

Abstract
Type 1 fimbriae and flagella, two surface organelles critical for colonization of the urinary tract by uropathogenic Escherichia coli (UPEC), mediate opposing virulence objectives. Type 1 fimbriae facilitate adhesion to mucosal cells and promote bacterial persistence in the urinary tract, while flagella propel bacteria through urine and along mucous layers during ascension to the upper urinary tract. Using a transposon screen of the E. coli CFT073 fim locked-ON (L-ON) mutant, a construct that constitutively expresses type 1 fimbriae and represses motility, we identified six mutants that exhibited a partial restoration of motility. Among these six mutated genes was mutS, which encodes a component of the methyl-directed mismatch repair (MMR) system. When complemented with mutS in trans, motility was again repressed. To determine whether the MMR system, in general, is involved in this reciprocal control, we characterized the effects of gene deletions of other MMR components on UPEC motility. Isogenic deletions of mutS, mutH, and mutL were constructed in both wild-type CFT073 and fim L-ON backgrounds. All MMR mutants showed an increase in motility in the wild-type background, and ΔmutH and ΔmutS mutations increased motility in the fim L-ON background. Cochallenge of the wild-type strain with an MMR-defective strain showed a subtle but significant competitive advantage in the bladder and spleen for the MMR mutant using the murine model of ascending urinary tract infection after 48 h. Our findings demonstrate that the MMR system generally affects the reciprocal regulation of motility and adherence and thus could contribute to UPEC pathogenesis during urinary tract infections.
INTRODUCTION
Flagella, surface organelles that drive motility, and fimbriae, surface appendages that mediate adherence, perform opposing functions, and yet both are important for some bacterial pathogens to infect their host (5, 43). Specifically, in uropathogenic Escherichia coli (UPEC), type 1 fimbriae are highly expressed in vivo in the murine model of urinary tract infections (UTI) and are considered necessary for the bladder colonization by UPEC in this model (19, 52). Whereas type 1 fimbriae allow establishment of infection in the bladder by binding to mannose-containing glycoproteins, flagella allow UPEC to ascend from the bladder to the kidneys (31, 40, 47). One may argue that a single bacterium should not be both adherent and motile simultaneously, and we have provided evidence that the expression of fimbriae and flagella is reciprocally regulated (3, 9, 18, 30, 34).
The promoter that controls type 1 fimbriae expression resides upstream from the major structural subunit gene fimA and is located on an invertible element that alternates between two orientations, phase ON and phase OFF (1). When the promoter is facing the fim operon (fimAICDFGH), it is in the phase ON orientation and type 1 fimbriae are expressed. Alternatively, when the promoter faces in the opposite direction of the fim operon, it is phase OFF and the transcription of fim genes is repressed (6, 15, 26, 37). Alternatively, flagellin expression is regulated in a hierarchal manner consisting of three classes with more than 40 genes expressed (8, 53). Transcription of the class I flagellar genes, flhDC, is necessary for the transcription of the class II flagellar genes, including fliA, the sigma factor that promotes expression of class III flagellar genes. Reciprocal control of adherence and motility explains that when it is advantageous for the bacteria to be motile, flagella are expressed and fimbriae are repressed because adherence would impede flagellar motility. Conversely, when it is advantageous for bacteria to be adherent, fimbriae are expressed and motility is repressed. Examples of such regulation have been observed in other pathogens, including Vibrio cholerae (18), Salmonella enterica serovar Typhimurium (9), Bordetella pertussis (3), and Proteus mirabilis (34).
A transposon screen of UPEC CFT073 mutants in which the fim promoter is locked in the phase ON orientation (CFT073 fim L-ON) identified six nonfimbrial genes involved in the repression of motility: one is mutS, a gene encoding a component of the methyl-directed mismatch repair (MMR) system (49). MMR is a well-characterized and widely conserved DNA repair system found in both eukaryotes and prokaryotes. The MMR pathway in E. coli is primarily composed of three proteins: MutS, MutL, and MutH (23, 24, 39). MutS recognizes mismatches in newly replicated chromosomal DNA; MutL forms a complex with MutS and recruits MutH in an ATP-dependent manner, allowing MutH to nick the unmethylated daughter strand at a hemimethylated GATC site (4, 21, 28, 56). Helicase II (UvrD), activated by the MutS/MutL complex, separates the daughter and the parent strands (12, 22, 57), while exonuclease Exo I, Exo VII, or RecJ degrades the mismatched containing DNA strand (10). After resynthesis of the gapped region, DNA ligase seals the nick to complete the repair process (28). MMR primarily corrects single base nucleotide mismatches, and small insertion/deletion mismatches introduced during DNA replication (for reviews, see references 27 and 46). MMR contributes ∼1,000-fold to the overall fidelity of the DNA replication pathway. In addition to correcting base-pairing errors, the MMR system in E. coli also prevents the recombination between divergent DNA sequences (44).
The striking role that we identified for MutS in the regulation of motility was confirmed by the construction of a clean isogenic mutS deletion in CFT073 that demonstrated increased motility (49). Complementation with mutS in trans restored motility and flagellin production to wild-type levels (49). We hypothesized that the entire MMR system and not just MutS mediated this reciprocal control of adherence and motility.
To test this hypothesis, we examined the contribution of three components of the methyl-directed mismatch repair system to the motility of wild-type CFT073 and CFT073 fim L-ON bacteria. FliC and FimA expression was directly assessed by Western blot, by immunofluorescence microscopy, and by determining the orientation of the type 1 fimbrial promoter by the invertible element assay and indirectly assessed by quantitative PCR (qPCR). Using the murine model of ascending UTI, we demonstrated that the loss of MMR decreased UPEC fitness only when type 1 fimbriae are expressed constitutively, whereas deletion of mutS in wild-type CFT073 increased the dissemination of bacteria into the bloodstream.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The prototype uropathogenic E. coli strain CFT073 was isolated from the urine and blood of a hospitalized patient suffering from acute pyelonephritis (38, 55). All E. coli strains and plasmids used in the present study are listed in Table 1. E. coli strains were cultured on Luria-Bertani (LB) agar or in LB broth and were incubated overnight at 37°C; liquid cultures were agitated at 200 rpm. Ampicillin (100 μg/ml), kanamycin (25 μg/ml), and chloramphenicol (20 μg/ml) were included as appropriate.
Table 1.
Relevant bacterial strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Descriptiona or sequence (5′–3′) | Source or reference |
|---|---|---|
| E. coli strains | ||
| CFT073 | Wild-type pyelonephritis isolate | 38 |
| fim L-ON | CFT073 fim IE phase locked ON; constitutive type 1 fimbrial expression | 19 |
| fiml-OFF | CFT073 fim IE phase locked OFF; blocking type 1 fimbrial expression | 19 |
| ΔmutS mutant | CFT073 ΔmutS:Kanr | 49 |
| ΔmutH mutant | CFT073 ΔmutH:Kanr | This study |
| ΔmutL mutant | CFT073 ΔmutL:Camr | This study |
| fim L-ON ΔmutS | CFT073 fim L-ON ΔmutS:Kanr | 49 |
| fim L-ON ΔmutH | CFT073 fim L-ON ΔmutH:Kanr | This study |
| fim L-ON ΔmutL | CFT073 fim L-ON ΔmutL:Kanr | This study |
| Plasmids | ||
| pGEN-MCS | Derivative of pGEN222 with a multiple cloning site replacing gfpuv (Ampr) | 31 |
| pGEN-mutS | CFT073 mutS cloned into BamHI-SphI sites of pGEN-MCS (Ampr) | 49 |
| pGEN-mutH | CFT073 mutH cloned into EcoRI-HindIII sites of pGEN-MCS (Ampr) | This study |
| pGEN-mutL | CFT073 mutL cloned into EcoRI-HindIII sites of pGEN-MCS (Ampr) | This study |
| Primers | ||
| Invertible element | ||
| IE-P1 foward | AGTAATGCTGCTCGTTTTGC | |
| IE-P2 reverse | GACAGAGCCGACAGAACAAC | |
| Mutant construction | ||
| mutS H1P1 | TGAAAGCCCAGCATCCCGAGATCCTGCTGTTTTACCGGATGTGTAGGCTGGAGCTGCTTC | |
| mutS H2P2 | TGCCGTGCGCGCTTAATAACCTCTTTTGGCACACCCGCCAATGGGAATTAGCCATGGTCC | |
| mutH H1P1 | ATGTCCCAACCTCGCCCACTGCTCTCTCCTCCCGGTGTAGGCTGGAGCTGCTGCTTC | |
| mutH H2P2 | ATCCGTTCGCCCCGGGCACCAATAGCTTCGGTCAGCATGGGAATTAGCCATGGTCC | |
| mutL L-ON H1P1 | GGGTGCAACGGCACAAACGGCCAGTACGGTGACGACGGTGTAGGCTGGAGCTGCTTC | |
| mutL L-ON H2P2 | GCGGACATAACCGTTCCACGTCCGCCAGCAGGGATGGGAATTAGCCATGGTCC | |
| mutL H1P1 | ACCGCCACAACTGGCGAACCAGATTGCCGCAGGTGTAGGCTGGAGCTGCTTC | |
| mutL H2P2 | TGGCCTGTGCCATTGACCACTGCGCATGTTCGCATGGGAATTAGCCATGGTCC | |
| Mutant screen | ||
| mutS forward | CCCGAGATCCTGCTGTTTTA | |
| mutS reverse | TGCGCGCTTAATAACCTCTT | |
| mutH forward | GTTACACTGCGAATATTCAGC | |
| mutH reverse | CAATCCTAGCCAATAGGTACG | |
| mutL forward | GATGCAATCTGCGCCGCAGG | |
| mutL reverse | CAGCTCAATGGCTAACGCCG | |
| Mutant complementation | ||
| pGEN-MCS forward | ACTTGCTCACGCTCTGTCAT | |
| pGEN-MCS reverse | AGAAGGACCATGTGGTCACG | |
| mutS forward | CAGCATGCGGACATGCTGCCTCCATACT | |
| mutS reverse | TGGATCCAGCAAAAGGCTATCGGGAAT | |
| mutH forward | ATATATGAATTCGGATAACATGGTGTAAGATCCTGTT | |
| mutH reverse | GTGCTGTGGGAAGCTTTGCCACCAC | |
| mutL forward | ATATATGAATTCGTTCTGCGTTCGCCGGATATTCCG | |
| mutL reverse | ATATATAAGCTTCTGCGACGGATCGCGAATATTCAG | |
| Sanger sequencing | ||
| mutH forward | GGCCGATACAAACCGGCTTCGTGTC | |
| mutH reverse | CGCCATGACGTAATCACTGGAACAC | |
| mutL forward | GTACCTTGCTCAAT | |
| mutL reverse | CGAACTGATGCAC | |
| Pap promoter forward | GCGTGAAGAGTATTTCCGGGCC | |
| Pap promoter reverse | CCATAGCTACCGCACCGGCA | |
| FlhDC promoter forward | TCTGTGAACTTCAGGTGAC | |
| FlhDC promoter reverse | TCAGCAACTCGGAGGTATGC | |
| qPCR | ||
| gapA forward | AAGTTGGTGTTGACGTTGTCGC | |
| gapA reverse | AGCGCCTTTAACGAACATCG | |
| fliC foward | ACAGCCTCTCGCTGATCACTCAA | |
| fliC reverse | GCGCTGTTAATACGCAAGCCAGA | |
| fimA forward | ACTCTGGCAATCGTTGTTCTGTCG | |
| fimA reverse | ATCAACAGAGCCTGCATCAACTGC |
Camr, chloramphenicol resistance; Ampr, ampicillin resistance; Kanr, kanamycin resistance.
Construction and complementation of isogenic mutants of E. coli CFT073 and CFT073 fim L-ON.
Deletion of MMR genes was accomplished in strains CFT073 and CFT073 fim L-ON using lambda red recombinase technology (13). To construct CFT073 ΔmutH, CFT073 fim L-ON ΔmutH (ΔmutH L-ON), and CFT073 fim L-ON ΔmutL (ΔmutL L-ON), using primers homologous to sequences flanking the 5′ and 3′ ends of the target gene, the kanamycin resistance gene was PCR amplified from plasmid pKD4 (the primers are listed in Table 1). To create CFT073 ΔmutL the same technology was used; however, the chloramphenicol resistance gene from template plasmid pKD3 was amplified (Table 1). Mutants were verified by using diagnostic PCR with the primers listed in Table 1. CFT073 ΔmutS and CFT073 fim L-ON ΔmutS (ΔmutS L-ON) mutants were constructed and verified as previously described (49).
To complement mismatch repair mutants, target genes with associated promoters were PCR- amplified (Table 1). The resulting PCR fragments were digested with EcoRI and HindIII, followed by ligation into a digested pGEN-MCS plasmid, a derivative of pGEN222 (17), with EcoRI and HindIII. The newly constructed pGEN plasmids with the inserted target gene were used to transform their respective mutant strains. Antibiotic resistance to ampicillin, as well as antibiotic resistance of the mutant strain to kanamycin or chloramphenicol, was used to select for transformants. Constructs were confirmed by PCR using the primer sets listed (Table 1).
Motility assay on soft agar.
Soft agar plates were used to evaluate the motility of bacterial strains and constructs, as previously described (32). Briefly, a sterile inoculating needle was dipped into a standardized (optical density at 600 nm [OD600] of 1.0), late-log-phase liquid culture, stabbed into the middle of a soft agar (0.25% [wt/vol]) plate, where touching the bottom of the plate was avoided. Motility diameters were measured after 16 h of incubation at 30°C. This was repeated at least in triplicate, and wild-type CFT073 and CFT073 fim L-ON were always included as controls. Ampicillin was added to the cultures and soft-agar plates when determining the motility of complemented mutants.
Capillary chemotaxis assay.
An alternative standardized quantitative measurement of random motility was conducted by using a modified capillary chemotaxis assay (2, 29, 42). Strains cultured on motility agar and then used to inoculate LB medium were incubated to mid-exponential phase (OD600 = 0.3 to 0.4), harvested by centrifugation to remove all spent medium, and resuspended and diluted (1:10) in phosphate-buffered saline (PBS). Samples of diluted, resuspended cultures (100 μl) were serially diluted and plated where CFU/ml was determined after an overnight incubation at 37°C. Bacterial suspensions (500 μl) were added to the wells of the chemotaxis chambers, followed by microcapillaries filled with sterile PBS. Chemotaxis chambers were incubated at 30°C for 90 min, and the contents of the capillaries were diluted and plated. The CFU/ml was determined after an overnight incubation at 37°C, and results are presented as the percentage of input bacteria present in the capillary. Assays were conducted in triplicate, and wild-type CFT073 and CFT073 fim L-ON were always included as controls.
Detection of flagellum and type 1 fimbrial protein production.
Western blotting was used to detect flagellin and type 1 fimbrial protein production, while qPCR was used to detect fliC and fimA transcript levels. For Western blotting, 15 μl of standardized (OD600 = 1.0), mid-log-phase cultures were added to 4 μl of 6× sodium dodecyl sulfate (SDS) loading buffer and boiled for 10 min. Lysates were electrophoresed on a 12.5% denaturing SDS-polyacrylamide (SDS-PAGE) gel and transferred to a polyvinylidene difluoride (PVDF) membrane (Immobilon-P; Millipore Corp.). The membrane was blocked with 2% milk in Tris-buffered saline plus 0.01% Tween 20 (TBST). FliC was detected with rabbit anti-FliC (1:40,000 dilution with TBST), followed by anti-rabbit-horseradish peroxidase (HRP) (1:20,000 dilution with TBST) (Sigma) and Amersham ECL Plus detection reagents (GE Healthcare Life Sciences).
Standardized whole-cell lysates were used to detect the FimA subunit of type 1 fimbriae by Western blotting. Bacterial pellets from 100 μl of culture were resuspended in 100 μl of acidified double-distilled H2O (pH 1.8) and boiled for 7 min. 15 μl of resuspended lysates were added to 10 μl of 2.5× SDS loading buffer, neutralized with 1 N NaOH (∼2.0 μl), electrophoresed on a SDS–12.5% PAGE gel, and transferred to a PVDF membrane blocked as previously described. FimA was detected using rabbit anti-FimA (1:10,000 dilution with TBST), followed by anti-rabbit-HRP (1:20,000 dilution with TBST), and Amersham ECL Plus detection reagents.
qPCR.
To extract total fliC and fimA RNA for qPCR, 500 μl of a standardized (OD600 = 1.0), late-log-phase culture was added to 65 μl of ice-cold phenol-ethanol stop solution (5% phenol in ethanol) and was collected by centrifugation. Using lysozyme, cells were lysed and RNA was extracted using the RNeasy kit (Qiagen) following the manufacturer's recommended protocol. Nucleic acid concentrations were determined by spectrophotometry (NanoDrop), and any DNA contamination was removed by incubation of the samples with 4 U of Turbo DNase (Ambion). RNA was recovered, quantified, and used as a template for PCR to confirm inactivation of any trace DNA. PCR-negative RNA was used for first-strand cDNA synthesis using SuperScript II reverse transcriptase (Invitrogen) according to manufacturer's protocol and using 50 ng of random hexamers. cDNA was purified on a QIAquick column (Qiagen), quantified, and diluted to 6 ng/μl with nuclease-free water. RNA transcripts were quantified on a MX3000P real-time PCR instrument (Stratagene) using Brilliant SYBR green qPCR mix (Stratagene) in 25-μl volumes containing 30 ng of cDNA. Optimal primer concentrations were determined empirically and are listed in Table 1.
For comparative, quantitative analysis, transcript levels were normalized to the level of gapA (glyceraldehyde 3-phosphate dehydrogenase A). Fold changes were determined using wild-type CFT073 and CFT073 fim L-ON as calibrators and the MXPro v 3.00 software package (Stratagene). Any transcript level comparisons greater than a 2-fold change (log2 = 1) were considered significant changes. Assays were conducted in duplicate. Wild-type CFT073 and CFT073 fim L-ON were always included as controls.
Indirect fluorescent antibody staining.
Sequential staining was used to quantify surface flagella. Standardized bacterial cultures (OD600 = 1.0) were diluted with LB (1:2), spotted, and dried onto glass microscope coverslips (Fisher), and fixed with 4% formaldehyde in PBS. Coverslips were washed with PBS three times, and flagella were detected with rabbit anti-FliC (1:1,000 dilution with 1% fetal bovine serum [FBS]) and anti-rabbit IgG (1:5,000 dilution with 1% FBS) (Alexa Fluor 488 conjugate; Molecular Probes). To detect nucleic acid, the slides were washed once with a dilution of 1.0 mg/ml propidium iodide (Sigma) in PBS (1:10,000), washed twice with PBS, and mounted using p-phenylenediamine onto glass microscope slides (25 by 75 by 1 mm; VWR). Slides were viewed on an Olympus BX60 upright microscope; images were quantified, and backgrounds were corrected using Metamorph Premier software (Molecular Devices, Inc.). Ratios of parental flagellar counts were calculated by dividing the ratio of flagella per mutant cell by the ratio of flagella per parental strain, quantified over three microscopic fields.
Invertible element PCR assay.
The orientation of the fim invertible element was determined using a PCR assay as previously described (35). Briefly, the primers IE-P1 and IE-P2 (Table 1) were used to amplify the 601-bp invertible element using bacteria from standardized (OD600 = 1.0) cultures. CFT073 and CFT073 fim L-ON were included as controls. Asymmetric digestion of the PCR products with SnaBI allowed determination of the orientation of the invertible element when electrophoresed on a 2% agarose–1× Tris-acetate-EDTA buffer (TAE) gel.
Measurement of growth rate.
Growth curves of the mutant constructs were generated by inoculating 3 ml of LB broth with 50 μl overnight culture, in duplicate, using μQuant (Bio-Tek Instruments, Inc. [KCJunior Software]). The OD600 of a culture sample (200 μl) was recorded every hour for 7 h. Wild-type CFT073 and CFT073 fim L-ON were always included as controls. Growth rates were determined using the equation μ = [(log10 N –log10 N0) 2.303]/(t − t0), where N is the concentration of cells (number of cells per unit volume) and t is time.
Measurement of spontaneous mutation frequency.
Spontaneous mutation frequencies were determined as described previously (48, 51). Briefly, 1.5 ml of late-log-phase cultures were standardized (OD600 = 1.0), centrifuged, and resuspended in 100 μl of 0.85% saline. The suspension was serially diluted and plated onto LB agar plates. The CFU/ml was determined after an overnight incubation at 37°C. The remainder of the suspension was plated onto LB agar plates containing rifampin (100 μg/ml). CFU of rifampin-resistant mutants were scored after an overnight incubation at 37°C, and spontaneous mutation frequencies were calculated by dividing the number of rifampin-resistant colonies by total viable cells.
CBA/J mouse model of ascending UTI.
Female CBA/J mice, 6 to 8 weeks old, were inoculated transurethrally as previously described (20). Briefly, pelleted overnight cultures (50 ml) were resuspended to an OD600 of ∼4.0 in sterile PBS. Diluted bacterial suspensions were mixed in a 1:1 ratio, and 50 μl containing 108 CFU was used to transurethrally inoculate each mouse. At 48 h or 7 days postinoculation, urine was collected, and the bladders, kidneys, and spleens were aseptically removed, weighed, and homogenized (Omni International). Serial dilutions were plated onto LB agar plates with or without kanamycin using a spiral plater (Autoplate 4000; Spiral Biotech) to quantify CFU of wild-type CFT073 (Kans) and mutant (Kanr), respectively. Competitive indices were calculated by dividing the ratio of wild-type to mutant at an endpoint CFU/ml by the ratio of wild-type to mutant input CFU/ml. A competitive index <1 indicates a fitness defect of the mutant strain.
Statistical analysis.
A paired Student t test was used to determine significant differences in motility between strains in motility assays (InStat; GraphPad Prism Software). For the murine model, statistically significant differences of in vivo colonization (P < 0.05) were calculated using a two-tailed, Wilcoxon signed-rank test where column medians were compared to a hypothetical value of zero (InStat; GraphPad Prism Software). Prism Software (GraphPad) was also used in linear regression analyses of motility data.
RESULTS
Deletions in the mismatch repair system increase the mutation rate of CFT073.
Deletion mutants were constructed for mutS, mutH, and mutL in both the wild-type CFT073 and CFT073 fim L-ON backgrounds. To verify that mutants were MMR defective, we measured the spontaneous mutation frequency by determining the occurrence of rifampin-resistant bacteria in culture (54). Rifampin resistance can be conferred by missense mutations in the rpoB (β subunit) gene of RNA polymerase. Defects in MMR cause an increase in spontaneous mutation frequency, which can be measured by an increase in the occurrence of rifampin-resistant bacteria (54).
Deletion of mutS, mutH and mutL resulted in an ∼100-fold increase in spontaneous mutation frequency in both the wild-type CFT073 and CFT073 fim L-ON genetic backgrounds. The frequency of mutant occurrence was nearly indistinguishable between the mutS, mutL, and mutH deletion constructs (Fig. 1). These results confirm that the MMR pathway is inactive in constructs deleted for mutS, mutH, and mutL (33).
Fig 1.
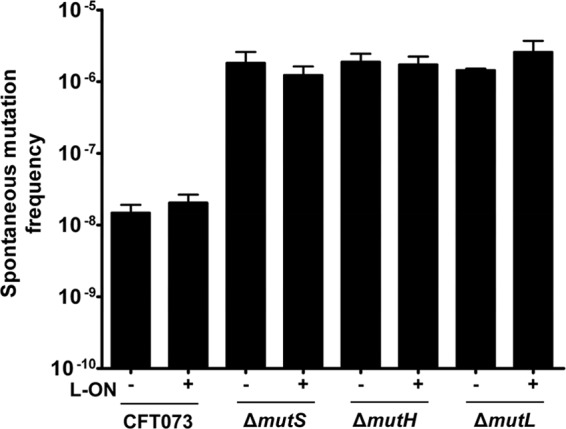
Frequency of rifampin-resistant mutants per number of isolates of motile CFT073 mismatch repair mutants. The “+” and “−” signs represent the fim L-ON status of the CFT073 background; “+” represents CFT073 fim L-ON, and “−” represents CFT073 wild type. Error bars represent the standard errors of the mean (SEM) of three independent cultures.
Deletion of mismatch repair genes in both wild-type CFT073 and fim L-ON backgrounds increases motility.
A previous study in our lab reported that MutS plays a role in the reciprocal control of motility and adherence in CFT073 (49). Deletion of mutS partially restored motility of a fim locked phase ON strain of CFT073 and increased motility of wild-type CFT073. Thus, we hypothesized that an intact MMR system was required to properly regulate motility and adherence. We predicted that mutation of mutH or mutL, two other genes encoding MMR components, would also result in increased motility. To test whether MutS alone or the entire MMR system plays a role in the reciprocal control of motility and adherence, the motilities of MMR deletion mutants mutS, mutH, and mutL, constructed in both CFT073 and CFT073 fim L-ON strains, were assessed on soft agar plates (Fig. 2A). CFT073 ΔmutS exhibited an average motility diameter increase of 9.0 mm ± 3.6 mm compared to the wild-type CFT073 parental strain (P = 0.034) (Fig. 2B). A significant increase in motility (6.6 mm ± 2.8 mm) was also observed by ΔmutS L-ON compared to the parental CFT073 fim L-ON strain (P = 0.039) (Fig. 2B). Deletion of mutH increased motility in both the wild-type CFT073 and the CFT073 fim L-ON, 13.2 mm ± 7.0 mm compared to wild-type CFT073 (P = 0.085) and 32.5 mm ± 3.1 mm compared to CFT073 fim L-ON (P < 0.001) (Fig. 2B), respectively. Interestingly, the motility increase of ΔmutH L-ON constitutes a full restoration of motility to CFT073 wild-type levels where only partial restoration of motility had been observed with ΔmutS L-ON (Fig. 2B). This unexpected result is examined below. Strain CFT073 ΔmutL also showed a significant increase in motility (13.2 mm ± 4.8 mm) compared to wild-type CFT073 (P = 0.017); however, no motility difference was observed between ΔmutL L-ON and CFT073 fim L-ON (0.9 mm ± 1.6 mm; P = 0.589) (Fig. 2B).
Fig 2.

Analysis of motility, percentage of motile bacteria within a population, correlation analysis between soft-agar motility, and percentage of motile bacteria within a population of CFT073 MMR mutants in wild-type and fim L-ON backgrounds and analysis of motility of mixed cultures with CFT073 fim L-ON and CFT073 fim L-OFF. (A) Motility of MMR mutants in soft agar are shown. (B) Bar graph representation of swimming motility diameters of motile deletion mutants. (C) Graphical representation of the percentage of motile bacteria within the populations of MMR mutants using the modified capillary assay. (D) Correlation between the average soft-agar swimming diameter and the average percentage of the population in a capillary. There is a positive correlation between soft agar motility and capillary motility, where the slope has a significant deviation from zero (P = 0.0037). (E) Measurement of swimming motility diameters from soft agar plates of mixed cultures starting with 100% nonmotile fim L-ON and 0% motile fim L-OFF strains and going up to 50% fim L-ON and 50% fim L-OFF. The line represents the best-fitting regression line. The slope has a significant deviation from zero (P = 0.0013). (B and C) The “+” and “−” signs indicate the fim L-ON status of the CFT073 background; “+” represents CFT073 fim L-ON, and “−” represents CFT073 wild type. The data represent the averages of at least three separate experiments. Error bars represent the SEM. Significant differences between MMR strains compared to parental strains were determined using a paired Student t test. *, P < 0.05.
An increase in the proportion of motile and flagellated CFT073 in a population correlates with increased motility.
Modified capillary assays were also used to verify the motility phenotypes of MMR deletion mutants where we determined the percentage of the population within a culture that would enter a capillary in 90 min (Fig. 2C). From the assay, ca. 6.9% ± 1.0% of the wild-type CFT073 population entered the capillary, while only 3.0% ± 0.8% of the CFT073 fim L-ON population did so (Fig. 2C). As expected, the ΔmutS, ΔmutH, and ΔmutL mutants all demonstrated a higher average motility increase compared to wild-type CFT073: 13.2% ± 4.0% (P = 0.014), 10.7% ± 14.0% (P = 0.459), and 9.6% ± 14.8% (P = 0.523), respectively (Fig. 2C). Also, as predicted, ΔmutS L-ON and ΔmutH L-ON strains demonstrated a significant increase in motility compared to CFT073 fim L-ON: 0.9% ± 0.5% (P = 0.041) and 6.4% ± 2.0% (P = 0.011), respectively, whereas ΔmutL L-ON did not show a significant difference in the percentage of motile population −0.2% ± 1.4% (P = 0.872) (Fig. 2C).
Using linear regression analysis (Fig. 2D), a strong positive correlation (r2 = 0.7785, P = 0.0037) between average diameters of motility determined by the soft agar motility assay (Fig. 2B) and average percentage of population expressing motility determined by the modified capillary assay (Fig. 2C) was observed.
To further verify whether the proportion of flagellated bacteria in a population correlates with the motility phenotype, CFT073 fim L-OFF (highly motile) and CFT073 fim L-ON (nonmotile) were mixed together in various ratios and motility was assessed in soft agar as shown in Fig. 2A. As predicted, an increase in the ratio of motile, non-type 1-fimbriated cells (CFT073 fim L-OFF) to nonmotile, type 1 fimbriated cells (CFT073 fim L-ON) within a population led to an increase in the diameter of swimming motility on soft agar (r2 = 0.7377, P = 0.0013) (Fig. 2E).
From these findings we conclude that motility diameters observed on soft agar plates have a strong positive correlation with motility measured in the standardized capillary assay conducted in Palleroni chambers (Fig. 2D). Furthermore, the observed motility of MMR mutants could be explained by the increased frequency of type 1 fimbrial promoter switching; as the increase in the population switches from fim ON to fim OFF, the population exhibits an increased motility (Fig. 2E).
Flagellin and type 1 fimbrial protein expression in CFT073 MMR deletion mutants constructed in wild-type and fim L-ON backgrounds correlate with motility studies.
To determine whether the increased diameter of motility correlates with increased flagellin production, FliC expression was measured using Western blotting with antibodies against FliC protein. As predicted, the levels of flagellin expression of motile CFT073 wild-type, ΔmutS, ΔmutH, ΔmutL, and ΔmutH L-ON strains were notably higher compared to the levels of nonmotile CFT073 fim L-ON, ΔmutS L-ON, and ΔmutL L-ON strains (Fig. 3, 60 kDa). Again, the motility behavior of ΔmutH L-ON was examined further (see below).
Fig 3.

Flagellin and fimbrial protein production of CFT073 MMR mutants in wild-type and fim L-ON backgrounds. Western blot analysis of flagellin production, FliC (60 kDa), and fimbrial production, FimA (15 kDa). The “+” and “−” signs indicate the fim L-ON status of the CFT073 background; “+” represents CFT073 fim L-ON, and “−” represents CFT073 wild type.
To determine whether the concept of reciprocal control holds true for the MMR mutants, the level of type 1 fimbrial structural subunit was measured using Western blotting against FimA protein. As predicted, the expression of FimA in motile CFT073 wild-type, ΔmutS, ΔmutH, ΔmutL, and ΔmutH L-ON strains was notably lower compared to the expression of nonmotile CFT073 fim L-ON, ΔmutS L-ON, and ΔmutL L-ON strains (Fig. 3, 15 kDa). Surprisingly, the hypermotile strain ΔmutH L-ON exhibited higher levels of FimA than comparably motile strains (see below).
qPCR was used to quantify the levels of fliC and fimA transcripts of MMR mutants constructed in both wild-type and fim L-ON backgrounds where transcript levels were then compared to parental strain levels (Fig. 4). As predicted, CFT073 fim L-ON, compared to the wild-type strain, had a 27.1-fold decrease of fliC transcript (log2 of −4.76) and a 172.5-fold increase (log2 of 7.43) in fimA transcript (Fig. 4). Motile ΔmutS and ΔmutL MMR mutants showed similar fliC and fimA transcript levels compared to the wild type; 1.6- and 1.6-fold (log2 of 0.66 and 0.64) and −2.0- and 1.0-fold (log2 of −0.99 and 0.05), respectively (Fig. 4). The ΔmutH strain, compared to the wild type, exhibited similar fliC transcript levels (1.2-fold, log2 of 0.23) and a slight increase in fimA transcript levels (2.8-fold, log2 of 1.48) (Fig. 4).
Fig 4.

Quantification of flagellar and type 1 fimbrial RNA transcript levels by qPCR of CFT073 MMR mutants in wild-type and fim L-ON backgrounds. RNA was isolated from wild-type CFT073, CFT073 fim L-ON, and MMR mutants cultured in LB. Transcript levels were measured in cDNA preparations from each strain and normalized to the gapA level. The results are shown as the log2 fold change relative to parental strain levels. Dashed lines represent a significant cutoff 2-fold change (log2 = 1), and bars indicate the SEM. The “+” and “−” signs represent the fim L-ON status of the CFT073 background; “+” represents CFT073 fim L-ON, and “−” represents CFT073 wild type.
ΔmutS L-ON exhibited similar quantities of fliC and fimA transcripts compared to CFT073 fim L-ON (1.9 [log2 of 0.92] and −1.2 [log2 of −0.24], respectively), whereas highly motile ΔmutH L-ON showed a 128.4-fold (log2 of 7.01) increase in fliC transcript levels compared to CFT073 fim L-ON (Fig. 4) and even a 4.4-fold increase (log2 of 2.14) compared to the wild type, as expected (data not shown) (see below). Also as predicted based on motility data, as well as Western blot data (Fig. 3, 15 kDa), the fimA transcript levels of ΔmutH L-ON were decreased 27.7-fold (log2 of −4.79) compared to the fim L-ON strain (Fig. 4) and increased 6.6-fold compared to the wild-type strain (log2 2.72) (data not shown) (see below). Lastly, nonmotile ΔmutL L-ON exhibited an unpredicted increase in fliC transcript levels of 6.5-fold (log2 of 2.70) and similar levels of fimA transcript (1.1, log2 of 0.81) (Fig. 4).
From these data, we can conclude that motile CFT073 wild-type, ΔmutS, ΔmutL, ΔmutH, and ΔmutH L-ON strains have a significantly greater quantity of flagellin transcripts and a reduced quantity of type 1 fimbrial transcripts compared to the nonmotile strains. The fliC and fimA transcript levels in these strains were also found to be relatively similar to one another, with the exception of ΔmutH L-ON, where increased levels of fliC and fimA transcripts were detected (see below), and ΔmutH, where a slight increase in fimA transcripts was detected; neither of these strain's fimA transcript levels however, reached CFT073 fim L-ON quantities. Nonmotile CFT073 fim L-ON, ΔmutS L-ON, and ΔmutL L-ON strains showed similar levels of fliC and fimA transcription with the exception of ΔmutL L-ON, which exhibited a rather large increase in fliC transcript levels; this increase, however, did not reach wild-type quantities.
Quantification of surface flagella correlates with motility.
Since whole-cell lysates were used to determine whether deletions in the mismatch repair system affected flagellin production, knowledge of the length, number, or presence of surface flagella was still unknown. Immunofluorescence microscopy was used to visualize and quantify flagella, and determine whether the increase in motility of mutants in both wild-type CFT073 and in the CFT073 fim L-ON backgrounds could be attributed to flagellar expression (Fig. 5). Individual bacteria were stained with propidium iodide, and surface flagella were detected with anti-FliC serum. When comparing CFT073 wild type to the ΔmutS, ΔmutH, and ΔmutL mutants, we observed flagella that appeared similar in size and morphology; however, we found that the MMR mutant UPEC produced a greater number of flagellated cells (Fig. 5A). When quantified, the average ratio of flagella/ΔmutS bacterium was (40.3 ± 48.9)-fold higher than the ratio of flagella/wild-type CFT073 bacterium, the ΔmutH strain was (89.5 ± 24.0)-fold higher than the wild type, and the ΔmutL strain was (137.1 ± 97.9)-fold higher than the wild type (Fig. 5A).
Fig 5.

Immunofluorescence analysis of surface flagellum expression of motile MMR mutants in wild-type CFT073 and CFT073 fim L-ON backgrounds as a ratio of the parental strain. (A) Bar graphs represent the average counts of flagella per MMR mutant bacterium in a CFT073 wild-type background as a ratio of average wild-type flagella per bacterium. (B) Graphical representation of average counts of flagella per mismatch repair mutant bacterium in CFT073 fim L-ON background as a ratio of average CFT073 fim L-ON flagella per bacterium is also shown.
We expected to observe few flagella on CFT073 fim L-ON because the strain constitutively expressed type 1 fimbriae and lacked motility. Indeed, few, if any, flagella were detected on the surface of CFT073 fim L-ON. When comparing ΔmutS L-ON and CFT073 fim L-ON, a slight increase in surface flagella was observed [(3.4 ± 1.3)-fold more; Fig. 5B], which was expected because ΔmutS L-ON showed a partial restoration of motility compared to CFT073 fim L-ON. Due to the increased motility of ΔmutH L-ON, we expected to observe more flagella than with CFT073 fim L-ON. Indeed, ΔmutH L-ON produced a greater number of flagella than CFT073 fim L-ON [(34.6 ± 0.6)-fold more; Fig. 5B] and (48.0 ± 41.6)-fold more flagella per bacterium than wild-type CFT073. Lastly, ΔmutL L-ON expressed (2.6 ± 0.01)-fold more flagella than CFT073 fim L-ON (Fig. 5B), which was unexpected since deletion of mutL failed to show a partial restoration of motility to the fim L-ON strain (Fig. 2B).
Deletion of mutH in the fim L-ON background reverts the “locked” invertible element back to the wild-type sequence.
The ΔmutH L-ON construct yielded numerous unexpected findings including hypermotility compared to other fim L-ON strains, increased levels of FimA and FliC from Western blot compared to other motile strains, and increased levels of fimA and fliC transcript levels from qPCR data compared to other motile strains. To understand the mechanism underlying these changes, the invertible element assay was used to determine the orientation of the type 1 fimbrial promoter to verify that the mutant remained “locked” in fim phase ON orientation (review construction in reference 19). We found, however, that the orientation of the type 1 fim promoter of ΔmutH L-ON was capable of switching, whereas all other fim L-ON mutants remained locked in the phase ON orientation (Fig. 6). The fim promoters of three independent ΔmutH L-ON constructs were sequenced and found to have reverted to the wild-type sequence in the left inverted repeat, which had originally been mutated to create the parental locked-ON strain for all MMR fim L-ON mutants (19). This result clearly explains the anomalous behavior of this mutant, including the large increase in motility and the increase in flagellin production. In addition, we sequenced the promoter of the electrocompetent cells prepared from CFT073 fim L-ON that were used to introduce the mutH knockout mutation and confirmed that this sequence was “locked” ON. We believe that the ability to revert to wild-type switching capability and reversion of the left repeat sequence can be attributed to the inadvertent introduction of a methylation site (GATC) to mutate one repeat within the invertible element during the construction of this strain, although the exact mechanism is not known. Therefore, with the exception of ΔmutH “L-ON”, the deletions of mismatch repair genes do not affect the orientation of the invertible fim promoter and is not the source of increased motility when type 1 fimbriae are constitutively expressed.
Fig 6.
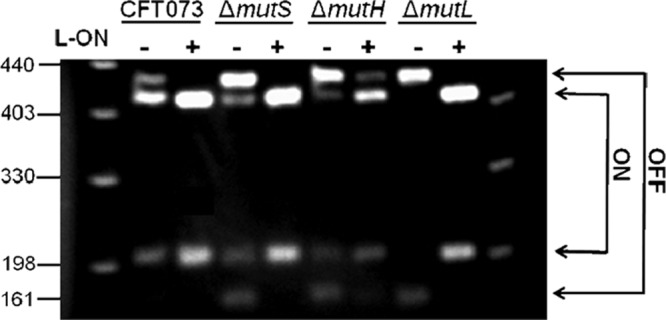
Orientation of the invertible fim promoter in CFT073 MMR mutants. The promoter orientation was determined by λ red PCR amplification of the invertible element, followed by digestion using SnaB1. Asymmetric cutting of SnaB1 revealed the orientation of the invertible element; phase ON is indicative of the expression of type 1 fimbriae, and phase OFF is indicative of blocked type 1 fimbrial expression. The “+” and “−” signs represent the fim L-ON status of CFT073 background; “+” represents CFT073 fim L-ON, and “−” represents CFT073 wild type.
Increased motility cannot be attributed to increased growth rate.
Since an increase in surface flagella was observed in strains exhibiting increased motility and, with the exception of ΔmutH “L-ON”, the “locked” fim invertible element was demonstrated to be stable; we sought to determine whether changes in motility could be attributed to an increased or decreased growth rate. The growth rate in LB medium for each mutant was compared to its parental strain to assess any growth deficiencies. No notable variations in growth rate were calculated, with the exception of the ΔmutH strain, where a slight growth deficiency was observed compared to its parental strain, wild-type CFT073 (data not shown). Therefore, we do not attribute the increased motility of the MMR mutant strains to in vitro growth.
The motility of MMR mutants can be restored to wild-type levels by complementation.
The motility of the complemented MMR mutants was assessed using soft agar and was compared to the motility of MMR mutants transformed with the vector alone and to the motility of parental wild-type CFT073 and CFT073 fim L-ON containing vector alone (Fig. 7A). The motility of the complemented ΔmutS strain was not different from wild-type CFT073 levels (P = 0.115), and complementation of the ΔmutS L-ON strain resulted in decreased motility to CFT073 fim L-ON levels (P < 0.001) (Fig. 7A). Complemented ΔmutH strain's motility was not different from wild-type CFT073 motility (P = 0.696); however, complemented ΔmutH “L-ON” was not able to reduce motility to CFT073 fim L-ON levels (P < 0.001) (Fig. 7A). This finding was expected due to the ΔmutH “L-ON” strain's ability to unlock itself from the phase ON orientation, as seen in Fig. 6. Therefore, as expected, the motility of the mutH “L-ON” mutant was not different from wild-type CFT073 levels (P = 0.096) (Fig. 7A). The motility of the complemented ΔmutL strain was also not different from wild-type CFT073 levels (P = 0.1466) (Fig. 7A). Complementation of ΔmutL L-ON motility was not measurable because the motilities of ΔmutL L-ON and CFT073 fim L-ON were indistinguishable.
Fig 7.
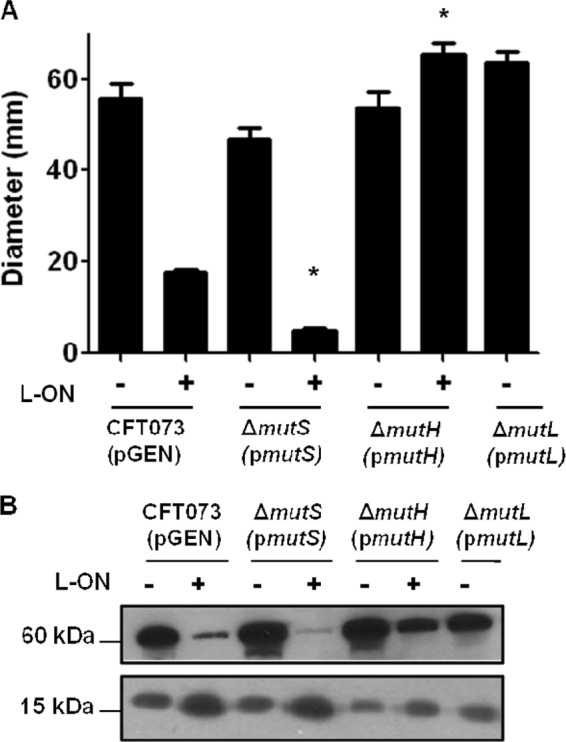
Analysis of motility, flagellin, and fimbrial production of CFT073 mismatch repair mutants in wild-type and fim L-ON backgrounds transformed with the complementing plasmid. (A) Graphical representation of swimming motility diameters of complemented motile deletion mutants. The “+” and “−” signs represent the fim L-ON status of the CFT073 background; “+” represents CFT073 fim L-ON, and “−” represents CFT073 wild type. The data represent averages of at least three separate experiments. Error bars represent the SEM. Significant differences in motility from parental strain motility were determined by using a paired Student t test. *, P < 0.05. (B) Western blot analysis of flagellin production, FliC (60 kDa), and fimbrial production, FimA (15 kDa). The “+” and “−” signs again represent the CFT073 fim L-ON status. The size in kilodaltons of both proteins is given.
Flagellin and FimA levels in the complemented mutant strains correlated well with motility; bacteria that produced more flagellin were more motile than bacteria that produced more FimA. As expected, the flagellin levels of CFT073 and the complemented ΔmutS, ΔmutH, ΔmutH “L-ON”, and ΔmutL strains were much higher than the nonmotile CFT073 fim L-ON and complemented ΔmutS L-ON strains (Fig. 7B, 60 kDa). The FimA levels of CFT073 and the complemented ΔmutS, ΔmutH, ΔmutH “L-ON”, and ΔmutL strains were much lower than the FimA levels of CFT073 fim L-ON and the complemented strain ΔmutS L-ON (Fig. 7B, 15 kDa). We conclude that the motilities of MMR deletion strains can be reduced to wild-type CFT073 levels by complementation, with the exception of ΔmutH “L-ON,” whose ability to unlock its type 1 fimbrial promoter has made full complementation a challenge.
The ΔmutS strain outcompetes the wild-type strain in the bladder and spleen during a 48-h ascending UTI.
Flagella and type 1 fimbriae are known to be important in the colonization of the murine urinary tract by UPEC (19, 31, 47, 52). From our study, we have determined that deletions in the MMR system play a surprising role in the reciprocal control of these two functions. To identify any contribution of MMR in vivo, we assessed host colonization using the murine model of ascending UTI. Using the ΔmutS strain as a representative mutant of the MMR system, in vivo colonization of the ΔmutS strain was measured in both a 48-h and 7-day cochallenge of the mutant with wild-type CFT073 (Fig. 8A and B). The ΔmutS mutant outcompeted wild-type CFT073 in both the bladder (P = 0.040) and the spleen (P < 0.001) during the 48-h infection (Fig. 8A). The ability to disseminate to the bloodstream and reach the spleen can possibly be attributed to the increase in motility of the ΔmutS mutant bacteria. A cochallenge of ΔmutS L-ON with CFT073 fim L-ON was also tested, and we found that the MMR mutant had a fitness defect, where the parental strain outcompeted the mutant in the bladder during the 48-h infection (P = 0.011) (Fig. 8C). Differences noted were subtle but nonetheless statistically significant. We also measured the fitness of the constructs over a longer period. No significant differences were detected from the 7-day infection of either of the cochallenges (Fig. 8B and D).
Fig 8.

Mice were infected transurethrally with ∼108 CFU of a 1:1 mixture of a ΔmutS mutant with its appropriate parental strain. After a 48 h or 7 days postinfection, urine, bladders, kidneys, and spleens were quantitatively cultured. Each symbol represents a datum from an individual mouse. Horizontal bars represent the median from each tissue. The limit of detection is 100 CFU/ml of urine or g of tissue. Significant differences in strain isolates cultured from organs were determined using a Wilcox signed-rank test. *, P < 0.05. (A) Cochallenge at 48 h with a 1:1 mix of CFT073 wild-type and ΔmutS strains in a wild-type background (n = 10). (B) Cochallenge at 7 days with a 1:1 mix of CFT073 wild-type and ΔmutS strains in a wild-type background (n = 14). (C) Cochallenge at 48 h with a 1:1 mix of CFT073 fim L-ON and ΔmutS fim L-ON (n = 10). (D) Cochallenge at 7 days with a 1:1 mix of CFT073 fim L-ON and ΔmutS fim L-ON (n = 14).
The loss of MMR does not cause mutations in the flhDC or pap operon promoters.
To identify a mechanism of MMR's involvement in the regulation of motility and adherence, we examined the most likely candidate genes that might affect these functions. The first of these was the promoter region for class I flagellar genes flhDC, which initiates the complex cascade of gene expression leading to flagellar expression (25). The second was the promoter region for the pap (pyelonephritis-associated pili) genes, which encode P fimbriae (7, 50). P fimbriae have also been shown to play a role in the reciprocal control of motility and adherence and are regulated by methylation of GATC sites in the pap promoter (7, 50). Primers were used to amplify the promoter regions of the MMR deletion strains in both fim L-ON and wild-type backgrounds (Table 1). The resulting PCR fragments were purified and sequenced for all bacterial constructs used in the present study. Sequences of the flhDC and pap promoters of all constructs were identical to the wild-type CFT073 sequence and thus cannot explain the observed increase in motility in the absence of MMR (data not shown).
DISCUSSION
In our previous study, we observed that motility was repressed when type 1 fimbrial genes (fim) were constitutively expressed in UPEC strain CFT073 (30). When the entire fim region was deleted in the CFT073 fim L-ON mutant, only a partial restoration of motility to wild-type levels was observed (30). This suggested that the repression of UPEC motility was not solely based on the expression or assembly of these surface organelles and additional factors must be considered. A screen of 12,000 transposon mutants in CFT073 fim L-ON identified six nonfimbrial genes that reversed the repression of motility (49). This suggested that these six genes play a role in the reciprocal control of adherence and motility. Among these six genes was mutS, which encodes a component of the mismatch repair (MMR) system. In the present study, we demonstrate that the MMR system itself, including MutS, MutH, and MutL, plays a role in the reciprocal regulation of motility and adherence in UPEC.
Deletion of the MMR genes mutS, mutH, and mutL significantly increased motility in both CFT073 wild-type and fim L-ON backgrounds (with the exception of strain ΔmutL L-ON, in which no increase in motility was observed). Along with motility, the FimA and FliC levels, in whole cells and expressed on the bacterial surface, were analyzed using Western blots and immunofluorescence, respectively. These assays revealed that the levels of the two proteins correlate well with motility; expressing more flagellin results in more motility and more FimA production occurs when motility is reduced: qPCR was used to confirm and quantify these observations. Motility, the levels of FimA, and the levels of flagellin, could be fully complemented for all strains except for ΔmutH “L-ON”; this exception was due to the unique ability of this strain to unlock the fim promoter in the fim L-ON background of CFT073.
The fim promoter, located on an invertible element, was analyzed to determine its orientation in the six MMR mutants. We found that the fim promoters of more motile strains were primarily in the phase OFF orientation. This could suggest that in the absence of MMR, the rate of recombination at the fim promoter repeats increases and thus, ON-OFF or OFF-ON switching is increased. Keeping in mind the theory of reciprocal control of adherence and motility, increasing the frequency at which recombination of the fim promoter occurs, switching from the phase ON orientation to the phase OFF orientation, results in less type 1 fimbriae being synthesized and, consequently, allows for increased motility. To further support this theory, the motility of a mixed population of CFT073 fim L-ON and CFT073 fim L-OFF was assessed and reveled a strong, positive correlation (r2 = 0.74) between the amount of CFT073 fim L-OFF in a culture and the diameter of swimming motility. These findings support our capillary assay data that demonstrate an increase in the percentage of motile bacteria within a MMR mutant population that also exhibit an increased swimming diameter on soft agar plates.
As a representative of MMR, the ΔmutS mutation was used to assess in vivo fitness during a cochallenge with wild-type CFT073 using the murine model of ascending UTI to determine whether the MMR effect on motility affects UPEC pathogenesis. We found that wild-type CFT073 was outcompeted by bacteria with mutS deleted in both the bladder and the spleen 48 h postinoculation. This relationship between MMR and UPEC pathogenesis may be supported, in part, by evidence that UTI isolates exhibit a higher occurrence of mutator strains than commensal E. coli or any other E. coli pathotype (14). This is remarkable given that >1% of food-borne E. coli pathogens exhibit high mutation rates as a result of defective MMR systems (33). Defects in the MMR system have also been identified and linked to virulent isolates of other pathogenic bacteria, including Salmonella enterica serovar Typhimurium (33), Neisseria meningitidis (45) and Pseudomonas aeruginosa (41). Overall, these associations are correlative, and some caution should be used in their interpretation.
To explain how MMR deficiencies could lead to increased virulence, we must explain how MMR assists in avoiding genetic change. It does this by two main pathways: first, by limiting genetic variation through the correction of errors in newly replicated DNA strands (23, 24, 39) and, second, by acting as a barrier between interspecies gene exchange (11, 16, 36, 44). Therefore, when an organism exhibits defects in its MMR system, it is more prone to genetic variations and interspecies horizontal gene transfer and, consequently, has an increased capacity to adapt to the host environment or acquire new virulence genes, respectively (11, 16, 36, 44).
The mutator phenotype allows bacteria to rapidly respond via genetic change when faced with challenges, such as the presence of antibiotics, attack from host immune cells, altered nutritional availability, and competition in the environment. In the urinary tract, an organism is exposed to many environmental changes as it ascends from the urethral opening, to the bladder, up the ureters, to the kidneys and eventually, if left untreated, to the bloodstream. This includes the exposure to iron-limited urine, neutrophil influx, diverse antibacterial defense molecules, and host cell types. The ability to survive such changes is essential for the organism's ability to successfully colonize the host. This survival strategy is consistent with our finding that the loss of MMR increases fitness during ascending UTI. It should be noted, however, that the motility phenotypes of MMR mutant UPEC cannot be directly explained by inheritable genetic mutations, other than the loss of MMR genes, since the mutant phenotypes can be restored to wild type by complementation.
In summary, our findings show a direct relationship between methyl-directed MMR systems and the reciprocal control of motility and adherence in UPEC. Deletion of three genes involved in the MMR system—mutS, mutH, and mutL—increased motility in wild-type CFT073 and in CFT073 fim L-ON, with the latter constitutively expressing type 1 fimbriae. A mechanism that satisfactorily explains this association remains to be determined; however, our study strengthens the connection between mutator isolates and virulence and suggests that MMR's effect on reciprocal control of motility and adherence contributes to UPEC pathogenesis.
ACKNOWLEDGMENTS
This study was funded in part by Public Health Service grants AI43363 and AI59722 from the National Institutes of Health (H.L.T.M.) and National Science Foundation grant MCB1050948 (L.A.S.).
We thank Elizabeth Cameron from the University of Michigan for constructing the CFT073 fim L-ON ΔmutL mutant during her rotation in our lab.
Footnotes
Published ahead of print 2 April 2012
REFERENCES
- 1. Abraham JM, Freitag CS, Clements JR, Eisenstein BI. 1985. An invertible element of DNA controls phase variation of type 1 fimbriae of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 82:5724–5727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adler J, Dahl MM. 1967. A method for measuring the motility of bacteria and for comparing random and non-random motility. J. Gen. Microbiol. 46:161–173 [DOI] [PubMed] [Google Scholar]
- 3. Akerley BJ, Cotter PA, Miller JF. 1995. Ectopic expression of the flagellar regulon alters development of the Bordetella-host interaction. Cell 80:611–620 [DOI] [PubMed] [Google Scholar]
- 4. Ban C, Yang W. 1998. Structural basis for MutH activation in Escherichia coli mismatch repair and relationship of MutH to restriction endonucleases. EMBO J. 17:1526–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beachey EH. 1981. Bacterial adherence: adhesion-receptor interactions mediating the attachment of bacteria to mucosal surfaces. J. Infect. Dis. 143:325–345 [DOI] [PubMed] [Google Scholar]
- 6. Blomfield IC, Kulasekara DH, Eisenstein BI. 1997. Integration host factor stimulates both FimB- and FimE-mediated site-specific DNA inversion that controls phase variation of type 1 fimbriae expression in Escherichia coli. Mol. Microbiol. 23:705–717 [DOI] [PubMed] [Google Scholar]
- 7. Braaten BA, et al. 1992. Leucine-responsive regulatory protein controls the expression of both the pap and fan pili operons in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 89:4250–4254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chilcott GS, Hughes KT. 2000. Coupling of flagellar gene expression to flagellar assembly in Salmonella enterica serovar Typhimurium and Escherichia coli. Microbiol. Mol. Biol. Rev. 64:694–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clegg S, Hughes KT. 2002. FimZ is a molecular link between sticking and swimming in Salmonella enterica serovar Typhimurium. J. Bacteriol. 184:1209–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cooper DL, Lahue RS, Modrich P. 1993. Methyl-directed mismatch repair is bidirectional. J. Biol. Chem. 268:11823–11829 [PubMed] [Google Scholar]
- 11. Cox EC. 1995. Recombination, mutation and the origin of species. Bioessays 17:747–749 [DOI] [PubMed] [Google Scholar]
- 12. Dao V, Modrich P. 1998. Mismatch-, MutS-, MutL-, and helicase II-dependent unwinding from the single-strand break of an incised heteroduplex. J. Biol. Chem. 273:9202–9207 [DOI] [PubMed] [Google Scholar]
- 13. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Denamur E, et al. 2002. High frequency of mutator strains among human uropathogenic Escherichia coli isolates. J. Bacteriol. 184:605–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dorman CJ, Bhriain NN. 1992. Thermal regulation of fimA, the Escherichia coli gene coding for the type 1 fimbrial subunit protein. FEMS Microbiol. Lett. 78:125–130 [DOI] [PubMed] [Google Scholar]
- 16. Elez M, Radman M, Matic I. 2007. The frequency and structure of recombinant products is determined by the cellular level of MutL. Proc. Natl. Acad. Sci. U. S. A. 104:8935–8940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Galen JE, et al. 1999. Optimization of plasmid maintenance in the attenuated live vector vaccine strain Salmonella typhi CVD 908-htrA. Infect. Immun. 67:6424–6433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gardel CL, Mekalanos JJ. 1996. Alterations in Vibrio cholerae motility phenotypes correlate with changes in virulence factor expression. Infect. Immun. 64:2246–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gunther IN, et al. 2002. Assessment of virulence of uropathogenic Escherichia coli type 1 fimbrial mutants in which the invertible element is phase-locked on or off. Infect. Immun. 70:3344–3354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hagberg L, et al. 1983. Ascending, unobstructed urinary tract infection in mice caused by pyelonephritogenic Escherichia coli of human origin. Infect. Immun. 40:273–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hall MC, Matson SW. 1999. The Escherichia coli MutL protein physically interacts with MutH and stimulated the MutH-associated endonuclease activity. J. Biol. Chem. 274:1306–1312 [DOI] [PubMed] [Google Scholar]
- 22. Hall MC, Jordan JR, Matson SW. 1998. Evidence for a physical interaction between the Escherichia coli methyl-directed mismatch repair proteins MutL and UvrD. EMBO J. 17:1535–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horst JP, Wu TH, Marinus MG. 1999. Escherichia coli mutator genes. Trends Microbiol. 7:29–36 [DOI] [PubMed] [Google Scholar]
- 24. Jiricny J. 1998. Replication errors: cha(lle)nging the genome. EMBO J. 17:6427–6436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Johnson JR. 1991. Virulence factors in Escherichia coli urinary tract infection. Clin. Microbiol. Rev. 4:80–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klemm P. 1986. Two regulatory fim genes, fimB and fimE, control the phase variation of type 1 fimbriae in Escherichia coli. EMBO J. 5:1389–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kunkel TA, Erie DA. 2005. DNA mismatch repair. Annu. Rev. Biochem. 74:681–710 [DOI] [PubMed] [Google Scholar]
- 28. Lahue RS, Au KG, Modrich P. 1989. DNA mismatch correction in a defined system. Science 245:160–164 [DOI] [PubMed] [Google Scholar]
- 29. Lane MC, Lloyd AL, Markyvech TA, Hagen EC, Mobley HL. 2006. Uropathogenic Escherichia coli strains generally lack functional Trg and Tap chemoreceptors found in the majority o f E. coli strains strictly residing in the gut. J. Bacteriol. 188:5618–5625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lane MC, Simms AN, Mobley HL. 2007. Complex interplay between type 1 fimbrial expression and flagellum-mediated motility of uropathogenic Escherichia coli. J. Bacteriol. 189:5523–5533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lane MC, Alteri CJ, Smith SN, Mobley HL. 2007. Expression of flagella is coincident with uropathogenic Escherichia coli ascension to the upper urinary tract. Proc. Natl. Acad. Sci. U. S. A. 104:16669–16674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lane MC, et al. 2005. Role of motility in the colonization of uropathogenic Escherichia coli in the urinary tract. Infect. Immun. 73:7644–7656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. LeClerc JE, Li B, Payne WL, Cebula TA. 1996. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 274:1208–1211 [DOI] [PubMed] [Google Scholar]
- 34. Li X, Rasko DA, Lockatell CV, Johnson DE, Mobley HL. 2001. Repression of bacterial motility by a novel fimbrial gene product. EMBO J. 20:4854–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lim JK, et al. 1998. In vivo phase variation of Escherichia coli type 1 fimbrial genes in women with urinary tract infection. Infect. Immun. 66:3303–3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matic I, Rayssiguier C, Radman M. 1995. Interspecies gene exchange in bacteria: the role of SOS and mismatch repair systems in evolution of species. Cell 80:507–515 [DOI] [PubMed] [Google Scholar]
- 37. McClain MS, Blomfield IC, Eisenstein BI. 1991. Roles of fimB and fimE in site-specific DNA inversion associated with phase variation of type 1 fimbriae in Escherichia coli. J. Bacteriol. 173:5308–5314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mobley HL, et al. 1990. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect. Immun. 58:1281–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Modrich P, Lahue R. 1996. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 65:101–133 [DOI] [PubMed] [Google Scholar]
- 40. Mulvey MA, et al. 1998. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 282:1494–1497 [DOI] [PubMed] [Google Scholar]
- 41. Oliver A, Canton R, Campo P, Baquero F, Blazquez J. 2000. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 88:1251–1254 [DOI] [PubMed] [Google Scholar]
- 42. Palleroni NJ. 1976. Chamber for bacterial chemotaxis experiments. Appl. Environ. Microbiol. 32:729–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ramos CH, Rumbo M, Sirard J-C. 2004. Bacterial flagellins: mediators of pathogenicity and host immune responses in mucosa. Trends Microbiol. 12:509–517 [DOI] [PubMed] [Google Scholar]
- 44. Rayssiquier C, Thaler DS, Radman M. 1989. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature 342:396–401 [DOI] [PubMed] [Google Scholar]
- 45. Richardson AR, Stojiljkovic I. 2001. Mismatch repair and the regulation of phase variation in Neisseria meningitidis. Mol. Microbiol. 40:645–655 [DOI] [PubMed] [Google Scholar]
- 46. Schofield MJ, Hsieh P. 2003. DNA mismatch repair: molecular mechanisms and biological function. Annu. Rev. Microbiol. 57:579–608 [DOI] [PubMed] [Google Scholar]
- 47. Schwan WR. 2008. Flagella allow uropathogenic Escherichia coli ascension into murine kidneys. Int. J. Med. Microbiol. 298:441–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Simmons LA, Davies BW, Grossman AD, Walker GC. 2008. Beta clamp directs localization of mismatch repair in Bacillus subtilis. Mol. Cell 29:291–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Simms AN, Mobley HL. 2008. Multiple genes repress motility in uropathogenic Escherichia coli constitutively expressing type 1 fimbriae. J. Bacteriol. 190:3747–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Simms AN, Mobley HL. 2008. PapX, a P fimbrial operon-encoded inhibitor of motility in uropathogenic Escherichia coli. Infect. Immun. 76:4833–4841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smith BT, Grossman AD, Walker GC. 2001. Visualization of mismatch repair in bacterial cells. Mol. Cell 8:1197–1206 [DOI] [PubMed] [Google Scholar]
- 52. Snyder JA, et al. 2004. Transcriptome of uropathogenic Escherichia coli during urinary tract infection. Infect. Immun. 72:6373–6381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spory A, Bosserhoff A, von Rhein C, Goebel W, Ludwig A. 2002. Differential regulation of multiple proteins of Escherichia coli and Salmonella enterica serovar Typhimurium by the transcriptional regulator SlyA. J. Bacteriol. 184:3549–3559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wehrli W. 1983. Rifampicin: mechanisms of action and resistance. Rev. Infect. Dis. 5:S407–S411 [DOI] [PubMed] [Google Scholar]
- 55. Welch RA, et al. 2002. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 99:17020–17024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Welsh KM, Lu AL, Clark S, Modrich P. 1987. Isolation and characterization of the Escherichia coli mutH gene product. J. Biol. Chem. 262:15624–15629 [PubMed] [Google Scholar]
- 57. Yamaguchi M, Dao V, Modrich P. 1998. Single-strand DNA-specific exonucleases in Escherichia coli: roles in repair and mutation avoidance. J. Biol. Chem. 273:9197–92019535910 [Google Scholar]