

Abstract
Pyrolysin-like proteases from hyperthermophiles are characterized by large insertions and long C-terminal extensions (CTEs). However, little is known about the roles of these extra structural elements or the maturation of these enzymes. Here, the recombinant proform of Pyrococcus furiosus pyrolysin (Pls) and several N- and C-terminal deletion mutants were successfully expressed in Escherichia coli. Pls was converted to mature enzyme (mPls) at high temperatures via autoprocessing of both the N-terminal propeptide and the C-terminal portion of the long CTE, indicating that the long CTE actually consists of the C-terminal propeptide and the C-terminal extension (CTEm), which remains attached to the catalytic domain in the mature enzyme. Although the N-terminal propeptide deletion mutant PlsΔN displayed weak activity, this mutant was highly susceptible to autoproteolysis and/or thermogenic hydrolysis. The N-terminal propeptide acts as an intramolecular chaperone to assist the folding of pyrolysin into its thermostable conformation. In contrast, the C-terminal propeptide deletion mutant PlsΔC199 was converted to a mature form (mPlsΔC199), which is the same size as but less stable than mPls, suggesting that the C-terminal propeptide is not essential for folding but is important for pyrolysin hyperthermostability. Characterization of the full-length (mPls) and CTEm deletion (mPlsΔC740) mature forms demonstrated that CTEm not only confers additional stability to the enzyme but also improves its catalytic efficiency for both proteineous and small synthetic peptide substrates. Our results may provide important clues about the roles of propeptides and CTEs in the adaptation of hyperthermophilic proteases to hyperthermal environments.
INTRODUCTION
Enzymes from hyperthermophiles are especially valuable for probing the stabilization mechanisms that allow proteins to function near the maximum temperature endurable by life forms (34, 36) and also offer insight into opportunities to greatly expand the reaction conditions of biocatalysis (1). The majority of extracellular proteases from hyperthermophiles belong to the superfamily of subtilisin-like serine proteases (subtilases) (37). Some of the subtilases from hyperthermophilic archaea are unusually large due to the presence of large insertions and/or long C-terminal extensions (CTE) and are grouped into the pyrolysin family of subtilases (28), including pyrolysin from Pyrococcus furiosus (35), stetterlysin from Thermococcus stetteri (36), and STABLE protease from Staphylothermus marinus (19). Besides these biochemically characterized enzymes, an increasing number of pyrolysin-like proteases have been identified in the genomes of hyperthermophiles, including T. onnurineus, T. gammatolerans, Thermococcus sp. AM4, T. barophilus, Thermococcus sp. 4557, P. yayanosii, and P. woesei.
P. furiosus, which was isolated from geothermally heated marine sediment (10), is highly proteolytic and requires peptides for growth (2, 5, 9, 30). Extensive studies conducted by de Vos and coworkers (8, 9, 35) indicate that pyrolysin from P. furiosus is a cell envelope-associated protease with a half-life of 4 h at 100°C, representing one of the most thermostable proteases. The precursor of pyrolysin (1,398 residues) contains a signal peptide (26 residues), an N-terminal propeptide (123 residues), a subtilisin-like catalytic domain, and a long CTE (∼740 residues). Two glycosylated, active forms without the prepropeptide, termed HMW and LMW pyrolysins, were purified from the P. furiosus cell envelope fraction (35). In view of their identical N-terminal sequences and the generation of LMW from HMW by prolonged incubation at 95 to 100°C, it was concluded that the HMW and LMW pyrolysins are different forms of pyrolysin that are generated by C-terminal proteolytic processing, but the maturation mechanism of pyrolysin remains to be clarified (8, 9, 35). In comparison with the Bacillus subtilisins, the catalytic domain of pyrolysin contains several extra surface loops as well as an unusually large insertion (147 residues) between the catalytic residues Asp and His (35, 36). Other members of pyrolysin family have a molecular architecture similar to that of pyrolysin. Interestingly, the long CTE is not limited only to pyrolysin-like proteases but is also conserved in putative thiol proteases of P. abyssi, P. horikoshii (8, 37), Pyrococcus sp. NA2, and T. kodakaraensis KOD1. The conservation of the long CTE in proteases from hyperthermophiles implies a common function; however, its role in enzyme function is unclear. Furthermore, precursors of pyrolysin-like proteases undergo a maturation process in order to achieve functionality. This maturation process is an indispensable property of enzyme adaptations to hyperthermal challenges. Therefore, our investigations into the maturation mechanism of pyrolysin should facilitate our understanding of adaptation strategies employed by hyperthermophilic proteases.
The purpose of this study was to investigate the maturation process of P. furiosus pyrolysin and to probe the role of the long CTE in the adaptation of the enzyme to hyperthermal environments. By using a recombinant proform of pyrolysin expressed in Escherichia coli, we found that the maturation of the enzyme involved autoprocessing of both the N-propeptide and a C-terminal propeptide. Thus, the long CTE can be dissected into a C-terminal propeptide and a C-terminal extension (CTEm) that is attached to the catalytic domain of the mature enzyme. Our results suggest that both of these propeptides assist in achieving pyrolysin hyperthermostability. In addition, several enzyme deletion mutants were constructed and biochemically characterized. These studies revealed that the CTEm is important for enzyme stability and activity.
MATERIALS AND METHODS
Strains and growth conditions.
E. coli DH5α and E. coli BL21-CodonPlus(DE3)-RIL were used as the hosts for cloning and expression, respectively. Bacteria were grown at 37°C in Luria-Bertani (LB) medium supplemented with ampicillin (50 μg/ml) or kanamycin (30 μg/ml) as needed.
DNA manipulation, plasmid construction, and mutagenesis.
The genomic DNA of P. furiosus DSM 3638 (Vc1) was obtained from the American Type Culture Collection (ATCC 43587D-5). The oligonucleotide sequences of the primers used in this study are listed in Table S1 in the supplemental material. The gene encoding the pyrolysin precursor was amplified from genomic DNA by PCR using primers P1 and P2 and KOD DNA polymerase (Toyobo), and the PCR product was inserted into the KpnI-SalI sites of pUC18 to construct the plasmid pUC18-pls. The genes encoding the proform of pyrolysin (Pls), the C-terminal truncation mutants (PlsΔC199, PlsΔC362, PlsΔC463, and PlsΔC740), the C-terminal fragments (PlsC740 and PlsC541), and the N-terminal propeptide (PlsN) were amplified by PCR from the pUC18-pls template with the primer pairs listed in Table S2 in the supplemental material. To facilitate DNA manipulation, the “megaprimer” method (23) was used to prepare the gene encoding the N-terminal propeptide deletion mutant (PlsΔN). Briefly, the 5′ end of the coding sequence was amplified with primer plsΔN-F and mutagenic primer plsΔNdeI-R, which changes the NdeI site in the mature region of the pyrolysin gene without changing the amino acid sequence, and the product was used as the megaprimer. The PlsΔN gene from the pUC18-pls template was then amplified with the megaprimer and primer pls-R. The amplified genes mentioned above were inserted into pET26b or pET15b to generate expression plasmids for target proteins (see Table S2 in the supplemental material). The QuikChange site-directed mutagenesis (SDM) method (21) was employed to construct the active site mutant PlsS441A by the use of primers plsS441A-F and plsS441A-R (see Table S1 in the supplemental material). The sequences of all recombinant plasmids were confirmed by DNA sequencing.
Expression, activation, and purification.
E. coli BL21-CodonPlus(DE3)-RIL cells that contained the recombinant plasmids were cultured at 37°C until the optical density at 600 nm (OD600) reached ∼0.6. The recombinant proteins were induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside, and the cultivation continued for 4 h at 37°C. Then, the harvested cells were suspended in 50 mM sodium phosphate buffer (pH 7.5) and disrupted by sonication. After centrifugation at 13,000 × g for 10 min, the soluble and insoluble fractions were collected. The soluble PlsN fused with a His tag was subjected to affinity chromatography on a Ni2+-charged chelating Sepharose Fast Flow resin (GE Healthcare) column equilibrated with 50 mM sodium phosphate buffer (pH 7.5). After the column was washed with 50 mM sodium phosphate buffer (pH 7.5) containing 40 mM imidazole, the bound PlsN was eluted with 50 mM sodium phosphate buffer (pH 7.5) containing 200 mM imidazole and then dialyzed against 50 mM sodium phosphate buffer (pH 7.5) overnight at 4°C to remove the imidazole. For the proteins that remained in the insoluble fractions (Pls, PlsS441A, PlsΔC199, PlsΔC362, PlsΔC463, PlsΔC740, PlsC740, PlsC541, and PlsΔN), the recovered pellets were dissolved in 50 mM sodium phosphate buffer (pH 7.5) containing 6 M urea, incubated at 4°C overnight, and then subjected to centrifugation at 13,000 × g for 10 min. In some cases, the resultant supernatants were dialyzed against 50 mM sodium phosphate buffer (pH 7.5) overnight at 4°C to remove the urea and were then used as crude protein samples. The purification procedure for these proteins was similar to that used for PlsN, except that 6 M urea was included in the washing and elution buffers. The eluted fractions were dialyzed against 50 mM sodium phosphate buffer (pH 7.5) to remove imidazole and urea and then stored at 4°C until use.
To prepare the mature enzymes, crude samples of the proforms (Pls and PlsΔC199) in 50 mM sodium phosphate buffer (pH 7.5) were incubated at 95°C for 3 h to activate the enzymes. The resultant mature enzymes (mPls and mPlsΔC199) were subjected to affinity chromatography on a bacitracin-Sepharose 4B (Amersham Biosciences, Sweden) column equilibrated with 50 mM sodium phosphate buffer (pH 7.5). After washing with 50 mM sodium phosphate buffer (pH 7.5) containing 6 M urea was performed, the bound enzymes were eluted with 50 mM sodium phosphate buffer (pH 7.5) containing 6 M urea and 20% isopropanol. It should be mentioned that pyrolysin is resistant to high concentrations of urea (8, 9, 35), and the 6 M urea was included in the washing and elution buffers to reduce nonspecific protein binding to the affinity resins. Finally, the eluted fractions containing the purified enzymes were dialyzed against 50 mM sodium phosphate buffer (pH 7.5) to remove urea and isopropanol. For preparation of the mature enzyme lacking the CTE (mPlsΔC740), the crude sample of PlsΔC740 was supplemented with a roughly equal molar concentration of purified PlsC740 and was incubated at 4°C overnight in 50 mM sodium phosphate buffer (pH 7.5). Thereafter, the enzyme was activated at 95°C and purified by bacitracin affinity chromatography as described above. The enzyme solution was concentrated with a Micron YM-3 centrifugal filter (Amicon, Beverly, MA) as needed. The protein concentrations of purified protein samples were determined using the Bradford method (3). The amount of target protein in the crude samples was estimated from band intensities on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels, using bovine serum albumin (BSA) as a standard.
SDS-PAGE, activity staining, and immunoblot analysis.
SDS-PAGE was performed with the glycine-Tris (16) or Tricine-Tris buffer systems (24). The samples for conventional SDS-PAGE analysis were precipitated with 20% (wt/vol) trichloroacetic acid (TCA), washed with acetone, solubilized in a loading buffer containing 8 M urea, and then subjected to electrophoresis without prior heat treatment. For activity staining of protease, the samples were directly mixed with the loading buffer without urea. The gelatin overlay assay and gelatin-containing SDS-PAGE analysis were performed according to the method described by Blumentals et al. (2), except that the proteolytic reactions were carried out at 95°C for 2 h (gelatin overlay assay) and at 30°C for 12 h (gelatin-containing SDS-PAGE). The His tag monoclonal antibody (Novagen) was used for immunoblot analysis as described previously (4).
Enzyme activity assay.
Unless otherwise indicated, the azocaseinolytic activity of the enzyme was assayed at 95°C for 30 min in 400 μl of reaction mixture containing 0.5% (wt/vol) azocasein (Sigma) and 200 μl of enzyme sample in 50 mM sodium phosphate buffer (pH 7.5). The reaction was terminated by the addition of 400 μl of 40% (wt/vol) TCA. After incubation at room temperature for 15 min, the mixture was centrifuged at 13,000 × g for 10 min, and the absorbance of the supernatant was measured at 335 nm in a 1-cm-light-path cell. One unit (U) of activity was defined as the amount of enzyme required to increase the A335 value by 0.01 unit (U) per min. The enzymatic activity of the protease on the substrate suc-AAPK-pNA (GL Biochem Ltd., Shanghai, China) was measured at 90°C in 50 mM sodium phosphate buffer (pH 7.5) containing 0.5 mM substrate. The activity was recorded by monitoring the initial velocity of suc-AAPK-pNA hydrolysis at 410 nm in a thermostated spectrophotometer (Cintra 10e; GBC, Australia). This velocity was calculated on the basis of an extinction coefficient for p-nitroaniline of 8,480 M−1 cm−1 at 410 nm (7). One unit (U) of enzyme activity was defined as the amount of enzyme that produced 1 μmol of pNA per min under the assay conditions. With suc-AAPK-pNA as the substrate, kinetic parameters were calculated from the initial velocity of hydrolysis at 90°C in 50 mM sodium phosphate buffer (pH 7.5) with a substrate concentration range of 0.1 to 15 mM. Km and kcat values were obtained using nonlinear regression Table Curve 2D software (Jandel Scientific; version 5.0).
N-terminal sequencing and mass spectrometry.
The proteins separated by SDS-PAGE were electroblotted onto a polyvinylidene difluoride membrane. After staining with Coomassie brilliant blue R-250 was performed, the target protein bands were excised and subjected to N-terminal amino acid sequence analysis using a Procise 492 cLC peptide sequencer (Applied Biosystems). For mass spectrometry, the target bands on SDS-PAGE gel were excised and subjected to in-gel digestion with trypsin (Promega, Madison, WI). The tryptic peptides were analyzed with a matrix-assisted laser desorption ionization–time of flight mass spectrometer (MALDI-TOF MS) (Voyager-DE STR; Applied Biosystems).
RESULTS
Construction, expression, and purification of recombinant proteins.
The sequence alignment of the long CTEs of pyrolysin-like proteases revealed substantial homology, especially within the extreme C-terminal region (see Fig. S1 in the supplemental material). The extreme C-terminal region (residues 1051 to 1249) of pyrolysin was named the C4 segment (Fig. 1; see also Fig. S1 in the supplemental material). In addition, a putative prepeptidase C-terminal domain (PPC; residues 787 to 887) was found within the long CTE of pyrolysin and was named the C2 segment (Fig. 1; see also Fig. S1 in the supplemental material). The predicted PPC domain exhibits ∼25% sequence identity with the β-jelly roll domain of Tk-SP subtilisin from T. kodakaraensis KOD1 (11). The other two regions connecting the catalytic domain, namely, the PPC domain and the extreme C-terminal region, were named the C1 (residues 510 to 786) and C3 (residues 888 to 1050) segments, respectively (Fig. 1; see also Fig. S1 in the supplemental material). In order to investigate the function of the long CTE, we constructed a recombinant proform of full-length pyrolysin (Pls) and sequential C-terminal truncation mutants (PlsΔC199, PlsΔC362, PlsΔC463, and PlsΔC740) as well as C-terminal fragments (PlsC740 and PlsC541). In addition, an active site mutant (PlsS441A), the N-terminal propeptide deletion mutant (PlsΔN), and the N-terminal propeptide (PlsN) were also constructed (Fig. 1A) to probe the maturation process of pyrolysin and the roles of the propeptide in enzyme function.
Fig 1.
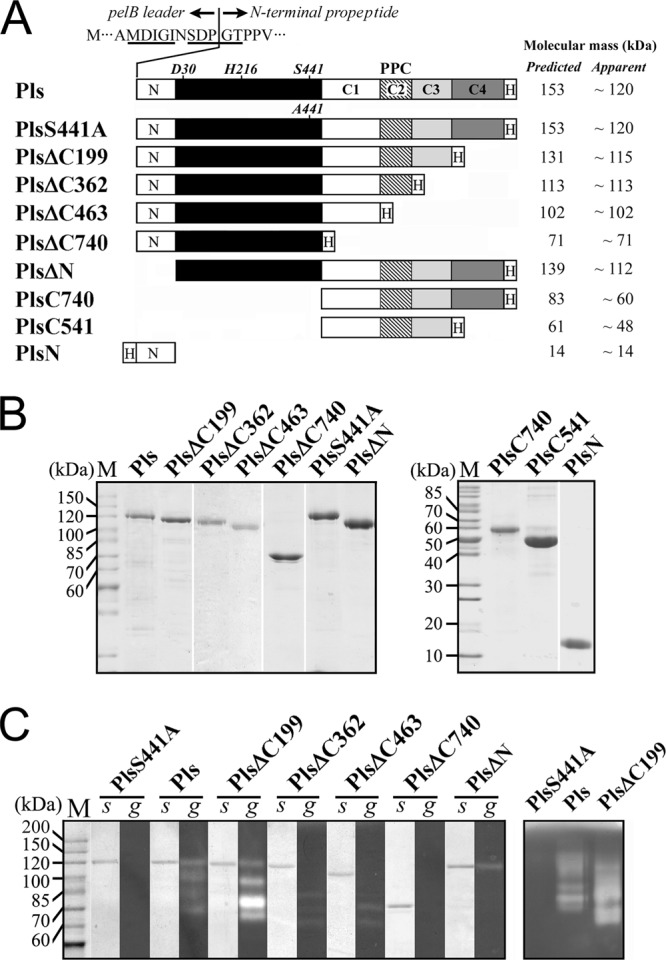
Schematic representation of the primary structures, SDS-PAGE analysis, and activity staining of purified Pls and its mutants. (A) The catalytic domain is indicated with a black box. The locations of the active-site residues are indicated. C1, C2 (PPC domain), C3, and C4 represent the four segments of the long CTE (see Fig. S1 in the supplemental material). N and H represent the N-terminal propeptide and the fused His tag, respectively. The residues around the cleavage site between the pelB signal peptide and the N-terminal propeptide of Pls, wherein the identified first five residues of recombinant Pls are underlined, are shown. The predicted molecular masses of the proteins were calculated based on their amino acid sequences, and the apparent molecular sizes were determined by SDS-PAGE analysis. (B) The purified proteins were precipitated with TCA and then subjected to SDS-PAGE analysis. (C) The purified enzyme samples were directly subjected to SDS-PAGE (s) followed by a gelatin overlay assay (g) at 95°C (left panel). Some samples were also subjected to gelatin-containing SDS-PAGE analysis at 30°C (right panel).
All of the expressed recombinant proteins, except PlsN, were found to reside mainly in the insoluble cellular fraction. Previously, native pyrolysin was reported to strongly and irreversibly bind to agarose-based resin. Thus, the enzyme was purified by preparative urea-PAGE and anion-exchange chromatography using acrylamide-based resin (8, 35). We found that, although the recombinant Pls and its mutants were able to bind to agarose-based resin, these proteins were easily purified by affinity chromatography using a Ni2+-charged chelating Sepharose Fast Flow resin column in the presence of 6 M urea. For some samples (e.g., Pls and PlsΔC199), a few minor protein bands, probably representing processed/degraded products, were detected following purification (Fig. 1B) as described below.
By N-terminal sequencing, the Pls band was found to consist of two classes of polypeptides that had MDIGI and SDPGT as the first five amino acids, indicating that the fused pelB signal peptide was cleaved from Pls at two positions by E. coli signal peptidase (Fig. 1 A). Pls was shown by SDS-PAGE analysis to have an apparent molecular size of 120 kDa (Fig. 1B), which is significantly less than its predicted molecular mass (153 kDa). This phenomenon was also observed for PlsS441A, PlsΔC199, PlsΔN, PlsC740, and PlsC541 (Fig. 1A and B). The possibility of C-terminal processing was ruled out not only by the fact that these proteins bound to the nickel column but also because they could be detected by anti-His tag immunoblot analysis (data not shown). This result implies that the proform was not fully unfolded after TCA/SDS treatment, as the partially unfolded proform migrated faster than the fully unfolded one due to a smaller radius of gyration. In contrast, the C-terminal truncation mutant that lacks the C3-C4 fragment (e.g., PlsΔC362, PlsΔC463, and PlsΔC740) displayed an apparent molecular size that was approximately the same as that calculated from its deduced amino acid sequence (Fig. 1A and B), implying that these mutants were fully unfolded. Together, these results suggest that the C3-C4 fragment of the long CTE forms a rigid and compact structure that is resistant to denaturation by urea, TCA, and SDS.
Detection of enzyme activity.
To determine whether the recombinant proteins can fold properly and exhibit activity after electrophoresis and incubation at high temperatures, the purified samples were subjected to a gelatin overlay assay at 95°C. Four proteolytic bands were resolved in the samples of Pls or PlsΔC199; the band with an apparent size of 120 kDa (Pls) or 115 kDa (PlsΔC199) corresponds to the partially unfolded proform of the enzyme (Fig. 1C). This finding indicates that, after electrophoresis, the partially unfolded proform refolded into a maturation-competent state and released proteolytic activity during the gelatin overlay assay and heat treatment. Unlike the bands for Pls and PlsΔC199, the band of the fully unfolded proforms of PlsΔC362 (113 kDa) and PlsΔC463 (102 kDa) did not exhibit proteolytic activity (Fig. 1C). Nevertheless, three proteolytic species that migrated much faster than the fully unfolded proform were detected in the PlsΔC362 and PlsΔC463 samples (Fig. 1C). To exclude the possibility that the multiple proteolytic bands resulted from the presence of contaminating E. coli proteases, we probed the proteolytic activity in purified samples with gelatin-containing SDS-PAGE analysis at 30°C. Indeed, multiple proteolytic species were detected in the Pls and PlsΔC199 samples but not in the PlsS441A sample purified using the same procedure (Fig. 1C), suggesting that the bands represented derivatives of the recombinant proteins rather than contaminating E. coli proteases. The proteolytic bands with sizes that were apparently reduced compared to those of the proforms were not clearly resolved on SDS-PAGE gels (Fig. 1C), perhaps due to the fact that the minor unfolded proforms may have folded into compact maturation-competent state(s) during enzyme preparation and/or electrophoresis. Therefore, the multiple proteolytic species detected in purified samples of Pls and its mutants are actually differently folded or processed forms of the enzymes.
Maturation process of pyrolysin.
The gelatin overlay assay revealed multiple proteolytic species in the Pls sample at both low and high temperatures. To identify the mature form of pyrolysin generated at high temperatures, Pls was subjected to heat treatment at 95°C followed by SDS-PAGE analysis. As shown in Fig. 2A, Pls was gradually converted to a 100-kDa mature form (mPls) via at least two intermediates, which were resolved in the purified sample that was incubated at 95°C for 0.5 h. One of these intermediates was detected in an unheated purified Pls sample that had been stored at 4°C for 1 day (Fig. 2A, lane 0 h) but was much less apparent in the freshly prepared sample (Fig. 1B, lane Pls). This finding is not surprising, as Pls may be activated at low temperatures (Fig. 1C, right panel). It was previously reported that the maturation of hyperthermophilic Tk-subtilisin is Ca2+ dependent (22). To examine whether the maturation of Pls is dependent on metal ions, Pls was subjected to heat treatment under chelating conditions. As shown in Fig. 2B (top panel), Pls was also converted to a 100-kDa mature form at 95°C in the presence of 5 mM EDTA, but the yield of the mature form was significantly lower than that seen in the absence of EDTA (Fig. 2A). The first five residues of mPls were identified as MYNST by N-terminal sequencing. This sequence is identical to that of native mature pyrolysin isolated from P. furiosus (35), indicating that the N-terminal propeptide had been processed. Interestingly, mPls appeared to be smaller (100 kDa) than the N-terminal propeptide deletion mutant PlsΔN (112 kDa) (Fig. 3), probably due to a C-terminal cleavage event during maturation. To test this possibility, the PlsΔC199 C4 segment deletion mutant that contained a His tag at the C terminus was subjected to heat treatment under identical conditions. This treatment generated a mature form (mPlsΔC199) with the same N-terminal end (MYNST) and apparent size as mPls (Fig. 2A and 3). Meanwhile, PlsΔN, but not mPls and mPlsΔC199, could be detected by anti-His tag immunoblot analysis (Fig. 3), suggesting that the fused His tag had been cleaved from the C terminus of the two mature forms. Since the sizes of mPls and mPlsΔC199, which also underwent C-terminal processing, were identical, cleavage occurred upstream of the C4 segment. Furthermore, the C-terminal tryptic peptide of mPls and mPlsΔC199 was identified as ENFNTLEK by MALDI-TOF MS (see Fig. S1 in the supplemental material), suggesting that the C-terminal cleavage occurred downstream of Lys1030. Therefore, the C-terminal cleavage site is likely located somewhere between Lys1030 and Leu1051 (see Fig. S1 in the supplemental material), and the C4 segment is actually a C-terminal propeptide. To distinguish the remaining C-terminal extension (C1-C3 segment) in the mature enzyme from the full-length CTE, the former was named CTEm (Fig. 2D).
Fig 2.
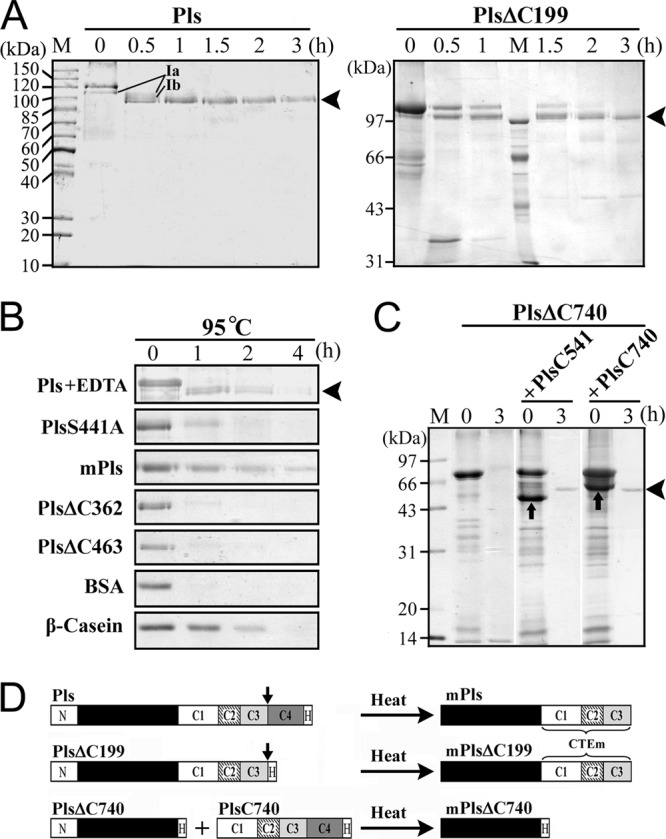
Maturation and degradation of Pls and its mutants. (A and B) Purified protein samples (10 μg/ml) were incubated at 95°C. At the time intervals indicated, samples were taken and subjected to TCA precipitation followed by SDS-PAGE analysis. The maturation process of Pls was investigated in the absence (A) and presence (B) of 5 mM EDTA. The amount of PlsΔ199 for sample loading was two times that of the other proteins. BSA and β-casein were used as controls. The two intermediates (Ia and Ib) detected during the maturation of Pls are indicated (A). The arrowheads indicate the positions of mPls (A, left panel, and B) and mPlsΔC199 (A, right panel) on the gels. (C) Crude samples of PlsΔC740 were incubated at 95°C for 3 h in the presence or absence of roughly equal molar concentrations of purified PlsC541 or PlsC740 (indicated by up arrows), followed by TCA precipitation and SDS-PAGE analysis. The arrowhead indicates the position of mPlsΔC740 on the gel. (D) Schematic representation of the maturation processes of Pls and its mutants. The down arrow indicates the possible C-terminal cleavage site. CTEm represents the C-terminal extension in mature enzymes.
Fig 3.
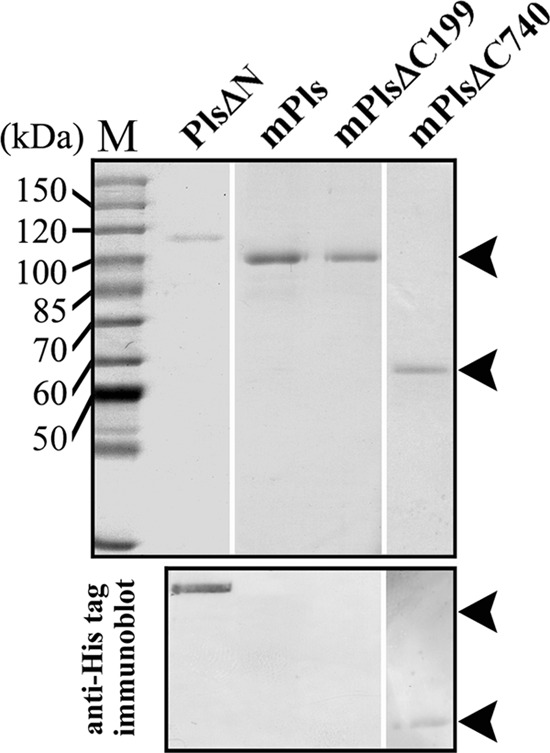
SDS-PAGE (upper panel) and immunoblot (lower panel) analyses of purified mature enzymes. Arrowheads indicate the positions of mature enzymes on the gel or membrane. PlsΔN was used as a control.
In contrast to Pls, the PlsS441A active site mutant was not converted to the mature form, as evidenced by SDS-PAGE analysis (Fig. 2B). This result indicates that the active site is involved in enzyme maturation. The PlsS441A bands were degraded completely after incubation at 95°C for 2 h (Fig. 2B). Meanwhile, other proteins that do not contain intrinsic proteolytic activity, such as BSA and β-casein, were also degraded to different extents under the same conditions (Fig. 2B). One reasonable explanation for this phenomenon is that these proteins suffered thermogenic hydrolysis of peptide bonds (6, 14, 34). Interestingly, mPls was degraded to a lesser extent than the PlsS441A proform, and ∼ 20% of mPls remained after incubation at 95°C for 4 h (Fig. 2B). Given that the degradation of active mPls at high temperatures results not only from thermogenic hydrolysis of peptide bonds but also from autoproteolysis, mPls is more stable than the proform in terms of resistance to thermogenic hydrolysis. In addition, the proform (e.g., Pls and PlsS441A) tends to aggregate more easily than mPls at higher protein concentrations (data not shown), and this finding probably reflects the structural differences between the two forms. Taken together, these results demonstrate that the proform of pyrolysin undergoes autoprocessing, including removal of both the N- and C-terminal propeptides, to yield a mature enzyme with high-temperature resistance.
N-terminal propeptide-mediated folding and inhibition of pyrolysin.
The N-terminal propeptides of bacterial subtilases usually function as intramolecular chaperones to assist the correct folding of the catalytic domain and as potent inhibitors of the mature enzyme (26, 27); however, the PlsΔN N-terminal propeptide deletion mutant displayed proteolytic activity in the gelatin overlay assay (Fig. 1C) and exhibited weak activity toward suc-AAPK-pNA (data not shown). These results imply that the propeptide is not essential for enzyme folding. Nevertheless, unlike Pls, which is capable of maturing to mPls, PlsΔN was degraded completely after incubation at 95°C for 30 min (Fig. 4A). Thus, in the absence of the N-terminal propeptide, PlsΔN seemingly folded into an active but unstable form that was highly susceptible to autoproteolysis and/or thermogenic hydrolysis at high temperatures.
Fig 4.
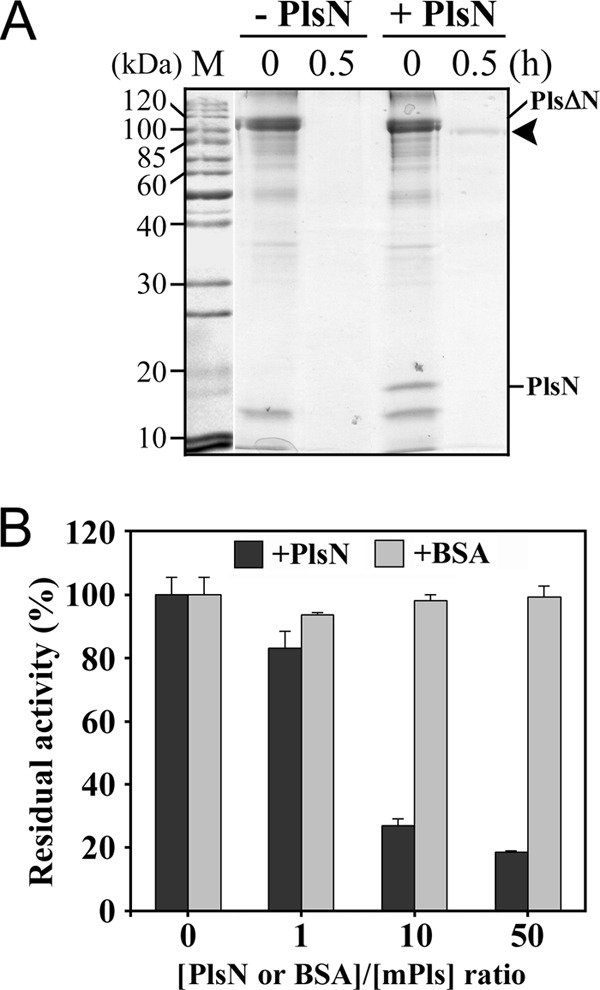
The effects of the N-terminal propeptide on the folding and activity of pyrolysin. (A) A crude sample of PlsΔN was incubated at 95°C in the presence (+) or absence (-) of roughly equal molar concentrations of purified PlsN, followed by TCA precipitation and SDS-PAGE analysis. The arrowhead indicates the position of the mature enzyme on the gel. (B) Purified mPls (1 μg/ml) was mixed with PlsN or BSA at different molar ratios as indicated and then subjected to an azocaseinolytic activity assay at 95°C. The values are expressed as means ± standard deviations (SD) (bars) of the results of three independent experiments.
To determine whether the N-terminal propeptide acts in trans, an equivalent molar amount of PlsN was added to the PlsΔN sample in 50 mM sodium phosphate buffer (pH 7.5) containing 6 M urea, and the mixture was subjected to incubation at 4°C overnight, removal of urea by dialysis, and heat treatment at 95°C. Through this procedure, the PlsΔN in the mixture was converted to a mature form that was the same size as mPls (100 kDa; Fig. 4A). These results indicate that both covalently and noncovalently linked N-terminal propeptides are able to assist pyrolysin folding into a stable mature conformation at high temperatures.
Next, we examined the inhibitory activity of the N-terminal propeptide. When different concentrations of PlsN were mixed with mPls, the residual activity decreased as the [PlsN]/[mPls] ratio increased (Fig. 4B), suggesting that the N-terminal propeptide has the ability to inhibit the activity of mPls.
The effects of the CTE on enzyme folding.
Despite lacking the C-terminal propeptide (C4 segment), PlsΔC199 was converted to mPlsΔC199 with the same size as mPls generated from Pls following heat treatment (Fig. 2A and 3), demonstrating that the C-terminal propeptide is not essential for the folding of pyrolysin. Nevertheless, mPlsΔC199 was less stable than mPls at high temperatures (Fig. 5A), implying that the presence of the C-terminal propeptide in the proform aids in the hyperthermostability of the enzyme.
Fig 5.
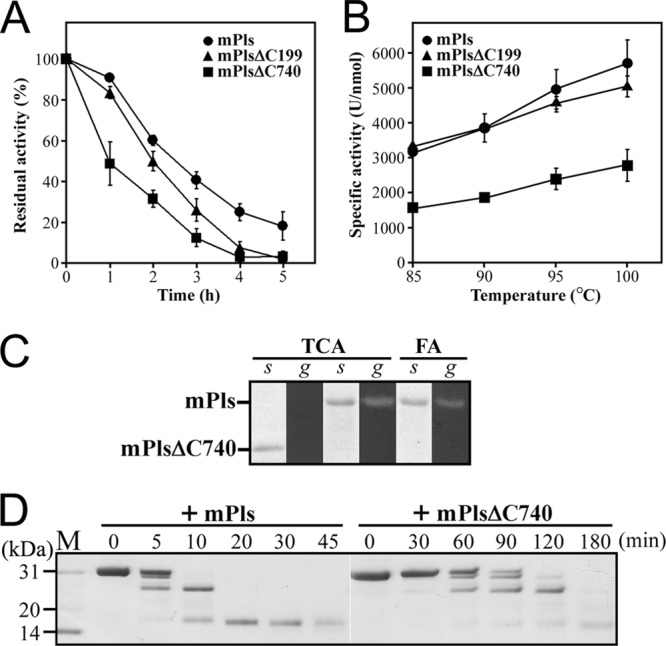
Properties of mature enzymes. (A) Thermal stability of mature enzymes. The enzymes (0.5 μg/ml) were incubated at 95°C for the indicated time periods and then subjected to an azocaseinolytic activity assay. The residual activity is expressed as a percentage of the original activity. The values are expressed as means ± SD (bars) of the results of three independent experiments. (B) Temperature dependence of enzyme activity. Using azocasein as the substrate, activity assays were performed for 10 min at the indicated temperatures. The values are expressed as means ± SD (bars) of the results of three independent experiments. (C) Acid resistance of the mature enzymes. The purified mature enzyme samples were precipitated with 20% TCA or treated with 5 M formic acid (FA) and then subjected to SDS-PAGE (s) and gelatin overlay assay (g) at 95°C. (D) Digestion patterns of β-casein cleaved by mature enzymes. The reaction was carried out at 85°C in 50 mM sodium phosphate buffer (pH 7.5) containing 0.5 mg/ml of β-casein (Sigma) and 2 nM enzyme for different time periods, and then the samples were subjected to Tricine–SDS-PAGE analysis.
Because PlsΔC362 and PlsΔC463 displayed proteolytic activity in the gelatin overlay assay (Fig. 1C), the enzyme clearly has the ability to convert to active forms in the absence of the C2-C4 segment; however, these two mutants were sensitive to heat treatment and were degraded completely after incubation at 95°C for 2 h (Fig. 2B), probably due to thermogenic hydrolysis and/or autoproteolysis. This result implies that the C2-C4 segment is not essential for the folding of pyrolysin but is important for conversion of the enzyme into a properly folded state with higher heat resistance.
In contrast to PlsΔC463, PlsΔC740 did not display proteolytic activity in the gelatin overlay assay (Fig. 1C), implying that the C1 segment is required for enzyme folding. After incubation at 95°C for 3 h, PlsΔC740 was degraded completely (Fig. 2C). Because PlsΔC740 is inactive, its degradation was not due to autoproteolysis. Instead, PlsΔC740 was likely unable to fold into a heat-resistant conformation in the absence of the CTE and thus was subject to thermogenic hydrolysis. Conversely, PlsΔC740 was converted to a 57-kDa mature form (mPlsΔC740) with the aid of exogenously added PlsC740 or PlsC541 (Fig. 2C). The first five residues of mPlsΔC740 were identified as MYNST by N-terminal sequencing, indicating that the N-terminal propeptide had been processed. Furthermore, mPlsΔC740 could be detected by immunoblotting with anti-His tag antibody (Fig. 3), suggesting that no further C-terminal processing occurred during the maturation of the PlsΔC740 that had been properly folded with the aid of PlsC740. These results indicate that the CTE is essential for pyrolysin function at high temperatures.
The importance of the CTE in enzyme activity and stability.
In comparison with mPlsΔC740, mPls and mPlsΔC199 have an additional CTEm (Fig. 2D), which allows us to investigate the effect of the CTEm on enzyme stability and activity. All three mature forms retained more than 90% of their original activities after incubation at 90°C for 2 h. When incubated at 95°C, mPlsΔC740 exhibited a shorter half-life than mPls and mPlsΔC199 (Fig. 5A), suggesting that the CTEm confers additional thermal stability to the enzyme. In the presence of 5 mM EDTA, less than 10% of the original activity remained after incubation of the three mature enzymes at 95°C for 1 h (data not shown). This loss of activity indicates that metal ions play an important role in stabilizing the enzyme. The effects of denaturing reagents on the activities of mPls and mPlsΔC740 were investigated (Table 1). When suc-AAPK-pNA was used as the substrate, the activities of the two enzymes decreased remarkably in the presence of 6 M urea and/or 2% SDS; however, both enzymes displayed azocaseinolytic activities in the presence of denaturing reagents similar to or higher than those seen in the absence of such reagents. This discrepancy may be ascribed to a higher level of denaturing reagent-induced conformational changes in protein substrates that result in the denaturing agents rendering the protein substrates more susceptible to proteolysis. In any case, mPls showed a slightly higher residual activity than mPlsΔC740 in the presence of denaturing reagents. Proteolytic activity was detected with a gelatin overlay assay in the TCA-treated sample of mPls but not in that of mPlsΔC740 (Fig. 5C), demonstrating that mPls is more resistant to acid treatment than mPlsΔC740. In addition, mPls retained proteolytic activity even after treatment with 5 M formic acid (Fig. 5C). The acid resistance of the enzyme appears to be responsible for the appearance of degraded products in TCA-treated Pls and PlsΔC199 (Fig. 1B). Minor acid-resistant mature forms may lead to the degradation of unfolded proforms during sample preparation and/or electrophoresis. Taken together, these results demonstrate that the presence of the CTEm increases the resistance of pyrolysin to heat treatment, denaturing reagents, and acid denaturation.
Table 1.
Effects of denaturing reagents on activities of mPls and mPlsΔC740
| Reagent | Relative activity (%)a |
|||
|---|---|---|---|---|
| mPls |
mPlsΔC740 |
|||
| Suc-AAPK-pNA | Azocasein | Suc-AAPK-pNA | Azocasein | |
| None | 100 | 100 | 100 | 100 |
| SDS (2%) | 21 | 103 | 19 | 88 |
| Urea (6 M) | 13 | 219 | 11 | 184 |
| SDS (2%) + urea (6 M) | 6 | 152 | 2 | 138 |
The enzyme activity on suc-AAPK-pNA and azocasein was measured at 90°C and 95°C, respectively. The relative activity values were calculated based on the measured activity in the absence of denaturing regent (defined as 100%). The values are expressed as means of the results of three independent experiments, and standard deviations were less than 10% of the means.
The azocaseinolytic activities of mPls, mPlsΔC199, and mPlsΔC740 increased with rising temperature up to 100°C (Fig. 5B). However, mPls and mPlsΔC199 exhibited activity ∼2-fold higher than that of mPlsΔC740 over the whole range of temperatures tested. Thus, the CTEm plays an important role in enzyme activity. The digestion patterns of β-casein cleaved by mPls and mPlsΔC740 were indistinguishable except that mPls demonstrated an increased hydrolysis rate (Fig. 5D), implying that the CTEm does not contribute significantly to the cleavage specificity of the enzyme. Furthermore, mPls also showed higher hydrolytic activity on small synthetic substrates than mPlsΔC740 (Table 2). Determination of the kinetic parameters of mPls and mPlsΔC740 against suc-AAPK-pNA at 90°C revealed that mPls has a kcat/Km value ∼2-fold higher than that of mPlsΔC740, mainly due to a higher hydrolysis rate constant (kcat; Table 2). These results demonstrate that the CTEm is beneficial for the catalytic efficiency of pyrolysin.
Table 2.
Kinetic parameters of mPls and mPlsΔC740 at 90°C
| Enzyme | Values (mean ± SD) for indicated kinetic parameter (suc-AAPK-pNA)a |
||
|---|---|---|---|
| Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) | |
| mPls | 3.32 ± 0.53 | 3128 ± 205 | 942 |
| mPlsΔC740 | 3.83 ± 0.43 | 1893 ± 162 | 494 |
The values are expressed as means ± standard deviations of the results of three independent experiments.
DISCUSSION
Maturation.
The data presented here indicate that the recombinant proform of pyrolysin is able to autoprocess its N- and C-terminal propeptides at high temperatures and convert to a 100-kDa mPls with the same N terminus as the glycosylated HMW (150 kDa) and LMW (130 kDa) pyrolysins purified from the P. furiosus cell envelope fraction (35). Whether mPls corresponds to HMW or LMW pyrolysin, however, remains unknown at this time, because their C termini have not yet been determined and the glycosylation of the HMW and LMW pyrolysins prevents determinations based on their molecular masses. Nevertheless, our results suggest that mPls is the major mature form of recombinant pyrolysin.
Efficient maturation is a prerequisite for almost all extracellular subtilases to release enzyme activity, but the maturation process greatly varies for different enzymes in terms of structural change during maturation, metal ion dependence, etc. For example, the proform of subtilisin E exists in a molten globule-like state, and the autoprocessing of the N-terminal propeptide leads to a structural reorganization that reduces hydrophobic surface area and increases the amount of tertiary structure of the mature enzyme (25). Albeit with an ∼30% lower yield of mature enzyme, the maturation of subtilisin E can also occur in the absence of Ca2+, and the stabilizing effect of Ca2+ is observed only after the completion of the autoprocessing of the propeptide (39). In contrast, the maturation of Tk-subtilisin is Ca2+ dependent, and the enzyme structure is not significantly altered during maturation in the presence of Ca2+. One unique Ca2+-binding site (Ca-7) is required to promote the autoprocessing reaction by stabilizing the autoprocessed form. The Ca2+-induced maturation and stabilization of the pro- and mature forms of the enzyme are considered important for Tk-subtilisin to adapt to a hyperthermal environment (22, 32, 33). Pyrolysin is able to mature in Ca2+-free 50 mM sodium phosphate buffer (pH 7.5) at 95°C, and the presence of EDTA in the buffer does not prevent its maturation but does reduce the yield of mature enzyme. This does not mean, however, that Ca2+ is not needed at all for maturation, because it is well known that it is difficult to remove metals from hyperthermophilic proteins, even with EDTA treatment at high temperatures (6, 14, 34). It remains to be elucidated whether pyrolysin contains high-affinity Ca2+-binding site(s). Nevertheless, the low maturation efficiency is closely related to self-degradation of the enzymes after maturation under chelating conditions, as evidenced by the destabilizing effect of EDTA on the mature enzyme. In this regard, the maturation of pyrolysin seems to be similar to that of bacterial subtilisin E.
The susceptibility of a protein to thermogenic hydrolysis is dependent upon the conformational integrity of the protein at that temperature (6). The proform (PlsS441A) was less resistant to degradation at high temperatures than the mature form (mPls), and this difference implies that pyrolysin undergoes a structural change during maturation in order to acquire a more stable conformation. Although the tertiary structures of the two forms remain to be determined, the evidence that the proform (e.g., Pls and PlsS441A) tends to aggregate more easily than mPls at higher protein concentrations leads us to postulate that the proform has an increased hydrophobic surface area. Our results indicate that the proform of pyrolysin (e.g., PlsS441A) suffers thermogenic hydrolysis at 95°C. Since pyrolysin originates from the hyperthermophilic archaeon P. furiosus that optimally grows at 100°C (10), the proform should be stable enough under hyperthermal conditions to maximize maturation efficiency. Notably, native pyrolysin is glycosylated and associated with the cell envelope. Earlier studies indicated that the half-life of pyrolysin in the cell envelope fraction of P. furiosus is 9 h at 95°C (9). Here, we found that the purified recombinant mPls showed a half-life of 2.5 h at 95°C. Thus, association with the cell envelope or glycosylation of pyrolysin or both plays an important role in stabilizing this enzyme. As a result, the proform of pyrolysin may also be stabilized by interactions with a cell envelope component(s) and/or by glycosylation in order to allow the proform to efficiently convert into mature enzyme at high temperatures. After maturation and structural reorganization, an additional increase in the stability of the mature form enables the enzyme to function under hyperthermal conditions.
The role of the N-terminal propeptide.
Propeptides are broadly classified into two categories: class I propeptides that directly catalyze correct folding and class II propeptides that are not directly involved in folding (26). The class I N-terminal propeptides from various proteins often act as intramolecular chaperones that facilitate the correct folding of their cognate functional domains (27). In some bacterial proteases (e.g., subtilisin E, subtilisin BPN′, and α-lytic protease) and eukaryotic proprotein convertases (PCs), the N-terminal propeptide is essential for enzyme folding (27). A thermolysin-like neutral protease (TLP-ste) from Bacillus stearothermophilus (18) and two subtilisin-like proteases (Tk-subtilisin [22] and Tk-SP subtilisin [12]) from T. kodakaraensis were recently found to fold into active forms in the absence of the N-terminal propeptides. In the case of Tk-subtilisin, the N-terminal propeptide is not required for correct folding of the catalytic domain but is required to accelerate this folding (31). Notably, the catalytic domain of Tk-subtilisin that was refolded in the absence of the propeptide, albeit with a low yield of ∼5%, is distinguishable from the mature enzyme derived from the proform in terms of enzyme properties (22) and structure (31). Our data show that PlsΔN possesses weak enzymatic activity, indicating that the N-terminal propeptide is not essential for folding of the enzyme into an active form; however, unlike Tk-subtilisin, which is capable of folding into its “native-like” mature form in the absence of the N-terminal propeptide, PlsΔN was completely degraded after heat treatment due to autoproteolysis and/or thermogenic hydrolysis. A heat-resistant mature pyrolysin was generated only in the presence of the N-terminal propeptide, although this N-terminal propeptide could be either covalently linked to the mature region or provided as a separate molecule. In this context, the N-terminal propeptide functions as an intramolecular chaperone that is required for the folding of pyrolysin into its native-like mature form, and thus, this molecule belongs to the class I propeptides (26). Meanwhile, the inhibitory activity of the N-terminal propeptide is useful for maximizing the yield of mature pyrolysin by minimizing the autoproteolysis of unstable folding intermediates at high temperatures.
The role of the long CTE.
In comparison with the CTEs of other subtilases, a unique feature of the pyrolysin CTE is its greater length. Our results demonstrate that the long CTE of pyrolysin contains at least two functional portions: the C-terminal propeptide and the CTEm. Both of these portions are important for the function of pyrolysin in hyperthermal environments.
The C-terminal propeptide of pyrolysin is not necessary for folding but is important for the mature enzyme to achieve its hyperthermostability, and thus, this propeptide may belong to the class II propeptides (26). The C-terminal propeptide is conserved not only within pyrolysin-like proteases (see Fig. S1 in the supplemental material) but also within the thiol proteases from hyperthermophilic archaea (8). Therefore, we propose that these conserved segments may function as C-terminal propeptides and contribute to the hyperthermostability of the cognate mature enzymes as well.
The CTEm is involved in enzyme folding, stability, and enzymatic activity. The fact that PlsΔC199, rather than PlsΔC740, was converted to the active mature form clearly demonstrates that the CTEm is indispensable for correct folding of the enzyme. In addition, PlsC541 as a separate molecule was able to assist PlsΔC740 conversion to the active mature form, suggesting that CTEm-mediated enzyme folding involves the interaction between the two polypeptides. As determined on the basis of the observation that PlsΔC463 exhibited proteolytic activity after heat treatment, the C1 segment immediately downstream of the catalytic domain likely participates in this interaction. Our results also show that the presence of the CTEm increases the half-life of the mature enzyme at high temperatures as well as increasing its resistance to denaturing reagents. These findings demonstrate that the CTEm contributes to the structural stability of the catalytic domain. The stabilizing effect of the CTE on the catalytic domain has been described for mammalian furin (13, 41), tomato subtilase 3 (20), halolysin SptA (38) Tk-SP subtilisin (11, 29), fervidolysin (15), and a subtilase from Pseudoalteromonas sp. SM9913 (40). The attachment of CTE to the C terminus of the catalytic domain appears to be a common strategy used by subtilases to acquire extra stability.
The involvement of CTE in enzymatic activity has been reported for some subtilases, although the mode of action of these CTEs depends on enzyme functions and also on environmental conditions (17, 38, 41). In the case of pyrolysin, the presence of the CTEm improves its catalytic efficiency with respect to both proteineous and small synthetic peptide substrates, mainly due to an increase in the hydrolysis rate (kcat). Given that pyrolysin is an extracellular protease of P. furiosus, which requires peptides for growth (30), the improved catalytic efficiency afforded by CTEm is likely important for the enzyme to fulfill its physiological role. Whether the effects of CTEm on the kinetic properties of pyrolysin are due to direct steric influence or to an indirect influence such as induction of conformational change in the active site, however, remains to be determined.
As one of the largest and most stable proteases, pyrolysin is distinguished from other known subtilases by the presence of a long CTE and large insertions (8, 35). With whole-genome sequence data quickly becoming available for hyperthermophiles, an increasing number of pyrolysin-like proteases have been found to contain a mosaic of domains. Our results provide important clues about enzyme maturation, the roles of the N-terminal propeptide, and the function of the long CTE in the adaptation of pyrolysin-like proteases to hyperthermal environments. Further studies to investigate the relationship of other structural elements, such as the large insertions, to enzyme function and to explore the mechanism of the association of the enzyme with the cell envelope may allow us to gain a better understanding of the adaptation mechanism of hyperthermophilic proteases.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Natural Science Foundation of China (30470019).
Footnotes
Published ahead of print 13 April 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Atomi H. 2005. Recent progress towards the application of hyperthermophiles and their enzymes. Curr. Opin. Chem. Biol. 9:166–173 [DOI] [PubMed] [Google Scholar]
- 2. Blumentals II, Robinson AS, Kelly RM. 1990. Characterization of sodium dodecyl sulfate-resistant proteolytic activity in the hyperthermophilic archaebacterium Pyrococcus furiosus. Appl. Environ. Microbiol. 56:1992–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 4. Cheng G, Zhao P, Tang X-F, Tang B. 2009. Identification and characterization of a novel spore-associated subtilase from Thermoactinomyces sp. CDF. Microbiology 155(Pt. 11):3661–3672 [DOI] [PubMed] [Google Scholar]
- 5. Connaris H, Cowan DA, Sharp RJ. 1991. Heterogeneity of proteinases from the hyperthermophilic archaeobacterium Pyrococcus furiosus. J. Gen. Microbiol. 137:1193–1199 [Google Scholar]
- 6. Daniel RM, Dines M, Petach HH. 1996. The denaturation and degradation of stable enzymes at high temperatures. Biochem. J. 317(Pt. 1):1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DelMar EG, Largman C, Brodrick JW, Geokas MC. 1979. A sensitive new substrate for chymotrypsin. Anal. Biochem. 99:316–320 [DOI] [PubMed] [Google Scholar]
- 8. de Vos WM, et al. 2001. Purification, characterization, and molecular modeling of pyrolysin and other extracellular thermostable serine proteases from hyperthermophilic microorganisms. Methods Enzymol. 330:383–393 [DOI] [PubMed] [Google Scholar]
- 9. Eggen R, Geerling A, Watts J, de Vos WM. 1990. Characterization of pyrolysin, a hyperthermoactive serine protease from the archaebacterium Pyrococcus furiosus. FEMS Microbiol. Lett. 71:17–20 [Google Scholar]
- 10. Fiala G, Stetter KO. 1986. Pyrococcus furiosus sp. nov. represents a novel genus of marine heterotrophic archaebacteria growing optimally at 100°C. Arch. Microbiol. 145:56–61 [Google Scholar]
- 11. Foophow T, et al. 2010. Crystal structure of a subtilisin homologue, Tk-SP, from Thermococcus kodakaraensis: requirement of a C-terminal beta-jelly roll domain for hyperstability. J. Mol. Biol. 400:865–877 [DOI] [PubMed] [Google Scholar]
- 12. Foophow T, Tanaka S, Koga Y, Takano K, Kanaya S. 2010. Subtilisin-like serine protease from hyperthermophilic archaeon Thermococcus kodakaraensis with N- and C-terminal propeptides. Protein Eng. Des. Sel. 23:347–355 [DOI] [PubMed] [Google Scholar]
- 13. Henrich S, et al. 2003. The crystal structure of the proprotein processing proteinase furin explains its stringent specificity. Nat. Struct. Biol. 10:520–526 [DOI] [PubMed] [Google Scholar]
- 14. Hensel R, Jakob I, Scheer H, Lottspeich F. 1992. Proteins from hyperthermophilic archaea: stability towards covalent modification of the peptide chain. Biochem. Soc. Symp. 58:127–133 [PubMed] [Google Scholar]
- 15. Kim JS, Kluskens LD, de Vos WM, Huber R, van der Oost J. 2004. Crystal structure of fervidolysin from Fervidobacterium pennivorans, a keratinolytic enzyme related to subtilisin. J. Mol. Biol. 335:787–797 [DOI] [PubMed] [Google Scholar]
- 16. King J, Laemmli UK. 1971. Polypeptides of the tail fibres of bacteriophage T4. J. Mol. Biol. 62:465–477 [DOI] [PubMed] [Google Scholar]
- 17. Kobayashi H, et al. 2009. Structural basis for the kexin-like serine protease from Aeromonas sobria as sepsis-causing factor. J. Biol. Chem. 284:27655–27663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mansfeld J, Petermann E, Dürrschmidt P, Ulbrich-Hofmann R. 2005. The propeptide is not required to produce catalytically active neutral protease from Bacillus stearothermophilus. Protein Expr. Purif. 39:219–228 [DOI] [PubMed] [Google Scholar]
- 19. Mayr J, et al. 1996. A hyperthermostable protease of the subtilisin family bound to the surface layer of the archaeon Staphylothermus marinus. Curr. Biol. 6:739–749 [DOI] [PubMed] [Google Scholar]
- 20. Ottmann C, et al. 2009. Structural basis for Ca2+-independence and activation by homodimerization of tomato subtilase 3. Proc. Natl. Acad. Sci. U. S. A. 106:17223–17228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Papworth C, Bauer J, Braman J, Wright D. 1996. Site-directed mutagenesis in one day with >80% efficiency. Strategies 9:3–4 [Google Scholar]
- 22. Pulido M, et al. 2006. Ca2+-dependent maturation of subtilisin from a hyperthermophilic archaeon, Thermococcus kodakaraensis: the propeptide is a potent inhibitor of the mature domain but is not required for its folding. Appl. Environ. Microbiol. 72:4154–4162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sarkar G, Sommer SS. 1990. The “megaprimer” method of site-directed mutagenesis. Biotechniques 8:404–407 [PubMed] [Google Scholar]
- 24. Schägger H, von Jagow G. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166:368–379 [DOI] [PubMed] [Google Scholar]
- 25. Shinde U, Inouye M. 1995. Folding pathway mediated by an intramolecular chaperone: characterization of the structural changes in pro-subtilisin E coincident with autoprocessing. J. Mol. Biol. 252:25–30 [DOI] [PubMed] [Google Scholar]
- 26. Shinde U, Inouye M. 2000. Intramolecular chaperones: polypeptide extensions that modulate protein folding. Semin. Cell Dev. Biol. 11:35–44 [DOI] [PubMed] [Google Scholar]
- 27. Shinde U, Thomas G. 2011. Insights from bacterial subtilases into the mechanisms of intramolecular chaperone-mediated activation of furin. Methods Mol. Biol. 768:59–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Siezen RJ, Leunissen JA. 1997. Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci. 6:501–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sinsereekul N, et al. 2011. An alternative mature form of subtilisin homologue, Tk-SP, from Thermococcus kodakaraensis identified in the presence of Ca2+. FEBS J. 278:1901–1911 [DOI] [PubMed] [Google Scholar]
- 30. Snowden LJ, Blumentals II, Kelly RM. 1992. Regulation of proteolytic activity in the hyperthermophile Pyrococcus furiosus. Appl. Environ. Microbiol. 58:1134–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tanaka S, et al. 2008. Crystal structure of Tk-subtilisin folded without propeptide: requirement of propeptide for acceleration of folding. FEBS Lett. 582:3875–3878 [DOI] [PubMed] [Google Scholar]
- 32. Tanaka S, Matsumura H, Koga Y, Takano K, Kanaya S. 2007. Four new crystal structures of Tk-subtilisin in unautoprocessed, autoprocessed and mature forms: insight into structural changes during maturation. J. Mol. Biol. 372:1055–1069 [DOI] [PubMed] [Google Scholar]
- 33. Tanaka S, et al. 2007. Crystal structure of unautoprocessed precursor of subtilisin from a hyperthermophilic archaeon: evidence for Ca2+-induced folding. J. Biol. Chem. 282:8246–8255 [DOI] [PubMed] [Google Scholar]
- 34. Vieille C, Zeikus GJ. 2001. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 65:1–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Voorhorst WG, et al. 1996. Isolation and characterization of the hyperthermostable serine protease, pyrolysin, and its gene from the hyperthermophilic archaeon Pyrococcus furiosus. J. Biol. Chem. 271:20426–20431 [DOI] [PubMed] [Google Scholar]
- 36. Voorhorst WG, Warner A, de Vos WM, Siezen RJ. 1997. Homology modelling of two subtilisin-like proteases from the hyperthermophilic archaea Pyrococcus furiosus and Thermococcus stetteri. Protein Eng. 10:905–914 [DOI] [PubMed] [Google Scholar]
- 37. Ward DE, et al. 2002. Proteolysis in hyperthermophilic microorganisms. Archaea 1:63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu Z, et al. 2011. Functional insight into the C-terminal extension of halolysin SptA from haloarchaeon Natrinema sp. J7. PLoS One 6:e23562 doi:10.1371/journal.pone.0023562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yabuta Y, Subbian E, Takagi H, Shinde U, Inouye M. 2002. Folding pathway mediated by an intramolecular chaperone: dissecting conformational changes coincident with autoprocessing and the role of Ca2+ in subtilisin maturation. J. Biochem. 131:31–37 [DOI] [PubMed] [Google Scholar]
- 40. Yan BQ, et al. 2009. Molecular analysis of the gene encoding a cold-adapted halophilic subtilase from deep-sea psychrotolerant bacterium Pseudoalteromonas sp. SM9913: cloning, expression, characterization and function analysis of the C-terminal PPC domains. Extremophiles 13:725–733 [DOI] [PubMed] [Google Scholar]
- 41. Zhou A, Martin S, Lipkind G, LaMendola J, Steiner DF. 1998. Regulatory roles of the P domain of the subtilisin-like prohormone convertases. J. Biol. Chem. 273:11107–11114 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.