

Abstract
Bacterial species such as Shewanella oneidensis MR-1 require extracellular nucleolytic activity for the utilization of extracellular DNA (eDNA) as a source of nutrients and for the turnover of eDNA as a structural matrix component during biofilm formation. We have previously characterized two extracellular nucleases of S. oneidensis MR-1, ExeM and ExeS. Although both are involved in biofilm formation, they are not specifically required for the utilization of eDNA as a nutrient. Here we identified and characterized EndA, a third extracellular nuclease of Shewanella. The heterologously overproduced and purified protein was highly active and rapidly degraded linear and supercoiled DNAs of various origins. Divalent metal ions (Mg2+ or Mn2+) were required for function. endA is cotranscribed with phoA, an extracellular phosphatase, and is not upregulated upon phosphostarvation. Deletion of endA abolished both extracellular degradation of DNA by S. oneidensis MR-1 and the ability to use eDNA as a sole source of phosphorus. PhoA is not strictly required for the exploitation of eDNA as a nutrient. The activity of EndA prevents the formation of large cell aggregates during planktonic growth. However, in contrast to the findings for ExeM, endA deletion had only minor effects on biofilm formation. The findings strongly suggest that the extracellular nucleases of S. oneidensis exert specific functions required under different conditions.
INTRODUCTION
Species of the genus Shewanella are Gram-negative, facultatively anaerobic gammaproteobacteria. Members of this genus are generally characterized by an enormous respiratory flexibility that allows them to use a wide array of alternative terminal electron acceptors under anaerobic conditions (21, 43, 44). The ability of shewanellae to reduce halogenated compounds as well as a range of metal ions, among which are numerous radionucleotides, allows them to be employed for the bioremediation of contaminated soils (reviewed in references 26 and 27) or in the production of microbial fuel cells (8, 37). In addition, several species of that genus have emerged as opportunistic pathogens (58). Shewanellae are commonly found in aquatic and sedimentary systems, chemically and redox-stratified environments to which they are well adapted due to their respiratory versatility (29, 64). Such environments are often limited in bioavailable anorganic phosphate (Pi) (3, 6, 30, 33). Thus, it has been speculated that bacterial species that thrive in these environments are capable of utilizing alternative organic phosphorus sources, such as phospholipids, phosphoproteins, and, in particular, extracellular DNA (eDNA) (48). Accordingly, Pinchuk et al. demonstrated that species of Shewanella exploit eDNA as a sole source of carbon, nitrogen, and phosphorus in media that lack these nutrients under both aerobic and anaerobic conditions (48). Notably, eDNA is not only a valuable nutrient for Shewanella and other species (41). We have recently demonstrated the presence of significant amounts of eDNA in S. oneidensis MR-1 biofilms, which are thought to be released by prophage-induced cell lysis (23). We demonstrated that treatment with DNase I strongly decreases the ability of S. oneidensis MR-1 to attach to a surface initially and that the subsequent formation of 3-dimensional structures is severely affected. Also, established biofilms release large amounts of biomass upon treatment with DNase I. Thus, eDNA serves as an important structural component and as an adhesion factor in the formation of surface-associated communities of S. oneidensis MR-1 (23). Similar observations have been reported for a wide range of bacterial species as well as for mixed communities (18, 19, 53), indicating a rather common role of eDNA in bacterial biofilm formation. On the other hand, high concentrations of eDNA are toxic for bacterial cells due to the chelating properties of DNA (40).
Utilization of eDNA as a nutrient, in particular as a source of phosphate, requires the presence of extracellular nucleases and phosphatases, and corresponding activity was readily detected in Shewanella cultures (22, 48). Recently, we characterized the role of two extracellular nucleases, ExeM and ExeS, in Shewanella oneidensis MR-1 (22). ExeS is predicted to be released into the medium supernatant, while ExeM appears to remain associated with the cell envelope (9, 48, 54). By mutant analysis, we demonstrated that both proteins actively degrade eDNA. Expression of both exeM and exeS was strongly induced upon phospholimitation and surface association, indicating a role in utilizing DNA as a nutrient and a role during biofilm formation. Accordingly, ExeS affected S. oneidensis MR-1 biofilm formation under static conditions. The role of surface-associated ExeM in biofilm formation was more pronounced than that of ExeS under both static and hydrodynamic conditions. In the absence of exeM, the cells failed to cover the surface, and large amounts of eDNA accumulated in the biofilm matrix. In contrast, the absence of both exeM and exeS had little or no effect on the use of eDNA as the sole source of phosphorus. In addition, a mutant lacking both enzymes still displayed substantial degradation of eDNA in the medium supernatant, indicating the presence of one or more additional proteins with nucleolytic function (22). That prompted us to screen for other potential extracellular nucleases in S. oneidensis MR-1.
In this study, we identified and characterized EndA, a third extracellular endonuclease present in Shewanella species. We demonstrate that this enzyme is highly active in the degradation of different types of nucleic acids and that it is essential for the utilization of eDNA as a nutrient by S. oneidensis MR-1. Surprisingly, EndA does not play a significant role in biofilm formation under hydrodynamic conditions, strongly indicating that the different nucleases exert different functions.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains used in this study are summarized in Table 1. For routine purposes, S. oneidensis and Escherichia coli strains were grown in LB medium at 30°C and 37°C, respectively. For E. coli strain WM3064, 2,6-diaminopimelic acid was added to the medium to a final concentration of 300 μM. For solidification, agar was added to a final concentration of 1.5% (wt/vol). For growth assays depending on the phosphorus source, S. oneidensis strains were grown in modified M1 mineral medium as described previously (22). NaH2PO4 at 0.86 mM or the indicated amount of salmon sperm DNA (Sigma, Deisenhofen, Germany) was added as the sole source of phosphorus. Prior to use, the DNA was purified by ethanol precipitation. When appropriate, media were supplemented with 100 μg · ml−1 ampicillin, 6 μg · ml−1 chloramphenicol, 10 μg · ml−1 gentamicin, and/or 50 μg · ml−1 kanamycin. To induce the overexpression of endA from pLacTac-endA, isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotypea | Source or reference |
|---|---|---|
| Bacterial strains | ||
| Escherichia coli | ||
| BL-21 Star (DE3) | F−ompT hsdSB(rB− mB−) gal dcm rne131 (DE3) | Invitrogen |
| DH5α λpir | ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 hsdR17 deoR thi-1 supE44 gyrA96 relA1/λpir | 39 |
| WM3064 | thrB1004 pro thi rpsL hsdS lacZΔM15 RP4-1360 Δ(araBAD)567 ΔdapA1341::[erm pir(wt)] | W. Metcalf, University of Illinois, Urbana |
| Shewanella oneidensis | ||
| MR-1 | Shewanella oneidensis MR-1 wild type | 59 |
| S198 | MR-1, tagged with eGfp in a mini-Tn7 construct; Cmr | 23 |
| S988 | MR-1 ΔexeM; deletion of the SO_1066 gene | 22 |
| S1015 | MR-1 ΔexeM, tagged with eGfp in a mini-Tn7 construct; Cmr | 22 |
| S989 | MR-1 ΔexeS; deletion of the SO_1844 gene | 22 |
| S2095 | MR-1 ΔendA; deletion of the SO_0833 gene | This work |
| S2303 | Wild type (complementation of ΔendA) | This work |
| S2373 | MR-1 ΔphoA; deletion of the SO_0831 gene | This work |
| Plasmids | ||
| pBBR1-MCS5 | oriT mobRK2 oriR lacZα; cloning vector; Gmr | 34 |
| pBBR1-MCS5-TT-Pmot-RBS-lux | oriT mobRK2 oriR luxCDABE under the control of the constitutive motAB promoter of S. oneidensis MR-1; Gmr | 22 |
| pLacTac | repA oriVpVSIoriVp15AoriT lacIq1-Ptac; Tcr | 55 |
| pLacTac-endA | endA in pLactTac | This work |
| pMal-P2X | pMB1 ori lacIqmalE, vector for overproduction of proteins fused N-terminally to the maltose-binding protein to be targeted to the periplasm; Apr | New England Biolabs |
| pMal-P2-0833-N | SO_0833 (endA) lacking the N-terminal signal sequence in pMal-P2X; Apr | This work |
| pNPTS138-R6KT | ori-R6K sacB, suicide plasmid for generating in-frame deletions; Kmr | 35 |
| pNPTS-R6K-dendA | Fragment for in-frame deletion of SO_0833 (endA) in pNPTS138-R6K; Kmr | This work |
| pNPTS-R6K-KI-endA | Fragment for endA complementation in pNPTS138-R6K; Kmr | This work |
| pNPTS-R6K-dphoA | Fragment for in-frame deletion of SO_0831 (phoA) in pNPTS138-R6K; Kmr | This work |
| pNPTS-R6K-KI-phoA | Fragment for phoA complementation in pNPTS138-R6K; Kmr | This work |
| pTNS2 | ori-R6K; encodes the TnsABC+D specific transposition pathway; Apr | 11 |
| pUC18-R6KT-miniTn7T-egfp | NotI-egfp-Cmr-NotI fragment from pBK-miniTn7-gfp3 in pUC18-R6KT-miniTn7T; Apr Cmr | 23 |
Apr, ampicillin resistance; Cmr, chloramphenicol resistance; Gmr, gentamicin resistance; Kmr, kanamycin resistance; Tcr, tetracyclin resistance.
Biofilms of S. oneidensis MR-1 were cultivated in plain LM medium supplemented with 0.5 mM lactate (47). DNase I (Serva Electrophoresis GmbH, Heidelberg, Germany) was used at a concentration of 30 μg · ml−1 in the medium. For staining of extracellular DNA in biofilms grown under hydrodynamic conditions, DDAO [7-hydroxy-9H-(1,3-dichloro-9,9-dimethylacridin-2-one)] was added to a final concentration of 4 μM.
Vector and strain constructions.
Chromosomal DNA was isolated as described previously (49), and DNA was manipulated according to standard protocols (50) or by following the manufacturers' instructions. Kits for the isolation of plasmids and the purification of PCR products were purchased from HiSS Diagnostics GmbH (Freiburg, Germany). The enzymes used in this study were purchased from New England Biolabs (NEB) (Frankfurt am Main, Germany) and Fermentas (St. Leon-Rot, Germany). The strains and plasmids used and generated in this study are listed in Table 1. Replicative plasmids were transferred into E. coli by transformation using chemically competent cells (31) and into S. oneidensis by conjugation from E. coli WM3064 or by electroporation.
Markerless in-frame deletion mutants of S. oneidensis were constructed as reported previously by using the suicide vector pNPTS138-R6KT (35); the appropriate PCR fragments were generated using the primers listed in Table S1 in the supplemental material. Successful deletion was confirmed by PCR using primers bracketing the deleted region. To determine whether the phenotypes observed were due to the absence of the deleted gene, the wild-type genotype was restored by reintegration of the deleted section by use of the same strategy used for gene deletion. In all cases, the wild-type phenotype was successfully restored. For overexpression of endA in S. oneidensis MR-1, the gene including an upstream ribosome-binding sequence was cloned into pLacTac after digestion with XhoI and PstI.
For biofilm studies, appropriate S. oneidensis MR-1 strains constitutively expressing gfp, encoding green fluorescent protein, were generated. To this end, we used a modified Tn7 delivery system as described previously (23).
Heterologous production and purification of EndA.
To construct a plasmid for the overexpression of endA, the gene was amplified without the sequence encoding the native signal peptide and was cloned into pMal-P2X using BamHI and SalI to result in an in-frame fusion to malE, encoding the maltose-binding protein (MBP). The primers used are listed in Table S1 in the supplemental material. The resulting vector, pMal-P2-0833-N, was transformed into E. coli BL21 Star (DE3) for heterologous overproduction of the fusion protein MBP-EndA. The fusion protein was overproduced and purified essentially as specified for the pMAL Protein Fusion and Purification System (NEB, Frankfurt am Main, Germany) using method II for periplasmic secretion of the fusion protein. Briefly, IPTG was added to a final concentration of 0.3 mM to exponentially growing cells (optical density at 600 nm [OD600], 0.5) in 500 ml SOB medium (Roth, Karlsruhe, Germany). After 2 h of incubation at 37°C, cells were harvested by centrifugation and were resuspended in 400 ml of 30 mM Tris-HCl–20% sucrose (pH 8.0). After the addition of 1 mM EDTA, the suspension was incubated for 10 min at room temperature. Subsequently, the cells were sedimented by centrifugation and were resuspended in 5 mM ice-cold MgSO4. After 10 min of shaking in an ice bath, the supernatant (osmotic shock fluid) was collected by centrifugation, and the pH was adjusted by the addition of 8 ml Tris-HCl (pH 7.4). The fusion protein was purified by binding to 5 ml amylose resin and was eluted in column buffer (20 mM Tris-HCl, 200 mM NaCl, 1 mM EDTA) containing 10 mM maltose (1.5-ml elution fractions). Elution fractions were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and protein concentrations were quantified using the Roti-Quant assay (Roth, Karlsruhe, Germany) or the Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific Inc., Rockford, IL). Fractions 2 to 9 were pooled, concentrated, and used for further analyses.
DNA degradation assays.
Qualitative nuclease assays in medium supernatants were carried out essentially as described previously (22, 48). Briefly, cells from an overnight LB preculture were incubated in fresh medium at an OD600 of 0.05 and were grown to an OD600 of 1.5. Aliquots (230 μl) of cell-free filter-sterilized supernatant were mixed with an appropriate nucleic acid sample at a final concentration of 5 μg · ml−1. The samples were incubated at 30°C; aliquots were removed at regular intervals; and the integrity of the DNA was analyzed by agarose gel electrophoresis. The assay was repeated in at least two independent experiments.
For a comparison of nuclease activity in culture supernatants and washed cells, cultures were grown to an OD600 of 1.5. In order to inhibit further protein synthesis, chloramphenicol was added to a final concentration of 30 μg · ml−1 and cultures were incubated for 20 min at 30°C. To determine the nuclease activity of washed cells, 230-μl aliquots were washed three times in LB medium containing chloramphenicol (30 μg · ml−1). For a comparable determination of nuclease activity in the respective culture supernatants, 230-μl aliquots of the same culture were used without washing in LB. Subsequently, cell suspensions were mixed with a 20-μl nucleic acid sample (833-bp PCR product) at a final concentration of 5 μg · ml−1. The samples were incubated at 30°C, and 20-μl aliquots were removed and centrifuged at regular intervals. A 20-μl volume of each supernatant was analyzed by agarose gel electrophoresis.
The nuclease activity of purified MBP-EndA (and, additionally, those of MBP and heat-inactivated MBP-EndA as controls) was determined by monitoring the decrease in the fluorescence of DNA probes treated with GelRed nucleic acid stain (Biotrend, Cologne, Germany) after the addition of the proteins. Each sample contained 0.02 μg of purified protein, 260 ng purified DNA [pBluescript II KS(+)], and 30 μl of 3× GelRed nucleic acid stain in a final volume of 100 μl in 10 mM Tris-HCl (pH 7.6) supplemented with the respective metal ion cofactors (2.5 mM) or dithiothreitol (DTT) (10 mM). Emission was monitored at 1-min intervals at 600 nm (excitation at 290 nm) and 30°C using a Tecan (Männedorf, Switzerland) Infinite M200 microplate reader. For this assay, we defined 1 U of enzyme activity as the amount of enzyme that completely degraded 1 μg of pBluescript DNA in 10 min at 30°C in a buffer supplemented with 2.5 mM Mg2+. To determine the degradation of eDNA in medium supernatants, 0.18 U MBP-EndA in 10 μl buffer (10 mM Tris-HCl [pH 7.6], 2.5 mM Mg2+) and 30 μl GelRed solution were added directly to 40 μl of medium supernatant after centrifugation and filtration. To harvest eDNA from biofilms, static biofilms were cultivated in petri dishes; the supernatant was discarded; and the cells were carefully scraped from the surface after the addition of fresh medium. Subsequent to centrifugation and filter sterilization, the eDNA in the supernatant was used for degradation assays as described for medium supernatants. Control assays ensured that the degradation of eDNA as measured by fluorescence corresponded to the disappearance of DNA as visualized by agarose gel electrophoresis (data not shown). Qualitative assays were carried out by the addition of 0.09 U MBP-EndA to 2.5 μg of the nucleic acid sample dissolved in 100 μl of 10 mM Tris-HCl (pH 7.6) supplemented with 2.5 mM Mg2+. Samples were taken at the desired time points and were separated by agarose gel electrophoresis.
Determination of eDNA concentration in planktonic cultures.
The concentration of eDNA in planktonic S. oneidensis MR-1 cultures was assayed by mixing 70 μl of culture supernatant with 30 μl of 3× GelRed nucleic acid stain (Biotium Inc., Hayward, CA). Emission at 600 nm (excitation at 290 nm) was quantified using the Tecan (Männedorf, Switzerland) Infinite M200 microplate reader. DNA concentrations in the supernatants were calculated based on a calibration curve prepared with appropriate amounts of S. oneidensis MR-1 chromosomal DNA in the corresponding medium.
Phosphatase assays.
Phosphatase activity was assayed by the addition of 10 μl of 100 mM p-nitrophenyl phosphate (NEB, Frankfurt am Main, Germany) to 90 μl membrane-filtered (pore size, 0.2 μm) culture supernatants. The accumulation of 4-nitrophenol (Sigma, Deisenhofen, Germany) was quantified by measuring the absorption at 418 nm at 30°C using a Tecan (Männedorf, Switzerland) Infinite M200 microplate reader. The phosphatase activity in the culture supernatants was determined by using a 4-nitrophenol calibration curve as a reference standard for product accumulation in correlation to the slope of the OD418 change over the first 10 min.
Killing assays.
Assays on the detrimental effect of high eDNA concentrations on strains of S. oneidensis MR-1 were performed as described previously (22, 28, 40). Overnight cultures of the appropriate strains carrying pBBR1-MCS5-TT-Pmot-lux were reinoculated into fresh medium and were grown to the mid-exponential-growth phase. The luminescence of 180 μl of culture was measured using a Tecan (Männedorf, Switzerland) Infinite M200 plate reader. Afterwards, 20 μl of herring sperm DNA dissolved in medium was added to the wells to yield the appropriate DNA concentration. After thorough mixing, the luminescence of the cells was measured at regular intervals for 500 ms; the loss of fluorescence served as a measure for a decrease in viability. Each measurement was performed in duplicate and was repeated in at least two independent experiments.
Total-RNA extraction and RT-PCR.
For quantitative reverse transcription-PCR (qRT-PCR) and operon mapping, total RNA was extracted from S. oneidensis MR-1 cells by using a hot-phenol method (1) as described previously (22). Cells from biofilm supernatants and planktonic cultures were harvested by centrifugation (5 min at 4,600 × g and 4°C). Cells for corresponding biofilm studies were harvested from static biofilm cultures incubated in petri dishes. Overnight cultures of S. oneidensis MR-1 strains grown in LM medium were diluted to an OD600 of 0.01 and were transferred to petri dishes for static biofilm formation. At appropriate time points, the medium supernatant was collected, and the surface-associated cells were collected in 1:10 stop solution (5% [vol/vol] phenol, 95% [vol/vol] ethanol [pH 7.4]) via scraping prior to RNA extraction. Residual contaminating DNA was removed by using the Turbo DNA-free kit (Applied Biosystems, Darmstadt, Germany) according to the manufacturer's instructions. The quality of the RNA was determined by agarose gel electrophoresis. The extracted total RNA was then applied as the template for random-primed first-strand cDNA synthesis using BioScript reverse transcriptase (Bioline, Luckenwalde, Germany) according to the manufacturer's instructions. The resulting cDNA was used as the template for qRT-PCR and operon mapping. qRT-PCR was carried out using a Real-Time 7300 PCR machine and a Sybr green detection system (both from Applied Biosystems, Darmstadt, Germany). The signals were standardized to that of recA. The cycle threshold (CT) was automatically determined by the Real-Time 7300 PCR software. The total number of cycles was set to 40, and all samples were assessed in duplicate. The efficiency of the corresponding primer pairs (see Table S1 in the supplemental material) was determined by using different amounts of S. oneidensis MR-1 chromosomal DNA (10, 1.0, 0.1, and 0.01 ng per reaction) as a template.
Operon mapping was carried out by PCR using cDNA as the template and appropriate primer pairs bracketing the gaps between the genes that were analyzed. A corresponding total-RNA sample taken before the reverse transcriptase reaction served as a negative control, and chromosomal DNA served as a positive control. The PCR products were analyzed by agarose gel electrophoresis.
Cultivation and imaging of biofilms.
Static biofilm formation by wild-type and mutant S. oneidensis MR-1 strains was quantified in polystyrene microtiter plates as described previously (23, 56). The assay was performed in eight wells per plate and was repeated in at least three independent experiments. To analyze biofilm formation under hydrodynamic conditions, wild-type and mutant strains constitutively expressing gfp were grown in custom-made flow chambers, essentially as described previously (23, 56). For DDAO staining, the medium flow was briefly arrested, and the medium in the corresponding bubble trap was replaced with a medium supplemented with DDAO at the appropriate concentration before the medium flow was resumed. This process took less than 1 min, and control channels ensured that the short arrest of the medium flow did not affect biofilm formation. Images were acquired after 1 h of exposure to DDAO.
Microscopic visualization of biofilms and image acquisition were conducted using an inverted Leica TCS SP5 confocal laser scanning microscope (CLSM; Leica Microsystems, Wetzlar, Germany) equipped with ×10/0.3 Plan-Neofluar and ×63/1.2W C-Apochromat objectives. The flow chambers were transferred to the microscope and were mounted on the microscopic stage without interrupting the medium flow. Images were taken at defined spots close to the medium inflow. Biofilms were not incubated further after staining with DDAO. To display biofilm images, CLSM image stacks were processed using the Imaris software package (Bitplane AG, Zürich, Switzerland).
RESULTS
Identification of EndA.
To identify further potential extracellular enzymes with nucleolytic activity, we performed bioinformatic analysis on the S. oneidensis MR-1 genome data and screened for potential orthologs of already characterized bacterial nucleases that might be secreted. One protein, SO_0833, annotated as endonuclease EndA, emerged as a promising candidate. The corresponding gene is 777 bp long and encodes a protein of 258 amino acids with a molecular mass of 29.1 kDa. The protein is predicted to have an N-terminal signal sequence that is likely to be cleaved between positions 30 and 31 (4). The deduced amino acid sequence has significant identities to the periplasmic nucleases EndA from E. coli (54% identity) and Vvn from Vibrio vulnificus (58%), as well as to the extracellular nucleases Dns from Vibrio cholerae (58%) and Aeromonas hydrophila (58%) (10, 20, 32, 36). endA is located in a potential operon comprising seven genes (Fig. 1). Notably, the SO_0830 gene, three genes downstream of endA, encodes a putative alkaline phosphatase. This gene, to which we refer as phoA, is 1,365 bp long and encodes a protein of 454 amino acids with a molecular mass of 48.5 kDa. Like EndA, PhoA is predicted to have a signal peptide likely to be cleaved between positions 23 and 24. To further determine whether endA and phoA are transcribed together, as suggested by the gene organization, we performed RT-PCR on total mRNA isolated from cells grown in mineral medium to the exponential growth phase. The results demonstrated that endA is organized in an operon with its downstream genes SO_0832, encoding a conserved hypothetical protein, SO_0831, encoding a putative glutathione synthetase (GshB), and phoA. Faint signals suggest that the transcript might also include SO_0834, upstream of endA, encoding an SprT-like protein, and SO_0828, downstream of phoA, encoding a conserved hypothetical protein. The organization of a putative extracellular endonuclease together with an extracellular alkaline phosphatase on a single transcriptional unit strongly suggested a role in the utilization of eDNA as a source of phosphorus.
Fig 1.

Genetic and transcriptomic organization of endA and phoA. (Top) Schematic representation of the genetic organization. Gene numbers or corresponding annotations are given. Arrows indicate the positions of the corresponding primer pairs. (Bottom) RT analysis of the transcript. −, negative control with no reverse transcriptase added to the reaction mixture; +, standard RT-PCR; c, control reaction with chromosomal DNA as the template. CHP, conserved hypothetical protein.
Characterization of EndA in vitro activity.
To determine whether EndA is an active nuclease, we overproduced the protein lacking its native signal peptide in E. coli as an N-terminal MBP fusion that is transported into the periplasm. Large amounts of soluble MBP-EndA were produced and subsequently were highly enriched by affinity chromatography using amylose resin (Fig. 2). To characterize the activity of the fusion protein, we determined the level of DNA degradation upon the addition of purified MBP-EndA. To this end, we measured the decrease in fluorescence due to the release of DNA-bound GelRed nucleic acid stain as a function of DNA degradation by using the pBluescript plasmid as the substrate (Fig. 2). The results were confirmed by visualization of the residual DNA by separation on agarose gels after incubation with MBP-EndA. As a negative control, we used the same concentration of MBP, produced and purified in parallel with MBP-EndA. These assays demonstrated that pBluescript was readily degraded by MBP-EndA, while no DNA degradation occurred when the same amount of MBP was added (Fig. 2B). Similarly to plasmids, linear DNA fragments, chromosomal DNA from Bacillus subtilis, S. oneidensis, and Caulobacter crescentus, high-molecular-weight eukaryotic DNA, and RNA were readily degraded (see Fig. S1 in the supplemental material). The homology to nucleases that have already been characterized, such as Vvn, strongly suggested the requirement of divalent ions for the function of the enzyme. Accordingly, no DNA degradation was observed when only Tris buffer was used in the sample or EDTA was added to the reaction mixture. DNA degradation occurred upon the addition of Mg2+, and significant enzyme activity was also observed with Mn2+. In contrast, supplementation of the reaction mixture with Ca2+, Fe2+, Zn2+, Ni2+, or Cu2+ failed to support DNA degradation or did so only weakly (Fig. 2; also data not shown). Shewanella EndA is not thermostable and was inactivated after heating to 70°C for 10 min. Treatment with DTT also rapidly inactivated the enzyme. According to the definition of 1 U of enzyme activity as the amount of MBP-EndA required to completely degrade 1 μg of the pBluescript vector within 10 min at 30°C in a reaction buffer supplemented with 2.5 mM Mg2+, the specific activity of purified MBP-EndA was as high as 9,100 U/mg.
Fig 2.

Purification and in vitro activity of MPB-EndA. (A) Polyacrylamide gel electrophoresis of proteins after enrichment using amylose resin. Unfused MBP was produced and enriched in parallel (center lane). MBP-EndA migrated at a position corresponding to a molecular mass of about 70 kDa, in close agreement with the estimated mass of 73.1 kDa. A single major contaminating band (marked by an asterisk), likely representing MBP (42 kDa), was visible after enrichment. (B) Activity of highly enriched MBP-EndA on the purified vector pBluescript. The activity was determined by the loss of fluorescence of DNA-bound GelRed nucleic acid stain due to DNA degradation. Several cofactors (Mg2+, Mn2+, Ca2+) were tested, and control assays were carried out either with no cofactor, after the addition of DTT, after heat inactivation of MBP-EndA, or with MBP that was analogously produced and purified. Only the addition of Mg2+ or Mn2+ as a cofactor to EndA resulted in rapid degradation of DNA.
Characterization of EndA in vivo activity.
To determine whether EndA plays a role in in vivo degradation of eDNA in S. oneidensis MR-1 and whether the enzyme occurs in the medium supernatant, we generated a markerless in-frame deletion in endA (ΔendA). Cell-free supernatants of cultures obtained from wild-type and ΔendA strains grown to exponential phase were tested for nucleolytic activity. In the absence of endA, no degradation of a defined linear DNA fragment was observed (Fig. 3C). Assays with washed cells in which protein synthesis was arrested by the addition of chloramphenicol revealed that DNA degradation occurred exclusively in the medium supernatant, not within the cell fraction (Fig. 3C). This observation strongly indicates that active EndA is released into the supernatant and is not localized in the periplasm or associated with the cell surface. The apparent absence of nucleolytic activity upon deletion of endA was a surprising finding, since at least two other Shewanella extracellular nucleases, ExeM and ExeS, were still present. Accordingly, supernatants of ΔendA mutant cultures were found to contain >2-fold and 4-fold higher concentrations of eDNA after 72 h in a complex medium and mineral medium, respectively (Fig. 3B). Reintegration of endA retained the wild-type phenotype, and overexpression of endA resulted in increased nuclease activity and, correspondingly, lower concentrations of eDNA in medium supernatants (see Fig. S2 in the supplemental material).
Fig 3.

In vivo activity of EndA in medium supernatants. (A) Flocculation phenotype of ΔendA mutants. Displayed are top views of cultures of the indicated strains grown for 72 h in M1 mineral medium. ΔendA mutants form aggregates (center) that are readily dispersed upon addition of DNase I (right). Heat-inactivated DNase I had no effect. Similar flocculation also occurred in complex LB medium. (B) Amounts of eDNA in medium supernatants of wild-type (filled bars) and ΔendA mutant (shaded bars) cultures grown in LB (left) or M1 (right) medium. Error bars, standard deviations. (C) Nuclease activities in medium supernatants of the wild type and the ΔendA mutant. The cultures were grown to exponential phase in LB medium, and production of new protein was blocked by the addition of chloramphenicol. Subsequently, an 833-bp DNA fragment was added directly to the supernatants of the cultures (SN) and to the supernatants of cultures in which the cell sediment had been washed prior to the addition of the PCR fragment (C). After the indicated times, aliquots were removed and analyzed by agarose gel electrophoresis. DNA degradation was observed only in the supernatants of unwashed wild-type cultures, strongly indicating that EndA is the main nuclease in the supernatant and likely is not associated with the cell envelope.
During growth experiments in mineral and complex media, we noticed that ΔendA mutants formed large cell aggregates after prolonged incubation (Fig. 3A). Such extensive aggregation was observed only with ΔendA mutants and never occurred in cultures of wild-type cells or of exeM and/or exeS mutants. DNase I treatment resulted in rapid dissolution of the aggregates, demonstrating that eDNA was the major cohesive factor. Although eDNA accumulation and cell aggregation were observed mainly during later stages of cultivation, qRT-PCR revealed that endA expression peaks during the early- and mid-exponential phases and is downregulated during stationary phase (Fig. 4A). phoA in the same operon is regulated similarly (see Fig. S3 in the supplemental material).
Fig 4.
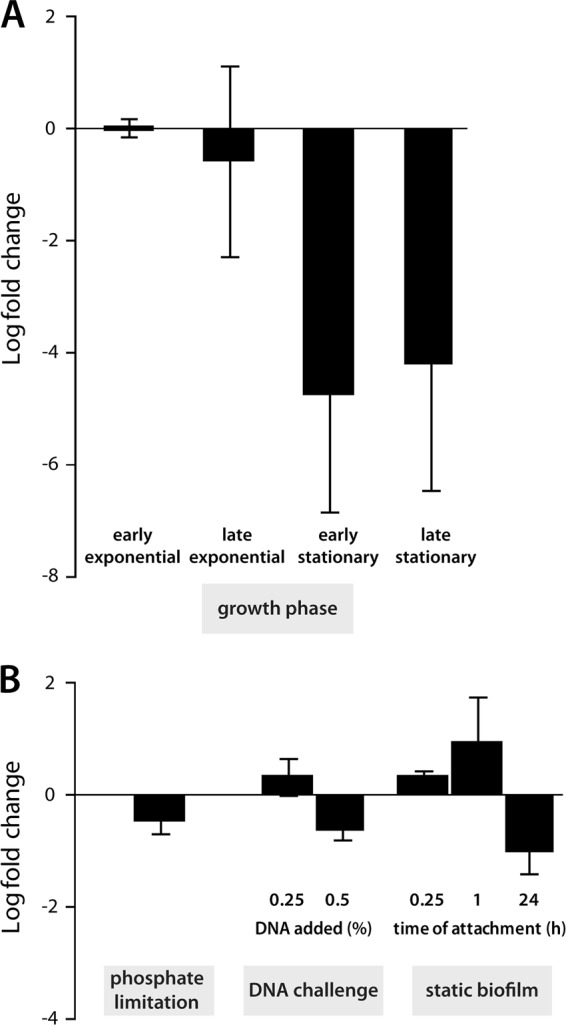
Regulation of endA. endA transcript levels were determined by qRT-PCR. (A) Growth-dependent regulation of endA. Displayed is the regulation of cells in the late-exponential-growth phase (OD600, 2.0) and in the early- and late-stationary-growth phases (OD600, 4.0 and 6.0) compared to that in the early-exponential-growth phase in LB medium (OD600, 0.6). (B) Regulation of endA in response to different environmental stimuli. (Left) Regulation during growth on DNA compared to growth on NaH2PO4; (center) regulation of endA 30 min after the addition of sublethal amounts of eDNA to exponentially growing cultures compared to nonchallenged cultures; (right) differential regulation of endA in planktonic cells and surface-associated cells harvested from static biofilm cultures at the indicated time points. Error bars, standard deviations.
Taken together, the results demonstrated that Shewanella EndA is an active nuclease that is likely transported into the medium supernatant. In addition, it is the dominant extracellular nuclease; it is crucial for the degradation of eDNA in S. oneidensis MR-1 under planktonic conditions and might prevent its accumulation to high levels. Thus, we further determined whether the nuclease might also confer short-term protection against the detrimental effects of eDNA due to its chelating properties (22, 40). To this end, we constructed wild-type and mutant strains constitutively expressing the lux operon of Photorhabdus luminescens from a plasmid. The loss of luminescence upon challenge with high concentrations of DNA was measured as a function of cell viability. No significant difference between the wild type and the mutant occurred at any of the DNA concentrations tested (data not shown), demonstrating that EndA does not protect cells from rapidly increasing concentrations of DNA. endA also was not observed to be significantly regulated when exponentially growing cells were challenged by the addition of sublethal amounts of DNA (Fig. 4).
EndA, but not PhoA, is required for the use of DNA as a phosphate source.
The absence of extracellular DNA degradation in the ΔendA mutant and the coexpression with a gene encoding a putative extracellular alkaline phosphatase strongly indicated that EndA is involved in, or even required for, the utilization of eDNA as a nutrient. We therefore determined the abilities of a ΔendA strain and a ΔphoA strain to grow when high-molecular-weight DNA was the only available source of phosphorus (Fig. 5A). While the wild type was able to grow using salmon sperm DNA in the absence of NaH2PO4, as has been reported previously, no growth could be observed with a ΔendA mutant. Accordingly, the eDNA was completely degraded in wild-type cultures, while it was still present in ΔendA cultures after 18 h (Fig. 5B). The wild type and the ΔendA mutant grew similarly well when NaH2PO4 was available and also during the initial stages of growth, when residual traces of phosphate were still available (see Fig. S4 in the supplemental material). Later stages of growth were harder to compare due to the clumping phenotype of the ΔendA mutant. Thus, the activity of EndA is critical for the utilization of eDNA as a sole source of phosphate. Reintegration of endA restored the capacity of the cells to grow on eDNA (see Fig. S5 in the supplemental material). In contrast to the ΔendA mutant, a ΔphoA mutant did not display a significantly decreased growth rate on eDNA as a phosphate source (Fig. 5C). To determine whether PhoA is an active extracellular phosphatase, we measured the corresponding activities in cell-free supernatants of exponentially growing cultures of the wild type and a phoA mutant (Fig. 5D). The phosphatase activity in supernatants of a phoA mutant was decreased to <40% of that in wild-type supernatants. Thus, PhoA is an active extracellular phosphatase that contributes significantly to the phosphatase activity in medium supernatants. However, under the conditions tested, substantial residual phosphatase activity remains, which is sufficient to release enough phosphate to enable growth on DNA.
Fig 5.

Contributions of EndA and PhoA to aerobic growth on eDNA as the sole source of phosphate. (A) Contribution of EndA to growth on eDNA. The growth of the wild type (filled squares) and the ΔendA mutant (shaded triangles) was followed for 62 h in M1 mineral medium supplemented with either 0.86 mM NaH2PO4 (dotted lines), salmon sperm DNA (0.5 g · liter−1) (solid lines), or no source of phosphorus (dashed lines). Error bars, standard deviations. (B) Degradation of eDNA during growth. Displayed is an agarose gel separation of added salmon sperm DNA after 18 h of incubation in plain M1 medium (control), with the wild type, and with a ΔendA mutant. In the absence of EndA, little DNA degradation occurred. (C) Contribution of PhoA to growth on eDNA as a sole source of phosphorus. The growth of the wild type (filled squares) and the ΔphoA mutant (shaded triangles) was followed for 62 h in M1 mineral medium supplemented with either 0.86 mM NaH2PO4 (dotted lines), salmon sperm DNA (0.5 g · liter−1) (solid lines), or no source of phosphorus (dashed lines). The utilization of eDNA as a source of phosphorus was not significantly affected in the ΔphoA mutant. (D) Activity of PhoA in medium supernatants. Phosphatase activity in M1 medium supernatants was determined for wild-type (filled triangles) and ΔphoA mutant (shaded squares) cultures. The graph displays the formation of 4-nitrophenol from p-nitrophenyl phosphate over time. Supernatants of wild-type cultures exhibit significantly higher phosphatase activity than supernatants of ΔphoA mutant cultures. Error bars, standard deviations.
The crucial role of EndA in the utilization of eDNA as a phosphate source and the organization of the gene in an operon together with phoA, encoding an active phosphatase, strongly suggested that the operon might be upregulated or specifically induced during phosphostarvation, as has been observed for exeM and exeS. To determine whether endA is regulated in response to phosphate levels, we quantified the corresponding transcript levels by qRT-PCR on total RNA prepared from cells growing in mineral medium supplemented with either NaH2PO4 or DNA as a source of phosphorus. Notably, the endA transcript levels under the two conditions were not significantly different (Fig. 4).
Role of EndA in biofilm formation.
We have demonstrated previously that eDNA is involved in mediating cell-cell and cell-surface interactions in Shewanella, both of which are affected by nucleolytic activity (23). These studies provided evidence that two other extracellular nucleases, ExeM and ExeS, influence biofilm formation under static conditions. Membrane-associated ExeM is also involved in structure formation under hydrodynamic conditions. This, together with the aggregation phenotype observed in planktonic cultures of ΔendA mutants, strongly suggested that EndA might also be involved in normal biofilm formation by S. oneidensis MR-1. To determine a potential role of this nuclease in S. oneidensis MR-1 biofilm formation, we performed static microtiter plate assays. Under these conditions, a ΔendA mutant accumulated significantly more surface-associated biomass than the wild type (Fig. 6). This phenotype was similar to that of the ΔexeS mutant, while a ΔexeM mutant displayed decreased biofilm formation. Surprisingly, addition of MBP-EndA had only a minor effect on the integrity of biofilms grown under static conditions. Also, the addition of excess Mg2+ did not improve the function of MBP-EndA (data not shown). Control experiments demonstrated that the enzyme is highly active at that concentration and readily degrades chromosomal DNA isolated from S. oneidensis MR-1 in the medium used for biofilm experiments. Also, eDNA present in planktonic culture supernatants and in the matrix of statically grown biofilms was rapidly degraded (see Fig. S6 in the supplemental material). In contrast to EndA addition, the addition of DNase I resulted in a significant loss of surface-associated cells, indicating that MBP-EndA is not active under those conditions. During static biofilm formation, the transcription levels of endA were not significantly different for planktonic and surface-associated cells, as determined by qRT-PCR (Fig. 4).
Fig 6.
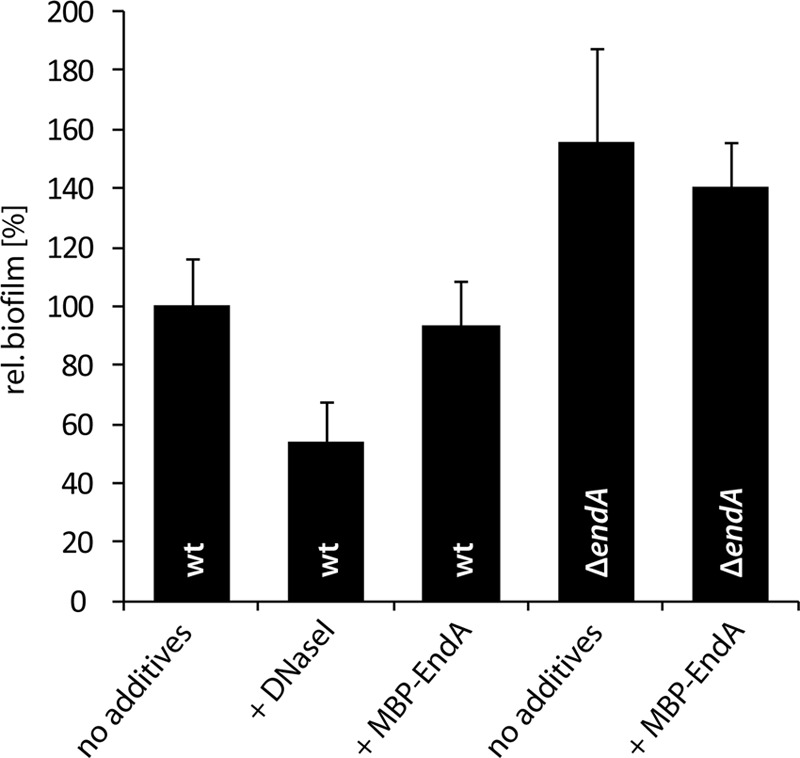
Role of EndA in biofilm formation under static conditions. Wild-type and ΔendA biofilms were grown in microtiter plates for 24 h, and DNase I or MBP-EndA was added as indicated. The values are means of three replicates. Error bars, standard deviations. rel., relative.
To enable the characterization of biofilm formation in hydrodynamic flow chambers, we generated a ΔendA mutant constitutively expressing gfp. Notably, no biofilm phenotype occurred with an endA mutant under these conditions, and the community formed was not distinguishable from that of wild-type cells (Fig. 7). Staining of eDNA by DDAO also revealed no differences between biofilms formed by the wild type versus the ΔendA mutant. In contrast, a biofilm developed by a ΔexeM mutant was characterized by tight cell-cell interactions and increased concentrations of eDNA accumulating in the matrix, as reported previously (22). Thus, EndA does not play a role in biofilm formation under hydrodynamic conditions. We hypothesized that this may be due to the fact that EndA is not produced under those conditions or that accumulation of the protein to significant levels is prevented by the constant medium flow. We therefore determined whether externally added MBP-EndA would lead to the detachment of biomass from established biofilms, as has been observed with DNase I treatment (23). However, MBP-EndA addition did not affect the integrity of flow chamber-grown biofilms, while the addition of DNase I resulted in the rapid release of large amounts of biomass (Fig. 7). The inability to disperse biofilms under static or hydrodynamic conditions and the lack of a significant biofilm phenotype under hydrodynamic conditions upon loss of endA strongly indicated that this nuclease only has a minor role, if any, in biofilm formation.
Fig 7.

Role of EndA in biofilm formation under hydrodynamic conditions. (A) (Left) The formation of biofilms by Gfp-tagged wild-type and ΔendA and ΔexeM nuclease mutant strains under hydrodynamic conditions was monitored by CLSM after 48 h of incubation. (Center) eDNA was then visualized by DDAO staining. (Right) The corresponding overlays are displayed. (B) A wild-type biofilm formed after 24 h in flow chambers was treated with MBP-EndA (center) or DNase I (right) for 45 min. The lateral edge of each micrograph measures 250 μm.
DISCUSSION
In this study, we identified and characterized the role of the extracellular nuclease EndA in S. oneidensis MR-1. This protein is highly homologous to other bacterial nucleases, such as V. vulnificus Vvn, E. coli EndA, V. cholerae Dns, and Aeromonas hydrophila Dns. All of these enzymes are exported from the cytoplasm and either remain in the periplasm, as do Vvn and E. coli EndA, or are released into the medium, as do the Dns enzymes (20, 32, 62). Our results strongly suggest that Shewanella EndA, like Dns, is an extracellular enzyme, but release through vesiculation or cell lysis cannot be ruled out (23, 24). EndA and its orthologs belong to the so-called ββα-Me superfamily, which is based on a conserved structural motif, a β-hairpin followed by an α-helix, and typically a single metal ion is crucial for function (63). Two enzymes of this class have been crystallized, and their structures have been determined (2, 36). Sequence alignments (Fig. 8) revealed that S. oneidensis MR-1 EndA harbors all the highly conserved amino acid residues that were previously identified as critical for the production of a functional enzyme. Most importantly, eight cysteine residues that form four disulfide bonds required for proper folding are present. Thus, these endonucleases are active only under oxidizing conditions, which might protect the cells from nuclease activity before the proteins are exported from the cytoplasmic space (36, 62). Accordingly, we have shown that the creation of a reducing environment by the addition of DTT readily inactivates S. oneidensis EndA. Also present in S. oneidensis EndA are the aspartic acid and glutamic acid residues that are involved in binding a catalytic divalent metal ion (36). Mg2+, Ca2+, and Zn2+ have been described as preferred catalytic metal ions of nucleases of that class (63). In this study, we have demonstrated that, in addition to Mg2+, not Ca2+ but Mn2+ supports the enzyme's function. For dissimilatory metal ion-reducing bacteria, such as Shewanella species, Mn2+ may be an abundant ion produced by bacterial reduction of manganese oxides and thus may be available in the organism's natural environments. Thus, for Shewanella and possibly other species, Mn2+ might have become a suitable cofactor for EndA. Nucleases homologous to EndA are nonspecific and degrade both DNA and RNA (62). Our results also indicate that both types of nucleic acids from various sources are subject to rapid degradation by S. oneidensis EndA.
Fig 8.

Sequence alignments of S. oneidensis MR-1 EndA with known nucleases. endA_S.on, S. oneidensis MR-1 EndA; Vvn_V.vul, V. vulnificus Vvn; endA_E.col, E. coli EndA; Dns_V.cho, V. cholerae Dns; Dns_A.hyd, A. hydrophila Dns. Areas of significant similarity are highlighted in gray and black. The gradient indicates the degree of conservation, from black (fully conserved) to light grey (less well conserved). The cysteine residues required for disulfide bond formation are highlighted in red, and residues involved in coordinating the metal cofactor are highlighted in green. The putative signal sequence of EndA is highlighted in yellow. The amino acid numbering of the longest peptide (Dns_A.hyd) is used, and every 10th position is indicated by either a number or an asterisk.
Extracellular DNA (eDNA) has been demonstrated to occur in many natural environments, sometimes at surprisingly high concentrations, as high as 20 mg · g−1, as determined for activated sludge in wastewater treatment plants (46). Substantial amounts of eDNA have also been observed in marine sediments, in which it may represent the major source of phosphorus (14–17). Marine sediments are typical environments from which dissimilatory metal ion-reducing bacteria, such as Shewanella species, can be isolated. Thus, the ability to utilize eDNA as a nutrient may be a crucial factor for the successful propagation of members of that genus in its natural environment. Our study has demonstrated that EndA is required in order for S. oneidensis to scavenge eDNA as a nutrient and is most highly expressed during exponential growth. This expression pattern might indicate that in the natural environment, phosphate might be limited and growth may be supported by eDNA degradation; however, we did not observe significant growth differences under the conditions tested. Some phenotypes occurring upon the loss of endA, such as eDNA accumulation and cell aggregation, were observed in later stages of cultivation, suggesting that a basal expression level is also required during the stationary phase. It might also be possible that EndA is stable in the supernatant over longer periods.
In Shewanella, endA is coexpressed with an extracellular phosphatase, phoA. Although PhoA contributes significantly to the phosphatase activity in culture supernatants, that enzyme, unlike EndA, is not essential for growth on eDNA. Unexpectedly, the operon including endA and phoA is not specifically upregulated upon phosphostarvation. Accordingly, in complex LB medium, the contribution of PhoA to the total phosphatase activity in the supernatant (>90% [data not shown]) was significantly higher than that in mineral medium, suggesting that additional phosphatases are activated under low-phosphate conditions. Several candidates for such extracellular enzymes can be readily identified in S. oneidensis MR-1, and the resulting activity is likely sufficient to scavenge enough phosphate from eDNA to support growth (48). The observation that endA is not regulated in response to a lack of phosphorus strongly indicates that EndA has another function in addition to the exploitation of eDNA as a nutrient source. Such a function might be to prevent the accumulation of high, and potentially lethal, eDNA concentrations in planktonic cultures. A further role that has been assigned to nucleases orthologous to EndA, such as V. cholerae Dns, V. vulnificus Vvn, and E. coli EndA, is to limit the uptake of eDNA by the cell (7, 20, 62). The observed endA expression pattern would suggest that such uptake is prevented during exponential growth. However, naturally occurring transformation of Shewanella has not been demonstrated directly so far, and our group is currently determining whether the loss of endA enhances the natural competence of S. oneidensis.
Another function of eDNA has become evident by a study by Whitchurch and coworkers in 2002, in which it was demonstrated that this molecule is an important factor in mediating cell-cell as well as cell-surface interactions in Pseudomonas aeruginosa biofilms (61). In the years that followed, similar observations were made for numerous other bacterial species, including S. oneidensis MR-1 (23). However, the exact role of eDNA in mediating such interactions is still unknown. Several studies have indicated that the molecule interacts directly or indirectly with the bacterial cell surface (12, 25, 60). This results in a change in cell surface properties, which might influence the interaction with other components of the biofilm matrix, such as polysaccharides, or the substratum (5, 12, 13, 25). A different mechanism for the effect of eDNA on biofilm formation has recently been described for P. aeruginosa. It was demonstrated that DNA effectively complexes divalent cations, such as Mg2+ and Ca2+, resulting in a cation-depleted cellular environment, which, in turn, is sensed by the cell and induces extracellular polysaccharide (EPS) production and antibiotic resistance (40, 42). The significance of eDNA in bacterial biofilm formation by many species strongly suggested that extracellular nucleases might play an important role in that process. Indeed, recent studies have demonstrated that nuclease activity impacts biofilm stability and integrity. For both Staphylococcus and Neisseria species, it was observed that the absence of an extracellular thermonuclease results in thicker biofilms characterized by elevated amounts of eDNA. Thus, the enzyme likely is involved in mediating the detachment of cells from the biofilm (38, 52). Similarly, an extracellular nuclease produced by Bacillus licheniformis has been shown to enable the dispersal of multispecies biofilms (45). In a previous study, we have characterized the roles of two extracellular nucleases, ExeM and ExeS, in Shewanella. Both are dispensable for the utilization of eDNA as a nutrient; however, membrane-associated ExeM in particular is required for normal biofilm formation under both hydrodynamic and static conditions (22). Although EndA was shown to prevent aggregation during planktonic growth, our study revealed that this enzyme does not play a role in Shewanella biofilm formation under hydrodynamic conditions. In addition, endA deletion did not result in differences in biofilm development or eDNA accumulation in the matrix, and external addition of MBP-EndA did not release biomass from biofilms under static conditions. The dominant activity of EndA in medium supernatants was a rather unexpected finding, which poses intriguing questions about why EndA does not degrade the eDNA required for cell-cell interactions in Shewanella biofilms. Since DNase I readily releases biomass from Shewanella biofilms, it is unlikely that EndA lacks access to this DNA, even as an MBP-EndA fusion. EndA might become inactive in biofilms, e.g., as a result of reducing conditions in anaerobic biofilm areas or because of the loss of the metal cofactor. However, in that case, we would still expect successive loss of biomass from the upper aerobic layers of the biofilms, which was not observed. Another possibility is that the eDNA mediating cohesive functions is protected from degradation by EndA. This may, for example, be caused by methylation of the corresponding DNA fragment, as has recently been demonstrated for the Neisseria gonorrhoeae thermonuclease Nuc (52). Notably, the majority of eDNA present in medium supernatants or obtained from the biofilm matrix was readily degraded by MBP-EndA. Thus, it will be interesting to characterize the eDNA directly mediating cohesive functions in Shewanella, which may represent only a small fraction of the overall eDNA, and to determine whether this DNA is modified or is not accessible to EndA.
Interestingly, a very recent study on Vibrio cholerae revealed that two nucleases, Dns and Xds, the orthologs of Shewanella EndA and ExeM, are both involved in utilizing eDNA as a nutrient and both affect biofilm formation (51), indicating a joint activity of the two enzymes. In sharp contrast, our results strongly indicate that the set of extracellular nucleases of Shewanella species has distinct functions. EndA is required for the utilization of eDNA as a source of phosphorus and might prevent the accumulation of eDNA to detrimental concentrations. Further functions, e.g., in transformation, are conceivable. In contrast, surface-associated ExeM mediates biofilm formation, particularly under hydrodynamic conditions, and might enable biofilm dispersal (22, 55, 57). The role of a third extracellular nuclease, ExeS, is less well defined. Like the other two nucleases, ExeS affects biofilm formation under static conditions. Further studies by our group will address the question of the interplay between the three nucleases and the nature of eDNA mediating cell-cell and cell-surface interactions. With regard to the important role of eDNA in biofilm formation by many bacteria and the wide distribution of extracellular nucleases, we expect that similar specific nucleolytic functions will be observed in other species as well.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the groups of Erhard Bremer and Martin Thanbichler for kindly providing Bacillus subtilis and Caulobacter crescentus chromosomal DNAs.
The study was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG, TH831/3-1) and the Max-Planck-Gesellschaft.
Footnotes
Published ahead of print 6 April 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Aiba H, Adhya S, de Crombrugghe B. 1981. Evidence for two functional gal promoters in intact Escherichia coli cells. J. Biol. Chem. 256:11905–11910 [PubMed] [Google Scholar]
- 2. Altermark B, Smalas AO, Willassen NP, Helland R. 2006. The structure of Vibrio cholerae extracellular endonuclease I reveals the presence of a buried chloride ion. Acta Crystallogr. D Biol. Crystallogr. 62:1387–1391 [DOI] [PubMed] [Google Scholar]
- 3. Baldwin DS. 1998. Reactive “organic” phosphorus revisited. Water Res. 32:2265–2270 [Google Scholar]
- 4. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340:783–795 [DOI] [PubMed] [Google Scholar]
- 5. Berne C, Kysela DT, Brun YV. 2010. A bacterial extracellular DNA inhibits settling of motile progeny cells within a biofilm. Mol. Microbiol. 77:815–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bjerrum CJ, Canfield DE. 2002. Ocean productivity before about 1.9 Gyr ago limited by phosphorus adsorption onto iron oxides. Nature 417:159–162 [DOI] [PubMed] [Google Scholar]
- 7. Blokesch M, Schoolnik GK. 2008. The extracellular nuclease Dns and its role in natural transformation of Vibrio cholerae. J. Bacteriol. 190:7232–7240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bretschger O, et al. 2007. Current production and metal oxide reduction by Shewanella oneidensis MR-1 wild type and mutants. Appl. Environ. Microbiol. 73:7003–7012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown RN, Romine MF, Schepmoes AA, Smith RD, Lipton MS. 2010. Mapping the subcellular proteome of Shewanella oneidensis MR-1 using Sarkosyl-based fractionation and LC-MS/MS protein identification. J. Proteome Res. 9:4454–4463 [DOI] [PubMed] [Google Scholar]
- 10. Chang MC, Chang SY, Chen SL, Chuang SM. 1992. Cloning and expression in Escherichia coli of the gene encoding an extracellular deoxyribonuclease (DNase) from Aeromonas hydrophila. Gene 122:175–180 [DOI] [PubMed] [Google Scholar]
- 11. Choi KH, et al. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2:443–448 [DOI] [PubMed] [Google Scholar]
- 12. Das T, Sharma PK, Busscher HJ, van der Mei HC, Krom BP. 2010. Role of extracellular DNA in initial bacterial adhesion and surface aggregation. Appl. Environ. Microbiol. 76:3405–3408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Das T, Sharma PK, Krom BP, van der Mei HC, Busscher HJ. 2011. Role of eDNA on the adhesion forces between Streptococcus mutans and substratum surfaces: influence of ionic strength and substratum hydrophobicity. Langmuir 27:10113–10118 [DOI] [PubMed] [Google Scholar]
- 14. Dell'Anno A, Corinaldesi C. 2004. Degradation and turnover of extracellular DNA in marine sediments: ecological and methodological considerations. Appl. Environ. Microbiol. 70:4384–4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dell'Anno A, Danovaro R. 2005. Extracellular DNA plays a key role in deep-sea ecosystem functioning. Science 309:2179. [DOI] [PubMed] [Google Scholar]
- 16. Dell'Anno A, Fabiano M, Duineveld GCA, Kok A, Danovaro R. 1998. Nucleic acid (DNA, RNA) quantification and RNA/DNA ratio determination in marine sediments: comparison of spectrophotometric, fluorometric, and high-performance liquid chromatography methods and estimation of detrital DNA. Appl. Environ. Microbiol. 64:3238–3245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dell'Anno A, Fabiano M, Mei ML, Danovaro R. 1999. Pelagic-benthic coupling of nucleic acids in an abyssal location of the northeastern Atlantic Ocean. Appl. Environ. Microbiol. 65:4451–4457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dominiak DM, Nielsen JL, Nielsen PH. 2011. Extracellular DNA is abundant and important for microcolony strength in mixed microbial biofilms. Environ. Microbiol. 13:710–721 [DOI] [PubMed] [Google Scholar]
- 19. Flemming HC, Wingender J. 2010. The biofilm matrix. Nat. Rev. Microbiol. 8:623–633 [DOI] [PubMed] [Google Scholar]
- 20. Focareta T, Manning PA. 1987. Extracellular proteins of Vibrio cholerae: molecular cloning, nucleotide sequence and characterization of the deoxyribonuclease (DNase) together with its periplasmic localization in Escherichia coli K-12. Gene 53:31–40 [DOI] [PubMed] [Google Scholar]
- 21. Fredrickson JK, et al. 2008. Towards environmental systems biology of Shewanella. Nat. Rev. Microbiol. 6:592–603 [DOI] [PubMed] [Google Scholar]
- 22. Gödeke J, Heun M, Bubendorfer S, Paul K, Thormann KM. 2011. Roles of two Shewanella oneidensis MR-1 extracellular endonucleases. Appl. Environ. Microbiol. 77:5342–5351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gödeke J, Paul K, Lassak J, Thormann KM. 2011. Phage-induced lysis enhances biofilm formation in Shewanella oneidensis MR-1. ISME J. 5:613–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gorby Y, et al. 2008. Redox-reactive membrane vesicles produced by Shewanella. Geobiology 6:232–241 [DOI] [PubMed] [Google Scholar]
- 25. Harmsen M, Lappann M, Knochel S, Molin S. 2010. The role of extracellular DNA during biofilm formation by Listeria monocytogenes. Appl. Environ. Microbiol. 76:2271–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hau HH, Gralnick JA. 2007. Ecology and biotechnology of the genus Shewanella. Annu. Rev. Microbiol. 61:237–258 [DOI] [PubMed] [Google Scholar]
- 27. Heidelberg JF, et al. 2002. Genome sequence of the dissimilatory metal ion-reducing bacterium Shewanella oneidensis. Nat. Biotechnol. 20:1118–1123 [DOI] [PubMed] [Google Scholar]
- 28. Hilpert K, Hancock RE. 2007. Use of luminescent bacteria for rapid screening and characterization of short cationic antimicrobial peptides synthesized on cellulose using peptide array technology. Nat. Protoc. 2:1652–1660 [DOI] [PubMed] [Google Scholar]
- 29. Höfle MG, Brettar I. 1996. Genotyping of heterotrophic bacteria from the Central Baltic Sea by use of low-molecular-weight RNA profiles. Appl. Environ. Microbiol. 62:1383–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hudson JJ, Taylor WD, Schindler DW. 2000. Phosphate concentrations in lakes. Nature 406:54–56 [DOI] [PubMed] [Google Scholar]
- 31. Inoue H, Nojima H, Okayama H. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 96:23–28 [DOI] [PubMed] [Google Scholar]
- 32. Jekel M, Wackernagel W. 1995. The periplasmic endonuclease I of Escherichia coli has amino-acid sequence homology to the extracellular DNases of Vibrio cholerae and Aeromonas hydrophila. Gene 154:55–59 [DOI] [PubMed] [Google Scholar]
- 33. Karl DM, Yanagi K. 1997. Partial characterization of the dissolved organic phosphorus pool in the oligotrophic North Pacific Ocean. Limnol. Oceanogr. 42:1398–1405 [Google Scholar]
- 34. Kovach ME, et al. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176 [DOI] [PubMed] [Google Scholar]
- 35. Lassak J, Henche AL, Binnenkade L, Thormann KM. 2010. ArcS, the cognate sensor kinase in an atypical Arc system of Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 76:3263–3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li CL, et al. 2003. DNA binding and cleavage by the periplasmic nuclease Vvn: a novel structure with a known active site. EMBO J. 22:4014–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Logan BE. 2009. Exoelectrogenic bacteria that power microbial fuel cells. Nat. Rev. Microbiol. 7:375–381 [DOI] [PubMed] [Google Scholar]
- 38. Mann EE, et al. 2009. Modulation of eDNA release and degradation affects Staphylococcus aureus biofilm maturation. PLoS One 4:e5822 doi:10.1371/journal.pone.0005822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170:2575–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mulcahy H, Charron-Mazenod L, Lewenza S. 2008. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 4:e1000213 doi:10.1371/journal.ppat.1000213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mulcahy H, Charron-Mazenod L, Lewenza S. 2010. Pseudomonas aeruginosa produces an extracellular deoxyribonuclease that is required for utilization of DNA as a nutrient source. Environ. Microbiol. 12:1621–1629 [DOI] [PubMed] [Google Scholar]
- 42. Mulcahy H, Lewenza S. 2011. Magnesium limitation is an environmental trigger of the Pseudomonas aeruginosa biofilm lifestyle. PLoS One 6:e23307 doi:10.1371/journal.pone.0023307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Myers CR, Nealson KH. 1988. Bacterial manganese reduction and growth with manganese oxide as the sole electron acceptor. Science 240:1319–1321 [DOI] [PubMed] [Google Scholar]
- 44. Nealson KH, Scott J. 2003. Ecophysiology of the genus Shewanella, p 1133–1151 In Dworkin M. (ed), The prokaryotes: an evolving electronic resource for the microbial community. Springer-NY, LLC, New York, NY [Google Scholar]
- 45. Nijland R, Hall MJ, Burgess JG. 2010. Dispersal of biofilms by secreted, matrix degrading, bacterial DNase. PLoS One 5:e15668 doi:10.1371/journal.pone.0015668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Palmgren R, Nielsen PH. 1996. Accumulation of DNA in the exopolymeric matrix of activated sludge and bacterial cultures. Water Sci. Technol. 34:233–240 [Google Scholar]
- 47. Paulick A, et al. 2009. Two different stator systems drive a single polar flagellum in Shewanella oneidensis MR-1. Mol. Microbiol. 71:836–850 [DOI] [PubMed] [Google Scholar]
- 48. Pinchuk GE, et al. 2008. Utilization of DNA as a sole source of phosphorus, carbon, and energy by Shewanella spp.: ecological and physiological implications for dissimilatory metal reduction. Appl. Environ. Microbiol. 74:1198–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pospiech A, Neumann B. 1995. A versatile quick-prep of genomic DNA from gram-positive bacteria. Trends Genet. 11:217–218 [DOI] [PubMed] [Google Scholar]
- 50. Sambrook K, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 51. Seper A, et al. 2011. Extracellular nucleases and extracellular DNA play important roles in Vibrio cholerae biofilm formation. Mol. Microbiol. 82:1015–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Steichen CT, Cho C, Shao JQ, Apicella MA. 2011. The Neisseria gonorrhoeae biofilm matrix contains DNA, and an endogenous nuclease controls its incorporation. Infect. Immun. 79:1504–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Steinberger RE, Holden PA. 2005. Extracellular DNA in single- and multiple-species unsaturated biofilms. Appl. Environ. Microbiol. 71:5404–5410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tang X, et al. 2007. Profiling the membrane proteome of Shewanella oneidensis MR-1 with new affinity labeling probes. J. Proteome Res. 6:724–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Thormann KM, et al. 2006. Control of formation and cellular detachment from Shewanella oneidensis MR-1 biofilms by cyclic di-GMP. J. Bacteriol. 188:2681–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Thormann KM, Saville RM, Shukla S, Pelletier DA, Spormann AM. 2004. Initial phases of biofilm formation in Shewanella oneidensis MR-1. J. Bacteriol. 186:8096–8104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Thormann KM, Saville RM, Shukla S, Spormann AM. 2005. Induction of rapid detachment in Shewanella oneidensis MR-1 biofilms. J. Bacteriol. 187:1014–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tsai MS, You HL, Tang YF, Liu JW. 2008. Shewanella soft tissue infection: case report and literature review. Int. J. Infect. Dis. 12:e119–e124 [DOI] [PubMed] [Google Scholar]
- 59. Venkateswaran K, et al. 1999. Polyphasic taxonomy of the genus Shewanella and description of Shewanella oneidensis sp. nov. Int. J. Syst. Bacteriol. 49:705–724 [DOI] [PubMed] [Google Scholar]
- 60. Vilain S, Pretorius JM, Theron J, Brözel VS. 2009. DNA as an adhesin: Bacillus cereus requires extracellular DNA to form biofilms. Appl. Environ. Microbiol. 75:2861–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. 2002. Extracellular DNA required for bacterial biofilm formation. Science 295:1487. [DOI] [PubMed] [Google Scholar]
- 62. Wu SI, Lo SK, Shao CP, Tsai HW, Hor LI. 2001. Cloning and characterization of a periplasmic nuclease of Vibrio vulnificus and its role in preventing uptake of foreign DNA. Appl. Environ. Microbiol. 67:82–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang W. 2011. Nucleases: diversity of structure, function and mechanism. Q. Rev. Biophys. 44:1–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ziemke F, Brettar I, Höfle MG. 1997. Stability and diversity of the genetic structure of a Shewanella putrefaciens population in the water column of the Central Baltic. Aquat. Microb. Ecol. 13:63–74 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.