

Abstract
Real-time quantitative PCR (qPCR) is a widely used technique in microbial community analysis, allowing the quantification of the number of target genes in a community sample. Currently, the standard-curve (SC) method of absolute quantification is widely employed for these kinds of analysis. However, the SC method assumes that the amplification efficiency (E) is the same for both the standard and the sample target template. We analyzed 19 bacterial strains and nine environmental samples in qPCR assays, targeting the nifH and 16S rRNA genes. The E values of the qPCRs differed significantly, depending on the template. This has major implications for the quantification. If the sample and standard differ in their E values, quantification errors of up to orders of magnitude are possible. To address this problem, we propose and test the one-point calibration (OPC) method for absolute quantification. The OPC method corrects for differences in E and was derived from the ΔΔCT method with correction for E, which is commonly used for relative quantification in gene expression studies. The SC and OPC methods were compared by quantifying artificial template mixtures from Geobacter sulfurreducens (DSM 12127) and Nostoc commune (Culture Collection of Algae and Protozoa [CCAP] 1453/33), which differ in their E values. While the SC method deviated from the expected nifH gene copy number by 3- to 5-fold, the OPC method quantified the template mixtures with high accuracy. Moreover, analyzing environmental samples, we show that even small differences in E between the standard and the sample can cause significant differences between the copy numbers calculated by the SC and the OPC methods.
INTRODUCTION
In microbial ecology, real-time quantitative PCR (qPCR) is used to measure the number of copies of a gene of interest in a community or an environmental sample (24, 25, 41, 42). The presence of specific phylogenetic groups can be quantified by targeting the 16S rRNA gene (1, 10). Moreover, functional genes have been targeted to quantify the genetic potential of a community to catalyze certain processes (23, 43, 50, 55).
Originally, the qPCR technique was developed as a tool in medical diagnostics targeting nonvariable regions (11, 27). However, for two reasons, the application of qPCR in microbial ecology needs to take into account genetic variations of the targeted region. First, although primers are designed to target a certain phylogenetic group, highly similar sequences in which primer target sites differ in only a few bases might be present in and coamplified from environmental samples (49). Second, to account for sequence variability within the target phylogenetic group, degenerate primers have frequently been used in qPCR assays (2, 17, 23, 50). Primer mismatches (3), as well as the use of degenerate primers differing in GC content (33), affect primer annealing kinetics and therefore might affect the accuracy of the qPCR assay.
The qPCR technique is based on real-time monitoring of amplicon formation by a reporter molecule (e.g., SYBR green dye) (5). Fluorescence (Y) is measured after each temperature cycle and is proportional to the amount of synthesized amplicon (N): N ∝ Y (Fig. 1A). The exponential growth of the amplicon concentration in the reaction mixture, NC, can be described as an exponential function of the template starting concentration, N0; the efficiency of the qPCR, E; and the number of qPCR cycles, C: NC = N0 × EC (5). Two parameters are essential for quantification: the threshold cycle, CT, and the qPCR E. The CT is the number of cycles necessary to reach a certain threshold fluorescence, YT. In one experimental setup, YT is the same for all samples. Since fluorescence is a relative measure of the DNA content, all samples contain the same number of amplicons, Nt, when passing the CT. E is a measure of amplification quality and depends on factors such as the primer GC content (33), primer mismatch (3), and the presence of PCR inhibitors (21). If E equals 2, the number of amplicons doubles per cycle, i.e., the efficiency is 100%. Two distinct methods can be used to estimate E. First, Efi can be estimated from the fluorescence increase using linear (30, 38) or nonlinear (26, 45) regression models (Fig. 1A). Second, Eds can be estimated from the slope of a dilution series (14, 35) (Fig. 1B).
Fig 1.
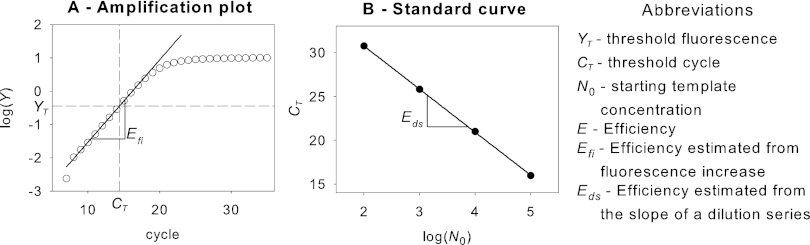
(A) Schematic amplification plot of log fluorescence increase over qPCR cycles, indicating the slope to estimate Efi by linear regression. (B) Schematic standard curve of a dilution series, plotting CT values over log template concentrations. The slope is used to estimate Eds.
Two major quantification methods have been developed and are widely used: relative quantification and absolute quantification. Relative quantification using the ΔΔCT method is the most common method used in gene expression analysis (48) but has also been applied in environmental microbiology (51). The method determines the gene expression ratio (or abundance ratio) of a target gene in a sample compared to a control, normalizing with the expression ratio of a reference gene. Target and reference genes often differ in their E values, and thus, the ΔΔCT method has been improved in order to account for differences in E (31).
Absolute quantification by the standard-curve (SC) method is frequently used in environmental microbiology. The SC method employs a dilution series of known template concentrations, N0, in the qPCR assay. Linear regression of log(N0) versus CT gives the standard curve, and this is then used to calculate template concentrations, N0, of the sample. This method assumes that E of the sample is the same as E of the standard.
In environmental microbiology, the SC method has been used to quantify the concentrations of target genes in diverse samples, e.g., microbial communities of biofilms (10), the rumen (1), and alpine soils (2). In most cases, the sample template is different from the standard template used to prepare the SC. The standards usually originate from pure cultures, while the sample is composed of a mixture of different species. Previous studies demonstrated that E estimates significantly affect quantification accuracy. It is therefore recommended that accurate E estimates be employed in quantification analysis (26, 34, 36, 38, 46). In spite of these recommendations, the SC method assumes a constant E for standard and sample. Although this introduces the possibility of increased quantification errors, it is still the method of choice in environmental microbiology.
Our first aim was to demonstrate the variability of qPCR E with the template source. As a model functional gene, we selected the nifH gene coding for the nitrogenase iron protein (NifH; EC 1.18.6.1) because it is the focus of numerous studies in ecology and therefore well characterized. In addition, the 16S rRNA gene was used to demonstrate the universality of the problem studied. Universal 16S rRNA gene targeting approaches are frequently used as a community reference in qPCR-based studies (1, 11, 54). We estimated Efi from the fluorescence increase of 19 bacterial strains covering Actinobacteria, Cyanobacteria, Firmicutes, and Proteobacteria, as well as nine environmental samples representing typical terrestrial and aquatic habitats. Moreover, we propose a novel absolute quantification method that accounts for differences in E. We demonstrate that the new method is less susceptible to quantification errors than the SC method by analyzing an artificial mixture of two templates that differ in E.
MATERIALS AND METHODS
Bacterial cultures. (i) Strains and cultivation conditions.
Nineteen bacterial strains containing the nifH gene were used in this study. The strains were selected to cover different phyla and to be genome sequenced, if possible. The strains listed in Table 1 were grown at room temperature in the medium specified by the Culture Collection of Algae and Protozoa (CCAP), the Pasteur Culture Collection (PCC), and the German Collection of Microorganisms and Cell Cultures (DSMZ) for the respective strain, except Methylosinus trichosporium and Methylococcus capsulatus, which were grown in nitrate mineral salts (NMS) medium (DSMZ medium 632 [52]) with 10% (vol/vol) methane in air in the headspace. The 16S rRNA and nifH genes were sequenced for strains that were taken from our laboratory collections (glycerol stocks) in order to verify the identities of the strains. Sequencing was performed at the Genetic Diversity Centre of ETH Zurich using BigDye Terminator v1.1 chemistry (Applied Biosystems, Foster City, CA) on a 3130xl Genetic Analyzer (Applied Biosystems).
Table 1.
Bacterial strains used in this study
| Species | Strain | Phylogenetic group |
|---|---|---|
| Frankia sp. | DSM 43829 | Actinobacteria |
| Cyanothece sp. | PCC 7425 | Cyanobacteria |
| Nostoc commune | CCAP 1453/33 | Cyanobacteria |
| Nostoc sp. | PCC 7120 | Cyanobacteria |
| Nostoc punctiforme | PCC 73102 | Cyanobacteria |
| Paenibacillus sabinae | DSM 17841 | Firmicutes |
| Azospirillum brasilense | DSM 1690 | Alphaproteobacteria |
| Bradyrhizobium japonicum | USDA 110 | Alphaproteobacteria |
| Methylosinus trichosporium | OB3ba | Alphaproteobacteria |
| Rhizobium leguminosarum | DSM 30132 | Alphaproteobacteria |
| Sinorhizobium meliloti | 1021b | Alphaproteobacteria |
| Sphingomonas azotifigens | DSM 18530 | Alphaproteobacteria |
| Xanthobacter autotrophicus | DSM 432 | Alphaproteobacteria |
| Acidithiobacillus ferrooxidans | DSM 14882 | Betaproteobacteria |
| Burkholderia xenovorans | DSM 17367 | Betaproteobacteria |
| Azotobacter vinelandii | DSM 85 | Betaproteobacteria |
| Methylococcus capsulatus | Batha | Betaproteobacteria |
| Pseudomonas stutzeri | DSM 4166 | Betaproteobacteria |
| Geobacter sulfurreducens | DSM 12127 | Deltaproteobacteria |
(ii) DNA extraction and quantification.
Bacterial colonies from agar plates or bacterial pellets from liquid cultures were washed three times in phosphate-buffered saline (pH 7.4) prior to DNA extraction with a Wizard Genomic DNA kit (Promega, Madison, WI). Extraction was performed according to the manufacturers' instructions, and DNA was eluted with 100 μl diethyl pyrocarbonate (DEPC)-treated water (Carl Roth, Karlsruhe, Germany). DNA purity was checked on a nanodrop spectrophotometer (NanoDrop Technologies, Wilmington, DE) and quantified with SYBR green I (28).
Environmental samples.
DNA extracts from three natural habitats, soil, plant, and lake, were used in this study to represent a broad range of samples typically studied in environmental microbiology. The DNA extracts were obtained in three previously conducted studies (2, 19, 20) and stored at −80°C until analysis. Samples “bulk soil, young” and “bulk soil, old” were taken from the forefield of the Damma glacier in Switzerland and represent more than 50 years and over 2,000 years of soil development, respectively (2). The soil samples were taken with shovels and frozen in the field on dry ice. Upon arrival in the laboratory, the soil samples were stored at −20°C. Rhizosphere and phyllosphere samples were obtained from rice plants grown at the International Rice Research Institute, Los Baños, Philippines (19). Samples from the eutrophic lake were taken at 10-, 80-, and 170-m depths at Lake Zug, Switzerland (20). The lake is meromictic and permanently anoxic below 160 m (20). Samples from the oligotrophic lake were taken at 5- and 200-m depths at Lake Brienz, Switzerland, which is fully oxic throughout the year (20). The lake water samples were stored in the dark on ice. Upon arrival in the laboratory, the samples were filtered through 0.2-μm polycarbonate filters following 5-μm prefiltration, and the filters were frozen in liquid nitrogen and stored at −80°C prior to DNA extraction (20). Details on sampling and DNA extraction are given in references 2, 19, and 20.
Real-time PCR assays.
All qPCR assays were performed on an ABI 7300 system (Applied Biosystems). The reaction volumes were 20 μl and contained 1-fold Kapa SYBR Fast PCR master mix (Kapa Biosystems, Cape Town, South Africa), DEPC-treated water (Carl Roth, Karlsruhe, Germany), and 200 nM both forward and reverse primers. Templates were added in 1-μl volume per reaction. Triplicates of no-template controls, containing DEPC-treated water, were included in each run. To amplify the nifH gene, the primer pairs nifHF/nifHR, PolF/PolR, and ForA/Rev were used. The 16S rRNA gene was amplified using the 27F/518R primer pair. All primers employed in this study were published previously and have been extensively used before. Details on primer sequences and thermal cycles are given in Table 2. In preliminary experiments, the annealing temperatures of all reactions were optimized for high specificity (crisp band; no smear) and high yield when amplifying the environmental samples. All qPCR templates were tested for amplification inhibition by dilution series. After each qPCR run, melting curve analysis was performed to verify the presence of the desired amplicon.
Table 2.
Primers and thermal qPCR profiles used
| Primer pair | Target gene | Annealing time and tempa | Primer sequenced (5′–3′) | Tme (°C) | Primer reference(s) |
|---|---|---|---|---|---|
| PolF/PolR | nifH | 25 s at 53°Cb | TGCGAYCCSAARGCBGACTC/ATSGCCATCATYTCRCCGGA | 60–64/62–68 | 32 |
| nifHF/nifHR | nifH | 25 s at 53°Cb | AAAGGYGGWATCGGYAARTCCACCAC/TTGTTSGCSGCRTACATSGCCATCAT | 76–82/78–80 | 37 |
| ForA/Rev | nifH | 60 s at 50°C | GCIWTITAYGGNAARGGNGG/GCRTAIABNGCCATCATYTC | 56–68/54–64 | 53 |
| 27F/518R | 16S rRNA gene | 30 s at 56°Cc | GAGTTTGATCMTGGCTCAG/ATTACCGCGGCTGCTGG | 56–58/60 | 16, 29 |
All reactions started with 300 s of initial denaturation at 95°C, followed by 40 cycles of 15 s of denaturation at 95°C, an annealing step, and 45 s of elongation at 72°C.
Touchdown cycle starting at 63°C with temperature decreases of 2°C per cycle.
Touchdown cycle starting at 63°C followed by 61, 59, 57, and 56°C annealing temperatures.
Primer sequences use IUPAC ambiguity codes, except that I stands for inosine.
Ranges of melting temperatures (Tm) of degenerate primers calculated as follows: Tm = 2 × (A + T) + 4 × (G + C) (39).
Determination of CT and Efi.
Raw data were exported from the ABI system and imported into the LinRegPCR program (v 11.4) (38). In the program settings, all samples of one qPCR run were treated as one amplicon group in order to set one common window of linearity. Then, the program automatically determined the fluorescence threshold for all samples and calculated the individual CT and Efi values (38). The results were exported, and the mean Efi of each sample was calculated as the arithmetic mean of all replicates, excluding as outliers any replicates that were 5% above or below the median efficiency of all replicates.
Quantification methods. (i) SC method.
The SC method is widely used in environmental microbiology and employs a dilution series of a defined target concentration. The target concentration (ctarget [copies μl−1]) was calculated from the DNA concentration (cDNA [ng μl−1]), the lengths of the DNA fragments (lDNA [bp]) (e.g., genome size), the number of targets per DNA fragment (ntarget [copies]), the Avogadro constant (NA) (6.022 × 1023 bp mol−1), and the average weight of a double-stranded base pair (Mbp) (660 g mol−1 = 6.6 × 1011 ng mol−1) (equation 1).
| (1) |
The linear regression of log(N0 standard) versus CT gives the constants intercept a and slope b of the standard curve (equation 2). The number of copies in the sample, N0 sample, can be calculated based on the regression. Slope b of the linear regression is used to estimate Eds (equation 3). For the individual experiments (see below), dilution series of Bradyrhizobium japonicum, Geobacter sulfurreducens, and Nostoc commune genomic DNAs were used to prepare standard curves.
| (2) |
| (3) |
(ii) OPC method.
We propose the one-point calibration (OPC) method as an alternative method for absolute quantification from qPCR data. The method accounts for template-related variability of E by correcting for differences in E between sample and standard.
OPC is performed by defining one standard containing a known number of template copies, N0 standard. Each standard point reaction mixture contained approximately 0.05 to 0.5 ng genomic DNA, equivalent to 104 to 106 copies (equation 1), and was replicated three or four times. The template concentration of a sample, N0 sample, was estimated from the cycle thresholds CT sample and CT standard and the efficiencies Esample and Estandard (equation 4).
| (4) |
We calculated Esample and Estandard as the mean Efi from individual replicates, because previous studies recommended using the means of Efi (9, 30, 40). Alternatively, Esample and Estandard could be estimated from dilution series, Eds (equation 3) (31). In addition, it is necessary to verify the linear range of quantification, for example, by analyzing a dilution series of a standard. Information about the coefficient of variation of the CT values of replicated standard dilutions can also be helpful in assessing the limit of detection (4, 6).
Efi of bacterial strains and environmental samples.
The amplification efficiencies of the bacterial strains and environmental samples were determined in qPCR assays and calculated from the fluorescence increase as described above (38); 0.1 to 0.01 ng of DNA per reaction was used. Each sample was run in four replicates. The number of samples prohibited the simultaneous amplification of all bacterial strains and environmental samples in a single run. Therefore, one qPCR run was performed to compare all bacterial strains, and one qPCR run was performed to compare all environmental samples. One-way analysis of variance (ANOVA) was performed in SigmaPlot (v 11.0; Systat Software) to test for significant differences (P < 0.01) between the bacterial strains and between the environmental samples.
Analysis of template mixtures from two species.
Serial dilutions of G. sulfurreducens and N. commune DNA templates were prepared. From the different dilution steps, template mixtures of G. sulfurreducens and N. commune were prepared. The template mixtures were prepared to have template ratios of approximately 100:1, 10:1, 1:1, 1:10, and 1:100 of G. sulfurreducens to N. commune. The dilution series and the template mixtures were run as triplicates in one qPCR assay, using nifHF/nifHR primers. Standard curves were calculated from G. sulfurreducens and N. commune dilution series. All samples were quantified using the SC and OPC methods. Each of the methods was calibrated once with G. sulfurreducens and once with N. commune as the standard.
Analysis of environmental samples.
Four replicates of each environmental sample and triplicates of a B. japonicum dilution series were amplified by qPCR using the nifHF/nifHR primer pair. This was the same run in which the Efi of the environmental samples was measured. All samples were quantified using the SC and OPC methods, using B. japonicum as the standard. The Mann-Whitney rank sum test (P < 0.01) was performed to test for significant differences between SC and OPC quantifications. B. japonicum was used for calibration, as it is heterotrophic and simple to maintain and therefore likely to be used for calibration in other laboratories.
RESULTS AND DISCUSSION
The efficiencies of qPCRs depend on the template.
The qPCR efficiencies of 19 bacterial strains covering four bacterial phyla were compared. The qPCR assays targeted the nifH gene using three independently developed and previously published primer pairs and targeted the 16S rRNA gene using one similarly well-established primer pair (Table 2). In all assays, the strains differed significantly in their efficiencies (Efi) (Fig. 2). In the assay employing the nifHF/nifHR primer pair, Efi values ranged from 1.67 in G. sulfurreducens to 1.77 in N. commune. In the assay targeting the 16S rRNA gene (primers 27F/518R), Efi values ranged from 1.84 in Pseudomonas stutzeri to 1.98 in Frankia sp. Some bacterial strains did not amplify when targeting the nifH gene with nifHF/nifHR and ForA/Rev primer pairs. In contrast, qPCR on nifH with PolF/PolR primers, as well as qPCR on the 16S rRNA gene (27F/518R), amplified DNA from all strains. The variation of Efi appeared to be independent of the phylogeny of the strains; however, we analyzed only a small subset of bacterial phyla and did not include Archaea or Eukarya in the analysis.
Fig 2.

Variability of qPCR efficiency, Efi, of bacterial strains (left) and environmental samples (right) targeting nifH and the 16S rRNA gene, estimated from the fluorescence increase. The error bars give the standard deviation of four replicates. The letters indicate significance groups by ANOVA (P < 0.01) tested for each panel individually. n.d., no amplification detected.
Similar to the observations made for bacterial strains, the Efis determined for environmental samples differed significantly (Fig. 2). Efis ranged from 1.72 for the oligotrophic lake (5 m) to 1.80 for the rhizosphere sample with the nifHF/nifHR primer pair. In the assay primed with PolF/PolR, Efis ranged from 1.77 in the eutrophic lake (10 m) to 1.99 in the bulk soil (old). Targeting the 16S rRNA gene, Efis ranged from 1.85 in the bulk soil (old) to 1.95 in the eutrophic lake (170 m). Interestingly, Efi was also variable for samples originating from within the same ecosystems. When amplified with the ForA/Rev primers, Efis in the oligotrophic lake sample ranged from 1.58 at 5-m depth to 1.83 at 200-m depth.
These results are supported by previous studies showing that E varies with the PCR template. Using the LinRegPCR method to calculate Efi, Čikos et al. (9) found that the interleukin 6 gene amplified with an Efi of 1.88, while the beta actin gene amplified with an Efi of 1.99. Moreover, they found that these differences between the genes remained regardless of the method used to determine Efi (9). Similarly, other studies found that E determined for the same PCR template is more constant (5, 9, 26). For example Bustin (5) reported a constant Efi of around 1.8 and low variation among the replicates for the amplification of the metalloproteinase inhibitor 1 gene. Furthermore, we could reproduce findings reported by Töwe and coworkers (47); we analyzed bulk soil from the Damma glacier and found that the Efi of bulk soil (1.75 to 1.76) was very similar to the Efi of Sinorhizobium meliloti (1.75) when using the nifHF/nifHR primer pair. Similarly, Töwe et al. (47) reported the same Efi (1.9) for both the sample from the Damma glacier and the standard from S. meliloti. The difference in Efi, 1.75 compared to 1.9, might be due to differing reaction conditions, i.e., assay chemistry, use of bovine serum albumin (BSA), and the thermocycler program. In summary, we demonstrated that over a range of bacterial strains and environmental samples tested, Efis differed significantly.
The differences in E can be caused by a number of mechanisms that have been previously addressed. The first studies on the problem emerged in environmental microbiology, as PCR was used to simultaneously amplify different templates (33, 44). The term PCR bias was coined to describe the preferential amplification of some templates over others, and the phenomenon has been studied extensively. The mechanisms identified as affecting PCR amplification and bias are as follows: the GC content of the primer binding site (33), primer mismatches (3), reannealing of the amplicon to templates (33), homoduplex formation during temperature decrease (22), the annealing temperature (15), PCR inhibitors (21, 54), and steric hindrance (13).
In our analysis, two mechanisms might have emphasized the significant differences in E: (i) the use of degenerate primers and (ii) the occurrence of primer mismatches. The use of degenerate primers has become widespread in qPCR analysis in order to target a broad set of organisms (12, 47, 50, 53). Degenerate primers are mixtures of highly similar primers that differ in at least one base, causing differences in the melting temperature (8). In the qPCR assay, we used primers with different degrees of degeneracy (2- to 128-fold) exhibiting wide ranges of melting temperatures (Table 2) that might have affected the kinetics of the qPCRs (15, 33) and so might have affected the E values of the individual assays. Similarly, primer mismatches significantly reduce E (3, 15). Despite the fact that we used degenerate primers (nifHF/nifHR), commonly reported in the literature, several of the bacterial strains exhibited one or more mismatches (see Table SA1 in the supplemental material). Although the number of mismatches was not correlated with E (n = 17; R2 = 5 × 10−5; P = 0.979), it might have accentuated the E differences. Nevertheless, the aim of our study was to demonstrate the effect of differences in E on the accuracy of the quantification, not to provide a detailed analysis of underlying mechanisms of primer bias.
Limitation of the SC method.
As demonstrated here, significant differences in qPCR E exist among bacterial strains and environmental samples. The SC quantification method does not account for the differences in E between the sample and the standard. Errors of up to orders of magnitude were observed in studies where the E values of the standard and the sample differed (3, 38). Bru et al. (3) observed that primer mismatches of a single base decrease E and can cause underestimation of the actual copy numbers by up to a factor of 1,000. A solution to this problem was found in gene expression analysis. The ΔΔCT method with correction for efficiency, also called the Pfaffl method or the comparative CT method with correction, was established (31, 48). It considers the differences in E of the analyzed genes to calculate the gene expression ratio (31). However, the ΔΔCT method is a relative quantification method only and cannot be used for absolute quantification.
Here, we propose the OPC method, which is based on the ΔΔCT method (31). The OPC method is an absolute quantification method. In contrast to the SC method, the OPC method accounts for template-related variability of E. This major advantage is achieved by considering the individual E values of the sample and the standard. One further advantage is that the OPC method does not necessarily rely on Eds estimates. Previous studies reported that Eds shows higher variability than Efi and demonstrated that Efi is less biased than Eds (18, 38, 30, 40). For example Karlen et al. (18) found that estimates of Efi are more robust than Eds estimates from dilution series. Similarly, in our analysis of five independently prepared dilution series, the overall variability was significantly higher for Eds than for Efi (see Fig. SA1 and Table SA2 in the supplemental material).
Hypothetical comparison of the SC and OPC methods.
Based on the efficiencies we detected (Fig. 2), we illustrate in a hypothetical example the errors that occur by not correcting for individual efficiency (Fig. 3). We quantify the nifH gene (PolF/PolR primers) in a hypothetical rhizosphere sample (CT sample = 21 and Esample = 1.99) using M. trichosporium as the standard (Estandard = 1.70). Using the SC method [CT = 33.382 − 4.34 × log(N0)], the estimate of the starting template concentration is as follows: log(N0 sample) = 2.85, i.e., N0 sample = 711 copies (Fig. 3). If we quantify the same sample using the OPC method (CT standard = 3; N0 standard = 1 × 107; Estandard = 1.70), we get N0 sample = N0 standard × EstandardCT standard × (EsampleCT sample)−1 = 107 × 1.703/1.9921 = 26. The estimate using the OPC method is 1 order of magnitude below the estimate by the SC method. This demonstrates the importance of correcting for the individual efficiencies.
Fig 3.

Quantification of a hypothetical sample (Esample = 1.99; CT = 21) by the SC and OPC methods using M. trichosporium (Estandard = 1.70) as a standard.
The bias due to ignoring the efficiency can be calculated as a ratio between OPC and SC quantifications (equation 5). It depends on the ratio of Estandard to Esample and the CT value of the sample. The more the ratio of Estandard to Esample deviates from 1, and the higher the CT value, the larger the bias will be. Therefore, the potential for error is particularly large when applying the SC method to samples with low template concentrations.
| (5) |
Experimental comparison of the SC and OPC methods.
Defined amounts of templates from two species were mixed in different ratios. The species were selected for their significant differences in E. G. sulfurreducens showed a lower Efi (1.69) than N. commune, which had an Efi of 1.77. We found that samples with a higher number of G. sulfurreducens templates had lower Efis than samples with a higher number of N. commune templates (Fig. 4).
Fig 4.

qPCR efficiency, Efi, of defined mixtures of G. sulfurreducens and N. commune templates amplified with nifHF/nifHR primers. The values are the means of three replicates; the error bars indicate the standard deviations.
Each of the defined mixtures was quantified by both the SC and OPC methods. Both methods were calibrated once with dilutions of G. sulfurreducens DNA and once with dilutions of N. commune DNA (Table 3). We considered a quantification to be accurate when the deviation of the estimate was within ±30% of the expected value. Using the SC method, the quantification of samples with a higher number of G. sulfurreducens templates was accurate when G. sulfurreducens was used as the standard (Fig. 5). Conversely, samples with a higher number of N. commune templates were accurately quantified when N. commune was used as the standard. SC method quantifications using the less suited standard were 3- to 5-fold above or below the expected copy numbers. In contrast, the OPC method accurately quantified samples, independent of the ratio between G. sulfurreducens and N. commune and independent of the standard used. Only one mixture (22:233) was quantified up to 110% above the expected copy number.
Table 3.
Parameters of SC and OPC methods used for sample quantification
| Template | Primer pair | SC method |
OPC method |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Slope | Intercept | R2 | nc | Eds | CT | N0 | Efi | ||
| G. sulfurreducensa | nifHF/nifHR | −4.249 | 40.86 | 0.993 | 8 | 1.72 | 22.2 | 22,160 | 1.69 |
| N. communea | nifHF/nifHR | −3.552 | 36.79 | 0.991 | 8 | 1.91 | 24.8 | 2,331 | 1.77 |
| B. japonicumb | nifHF/nifHR | −4.008 | 35.82 | 0.991 | 15 | 1.78 | 15.9 | 74,945 | 1.77 |
Used to quantify two-species mixtures of G. sulfurreducens and N. commune.
Used to quantify environmental samples.
Number of points in calibration.
Fig 5.

Copy numbers of defined mixtures of G. sulfurreducens (G. sul) and N. commune (N. com) templates estimated using the SC and OPC methods that were calibrated using both strains. The assays used nifHF/nifHR primers. The dashed lines give the expected copy numbers. The error bars give the standard deviations.
This experiment demonstrates that correcting for the individual E of each sample improves the accuracy of absolute quantification. The two bacterial strains amplified with different E values. Detecting the differences in E and accounting for them in the OPC method improved quantification accuracy independent of the calibration standard used and across a wide range of template mixtures amplifying with different E values. Therefore, the OPC method allows differences in the qPCR kinetics, which might have been introduced, e.g., by primer mismatches (3) or primer degeneracy (8), to be corrected.
Quantification of environmental samples by SC and OPC methods.
Nine environmental samples were quantified by SC and OPC methods using B. japonicum as the standard (Fig. 6). For the samples that had an Efi (Fig. 2) similar to the Eds of the standard (Table 3), differences between SC and OPC quantifications were not significant according to the Mann-Whitney rank sum test (P < 0.01). However, for the rhizosphere sample, copy numbers calculated by the SC and OPC methods were significantly different. This is because the Efi of the rhizosphere (1.80) was higher than the Eds of the SC (1.78). In this case, therefore, the SC method overestimated the copy number in the rhizosphere sample compared to the OPC method. On the other hand, the samples from the oligotrophic lake had an Efi lower than the Eds of the SC; therefore, the SC method significantly underestimated the copy numbers compared to the OPC method (Fig. 6). Quantification of the 16S rRNA gene in the environmental samples showed significant differences between the SC and OPC methods, too (data not shown). These results demonstrate the necessity of also considering sample and standard E values in comparative qPCR analysis of microbial communities, i.e., even if the absolute copy number is not of primary concern.
Fig 6.

Comparison of the OPC and SC methods for quantifying nifH gene copy numbers in environmental samples using nifHF/nifHR primers. The error bars give the standard deviations. The asterisks mark significant differences between the two methods by the Mann-Whitney rank sum test (P < 0.01).
Limitations of the OPC method.
The OPC method is an absolute quantification method that is simple to use. It corrects copy number estimates for the E of the individual sample and is compatible with any method to determine E, i.e., Eds (14, 35) or Efi (26, 30, 38) can be used.
Although the OPC method corrects for differences in E, it cannot overcome principle biases or limitations of the underlying PCR. For example, primer bias might introduce significant errors in the analysis of samples of unknown composition (44). Abundant templates and templates with the optimal primer kinetics might be preferentially amplified compared to templates with low abundance and suboptimal primer kinetics (33, 44). To some extent, these differences are accounted for because the OPC method corrects for differences in E. However, using degenerate primers, one specific template that matches one of the primers in the primer mixture might be preferentially amplified with the PCR program because of differences in GC content (33). Lowering the annealing temperature has been suggested to solve this problem (15). However, this may in turn increase nonspecific amplification, which is undesirable, specifically in SYBR green-based qPCR assays. Solid testing of the specificity and universality of the primers and the PCR protocol is therefore very important. Assays based on TaqMan are intrinsically less prone to reporting nonspecific amplification. In principle, the OPC method could be applied to TaqMan qPCR, and the LinReg program can be used to derive correct Efi estimates from TaqMan data, but this has not been tested.
The OPC method requires reliable E estimates, which might depend on the template concentration. Results by Bustin (5) and our own results (data not shown) do not support this hypothesis. However, other studies found indications that high template concentrations (>106 copies per reaction) had an inhibitory effect (46, 47). Furthermore, according to the model presented by Suzuki and Giovannoni (44), an inhibitory effect of high template concentrations on E would be expected. This problem should be addressed in future studies.
Since quantification by the OPC method relies on a single standard, care should be taken that CT and E for the standard are determined correctly, e.g., by using a larger number of replicates. The extent to which the OPC method can reliably correct for variation in E has not been determined. Samples with very low E values or irregularly shaped amplification plots should be carefully considered. The method also cannot address cases in which low template concentration, inhibition, or poorly optimized PCR conditions lead to negative results. In such cases, additional tests might be required.
Conclusion.
Currently, the gold standard for qPCR analysis in environmental microbiology is based on the SC method. This method assumes that the calibration standard and the sample have similar E values, although the SC method itself does not allow this assumption to be validated through testing. While the E of the standard is routinely assessed and is considered a quality criterion, few studies have tested if the E values of the sample and standard are similar (41, 47); however, even small differences can cause considerable errors (3).
Here, we demonstrated that significant differences in E can occur between bacterial strains and environmental samples, specifically when amplifying broad ranges of templates using, e.g., degenerate primers, which is a common practice in microbial ecology. Not considering those differences has the potential to severely diminish the accuracy of the quantification.
To solve the problem, we propose the OPC method, which corrects for the E values of individual samples. Moreover, we demonstrated experimentally that by correcting for the E of a sample by this method, the accuracy of the quantification increases.
Based on the results and considerations presented here, E needs to be considered in qPCR analysis when working with samples of unknown template composition. The E of each sample can be calculated from the increase in fluorescence units using freely available tools (26, 30, 38). If the sample and standard have the same E, the SC method can be used, although we see no principle advantage over the OPC method.
Supplementary Material
ACKNOWLEDGMENTS
This study is part of Transregional Collaborative Research Centre 38 (SFB/TRR 38).
The work of R.B. and J.Z. was financially supported by the Deutsche Forschungsgemeinschaft (DFG) (Bonn, Germany) and the Brandenburg Ministry of Science, Research and Culture (MWFK) (Potsdam, Germany). The research of N.B. was supported by the Marie Heim-Vögtlin program of the Swiss National Science Foundation.
We thank Julia Vorholt for kindly providing bacterial strains and DNA extracts from phyllosphere and rhizosphere; Krista Köllner for providing DNA extracts from lake water samples; Silvina Pombo for cultivating Geobacter sulfurreducens; Anna Lazzaro, Alessandro Franchini, and Marco Meola for preparing dilution series; and two anonymous reviewers and, in particular, Marcelino T. Suzuki for their constructive comments on the manuscript.
Footnotes
Published ahead of print 6 April 2012
Supplemental material for this article may be found at http://aem.asm.org.
REFERENCES
- 1. Bekele AZ, Koike S, Kobayashi Y. 2011. Phylogenetic diversity and dietary association of rumen Treponema revealed using group-specific 16S rRNA gene-based analysis. FEMS Microbiol. Lett. 316:51–60 [DOI] [PubMed] [Google Scholar]
- 2. Brankatschk R, Töwe S, Kleineidam K, Schloter M, Zeyer J. 2011. Abundances and potential activities of nitrogen cycling microbial communities along a chronosequence of a glacier forefield. ISME J. 5:1025–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bru D, Martin-Laurent F, Philippot L. 2008. Quantification of the detrimental effect of a single primer-template mismatch by real-time PCR using the 16S rRNA gene as an example. Appl. Environ. Microbiol. 74:1660–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burns M, Valdivia H. 2008. Modelling the limit of detection in real-time quantitative PCR. Eur. Food Res. Technol. 226:1513–1524 [Google Scholar]
- 5. Bustin SA. 2004. Quantification of nucleic acids by PCR, p 3–48 In Bustin SA. (ed), A-Z of quantitative PCR. IUL biotechnology series, vol 5 International University Line, La Jolla, CA [Google Scholar]
- 6. Bustin SA, et al. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55:611–622 [DOI] [PubMed] [Google Scholar]
- 7. Capela D, et al. 2001. Analysis of the chromosome sequence of the legume symbiont Sinorhizobium meliloti strain 1021. Proc. Natl. Acad. Sci. U. S. A. 98:9877–9882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen H, Zhu G. 1997. Computer program for calculating the melting temperature of degenerate oligonucleotides used in PCR or hybridization. Biotechniques 22:1158–1160 [DOI] [PubMed] [Google Scholar]
- 9. Čikos S, Bukovská A, Koppel J. 2007. Relative quantification of mRNA: comparison of methods currently used for real-time PCR data analysis. BMC Mol. Biol. 8:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Gregoris TB, Aldred N, Clare AS, Burgess JG. 2011. Improvement of phylum- and class-specific primers for real-time PCR quantification of bacterial taxa. J. Microbiol. Methods 86:351–356 [DOI] [PubMed] [Google Scholar]
- 11. Espy MJ, et al. 2006. Real-time PCR in clinical microbiology: applications for a routine laboratory testing. Clin. Microbiol. Rev. 19:165–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geets J, et al. 2007. Real-time PCR assay for the simultaneous quantification of nitrifying and denitrifying bacteria in activated sludge. Appl. Microbiol. Biotechnol. 75:211–221 [DOI] [PubMed] [Google Scholar]
- 13. Hansen MC, Tolker-Nielsen T, Givskov M, Molin S. 1998. Biased 16S rDNA PCR amplification caused by interference from DNA flanking the template region. FEMS Microbiol. Ecol. 26:141–149 [Google Scholar]
- 14. Higuchi R, Fockler C, Dollinger G, Watson R. 1993. Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Nat. Biotechnol. 11:1026–1030 [DOI] [PubMed] [Google Scholar]
- 15. Ishii K, Fukui M. 2001. Optimization of annealing temperature to reduce bias caused by a primer mismatch in multitemplate PCR. Appl. Environ. Microbiol. 67:3753–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Janssen PH, Yates PS, Grinton BE, Taylor PM, Sait M. 2002. Improved culturability of soil bacteria and isolation in pure culture of novel members of the divisions Acidobacteria, Actinobacteria, Proteobacteria, and Verrucomicrobia. Appl. Environ. Microbiol. 68:2391–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kandeler E, Deiglmayr K, Tscherko D, Bru D, Philippot L. 2006. Abundance of narG, nirS, nirK, and nosZ genes of denitrifying bacteria during primary successions of a glacier foreland. Appl. Environ. Microbiol. 72:5957–5962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karlen Y, McNair A, Perseguers S, Mazza C, Mermod N. 2007. Statistical significance of quantitative PCR. BMC Bioinform. 8:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Knief C, et al. Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice. ISME J. doi:10.1038/ismej.2011.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Köllner K, et al. Bacterial chitin hydrolysis in two lakes of contrasting trophic status. Appl. Environ. Microbiol. 78:695–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kontanis EJ, Reed FA. 2006. Evaluation of real-time PCR amplification efficiencies to detect PCR inhibitors. J. Forensic Sci. 51:795–804 [DOI] [PubMed] [Google Scholar]
- 22. Kurata S, et al. 2004. Reevaluation and reduction of a PCR bias caused by reannealing of templates. Appl. Environ. Microbiol. 70:7545–7549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leininger S, et al. 2006. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442:806–809 [DOI] [PubMed] [Google Scholar]
- 24. Liebner S, Schwarzenbach S, Zeyer J. 2011. Methane emissions from an alpine fen in central Switzerland. Biogeochemistry doi:10.1007/s10533-011-9629-4 [Google Scholar]
- 25. Liebner S, et al. 2011. Methane oxidation associated with submerged brown mosses reduces methane emissions from Siberian polygonal tundra. J. Ecol. 99:914–922 [Google Scholar]
- 26. Liu W, Saint D. 2002. A new quantitative method of real time reverse transcription polymerase chain reaction assay based on simulation of polymerase chain reaction kinetics. Anal. Biochem. 302:52–59 [DOI] [PubMed] [Google Scholar]
- 27. Mackay IM, Arden KE, Nitsche A. 2002. Real-time PCR in virology. Nucleic Acids Res. 30:1292–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsui K, Ishii N, Honjo M, Kawabata Z. 2004. Use of the SYBR Green I fluorescent dye and a centrifugal filter device for rapid determination of dissolved DNA concentration in fresh water. Aquat. Microb. Ecol. 36:99–105 [Google Scholar]
- 29. Muyzer G, Dewaal EC, Uitterlinden AG. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S ribosomal RNA. Appl. Environ. Microbiol. 59:695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peirson SN. 2003. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 31:e73 doi:10.1093/nar/gng073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45 doi:10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Poly F, Monrozier LJ, Bally R. 2001. Improvement in the RFLP procedure for studying the diversity of nifH genes in communities of nitrogen fixers in soil. Res. Microbiol. 152:95–103 [DOI] [PubMed] [Google Scholar]
- 33. Polz MF, Cavanaugh CM. 1998. Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 64:3724–3730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ramakers C, Ruijter JM, Deprez RHL, Moorman AFM. 2003. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 339:62–66 [DOI] [PubMed] [Google Scholar]
- 35. Rasmussen R. 2001. Quantification on the LightCycler, p 21–34 In Meuer SC, Wittwer C, Nakagawara K. (ed), Rapid cycle real-time PCR. Methods and applications. Springer, Berlin, Germany [Google Scholar]
- 36. Ritz C, Spiess A-N. 2008. qpcR: an R package for sigmoidal model selection in quantitative real-time polymerase chain reaction analysis. Bioinformatics 24:1549–1551 [DOI] [PubMed] [Google Scholar]
- 37. Rösch C, Mergel A, Bothe H. 2002. Biodiversity of denitrifying and dinitrogen-fixing bacteria in an acid forest soil. Appl. Environ. Microbiol. 68:3818–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ruijter JM, et al. 2009. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 37:e45 doi:10.1093/nar/gkp045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 40. Schefe JH, Lehmann KE, Buschmann IR, Unger T, Funke-Kaiser H. 2006. Quantitative real-time RT-PCR data analysis: current concepts and the novel “gene expression's CT difference” formula. J. Mol. Med. 84:901–910 [DOI] [PubMed] [Google Scholar]
- 41. Schubert CJ, et al. 2006. Aerobic and anaerobic methanotrophs in the Black Sea water column. Environ. Microbiol. 8:1844–1856 [DOI] [PubMed] [Google Scholar]
- 42. Smith CJ, Nedwell DB, Dong LF, Osborn AM. 2007. Diversity and abundance of nitrate reductase genes (narG and napA), nitrite reductase genes (nirS and nrfA), and their transcripts in estuarine sediments. Appl. Environ. Microbiol. 73:3612–3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Smith CJ, Osborn AM. 2009. Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol. Ecol. 67:6–20 [DOI] [PubMed] [Google Scholar]
- 44. Suzuki MT, Giovannoni SJ. 1996. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 62:625–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tichopad A, Dilger M, Schwarz G, Pfaffl MW. 2003. Standardized determination of real-time PCR efficiency from a single reaction set-up. Nucleic Acids Res. 31:e122 doi:10.1093/nar/gng122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tichopad A, et al. 2010. Quality control for quantitative PCR based on amplification compatibility test. Methods 50:308–312 [DOI] [PubMed] [Google Scholar]
- 47. Töwe S, Kleineidam K, Schloter M. 2010. Differences in amplification efficiency of standard curves in quantitative real-time PCR assays and consequences for gene quantification in environmental samples. J. Microbiol. Methods 82:338–341 [DOI] [PubMed] [Google Scholar]
- 48. VanGuilder HD, Vrana KE, Freeman WM. 2008. Twenty-five years of quantitative PCR for gene expression analysis. Biotechniques 44:619–626 [DOI] [PubMed] [Google Scholar]
- 49. von Wintzingerode F, von Gobel UB, Stackebrandt E. 1997. Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol. Rev. 21:213–229 [DOI] [PubMed] [Google Scholar]
- 50. Wallenstein MD, Vilgalys RJ. 2005. Quantitative analyses of nitrogen cycling genes in soils. Pedobiologia 49:665–672 [Google Scholar]
- 51. Walsh F, et al. 2011. Real-time PCR methods for quantitative monitoring of streptomycin and tetracycline resistance genes in agricultural ecosystems. J. Microbiol. Methods 86:150–155 [DOI] [PubMed] [Google Scholar]
- 52. Whittenbury R, Phillips KC, Wilkinson JF. 1970. Enrichment, isolation and some properties of methane-utilizing bacteria. J. Gen. Microbiol. 61:205–218 [DOI] [PubMed] [Google Scholar]
- 53. Widmer F, Shaffer BT, Porteous LA, Seidler RJ. 1999. Analysis of nifH gene pool complexity in soil and litter at a Douglas fir forest site in the Oregon Cascade Mountain Range. Appl. Environ. Microbiol. 65:374–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wiedbrauk DL, Werner JC, Drevon AM. 1995. Inhibition of PCR by aqueous and vitreous fluids. J. Clin. Microbiol. 33:2643–2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zehr JP, et al. 2008. Globally distributed uncultivated oceanic N2-fixing cyanobacteria lack oxygenic photosystem II. Science 322:1110–1112 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.