed] [Google Scholar]
- 35. Pendergrast M. 2001. Uncommon grounds: the history of coffee and how it transformed our world. Texere, London, United Kingdom [Google Scholar]
- 36. Posada D, Crandall KA. 2001. Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc. Natl. Acad. Sci. U. S. A. 98:13757–13762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Prado S, Lopes JRS, Demetrio C, Borgatto A, Almeida RPP. 2008. Host colonization differences between citrus and coffee isolates of Xylella fastidiosa in reciprocal inoculation. Sci. Agricola 65:251–258 [Google Scholar]
- 38. Purcell AH. 1990. Homopteran transmission of xylem-inhabiting bacteria. Adv. Dis. Vector. Res. 6:243–266 [Google Scholar]
- 39. Qin X, Miranda VS, Machado MA, Lemos EGM, Hartung JS. 2001. An evaluation of the genetic diversity of Xylella fastidiosa isolated from diseased citrus and coffee in São Paulo, Brazil. Phytopathology 91:599–605 [DOI] [PubMed] [Google Scholar]
- 40. Redak RA, et al. 2004. The biology of xylem fluid-feeding insect vectors of Xylella fastidiosa and their relationship to disease epidemiology. Annu. Rev. Entomol. 49:243–270 [DOI] [PubMed] [Google Scholar]
- 41. Scally M, Schuenzel EL, Stouthamer R, Nunney L. 2005. Multilocus sequence type system for the plant pathogen Xylella fastidiosa and relative contributions of recombination and point mutation to clonal diversity. Appl. Environ. Microbiol. 71:8491–8499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schaad NW, Postnikova E, Lacy G, Fatmi M, Chang CJ. 2004. Xylella fastidiosa subspecies: X. fastidiosa subsp piercei, subsp. nov., X. fastidiosa subsp. multiplex subsp. nov., and X. fastidiosa subsp. pauca subsp. nov. System. Appl. Microbiol. 27:290–300 [DOI] [PubMed] [Google Scholar]
- 43. Schuenzel EL, Scally M, Stouthamer R, Nunney L. 2005. A multigene phylogenetic study of clonal diversity and divergence in North American strains of the plant pathogen Xylella fastidiosa. Appl. Environ. Microbiol. 71:3832–3839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Simpson AJG, et al. 2000. The genome sequence of the plant pathogen Xylella fastidiosa. Nature 406:151–159 [DOI] [PubMed] [Google Scholar]
- 45. Webber HJ, Batchelor LD. (ed). 1943. The citrus industry: history, botany, and breeding, University of California Press, San Diego, CA [Google Scholar]
- 46. Wells JM, et al. 1987. Xylella fastidiosa gen. nov, sp. Nov: gram-negative, xylem-limited, fastidious plant bacteria related to Xanthomonas spp. Int. J. Syst. Bacteriol. 37:136–143 [Google Scholar]
- 47. Wickert E, Machado MA, Lemos EG. 2007. Evaluation of the genetic diversity of Xylella fastidiosa strains from citrus and coffee hosts by single-nucleotide polymorphism markers. Phytopathology 97:1543–1549 [DOI] [PubMed] [Google Scholar]
- 48. Yang Z. 2007. PAML4: a program package for phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24:1586–1591 [DOI] [PubMed] [Google Scholar]
- 49. Yuan XL, et al. 2010. MLST analysis of Xylella fastidiosa isolates causing Pierce's disease of grape and oleander leaf scorch across the United States. Phytopathology 100:601–611 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.