

Abstract
Shiga toxins (Stxs) are cytotoxins produced by the enteric pathogens Shigella dysenteriae serotype 1 and Shiga toxin-producing Escherichia coli (STEC). Stxs bind to a membrane glycolipid receptor, enter cells, and undergo retrograde transport to ultimately reach the cytosol, where the toxins exert their protein synthesis-inhibitory activity by depurination of a single adenine residue from the 28S rRNA component of eukaryotic ribosomes. The depurination reaction activates the ribotoxic stress response, leading to signaling via the mitogen-activated protein kinase (MAPK) pathways (Jun N-terminal protein kinase [JNK], p38, and extracellular signal-regulated kinase [ERK]) in human epithelial, endothelial, and myeloid cells. We previously showed that treatment of human macrophage-like THP-1 cells with Stxs resulted in increased cytokine and chemokine expression. In the present study, we show that individual inactivation of ERK, JNK, and p38 MAPKs using pharmacological inhibitors in the presence of Stx1 resulted in differential regulation of the cytokines tumor necrosis factor alpha and interleukin-1β (IL-1β) and chemokines IL-8, growth-regulated protein-β, macrophage inflammatory protein-1α (MIP-1α), and MIP-1β. THP-1 cells exposed to Stx1 upregulate the expression of select dual-specificity phosphatases (DUSPs), enzymes that dephosphorylate and inactivate MAPKs in mammalian cells. In this study, we confirmed DUSP1 protein production by THP-1 cells treated with Stx1. DUSP1 inhibition by triptolide showed that ERK and p38 phosphorylation is regulated by DUSP1, while JNK phosphorylation is not. Inhibition of p38 MAPK signaling blocked the ability of Stx1 to induce DUSP1 mRNA expression, suggesting that an autoregulatory signaling loop may be activated by Stxs. Thus, Stxs appear to be capable of eliciting signals which both activate and deactivate signaling for increased cytokine/chemokine production in human macrophage-like cells.
INTRODUCTION
Shiga toxins (Stxs) are bacterial cytotoxins produced by Shigella dysenteriae serotype 1 and Shiga toxin-producing Escherichia coli (STEC), the causative agents of bacillary dysentery and hemorrhagic colitis, respectively. Bacillary dysentery is prevalent in developing countries, where contaminated water supplies are the main source of infection. STEC infections, in contrast, are primarily observed in developed countries, where the sources of infection include undercooked ground beef, unpasteurized milk, or improperly washed vegetables contaminated with STEC (10). A subset of patients infected with these organisms develops life-threatening complications such as the hemolytic-uremic syndrome (HUS), which is characterized by acute renal failure, thrombocytopenia, and microangiopathic hemolytic anemia (45). Stxs are the major virulence factors associated with the development of HUS. STEC produces one or more antigenically related Stxs that may be divided into two categories, Shiga toxin type 1 (Stx1) and Stx2, based on their similarity to the prototypical Shiga toxin expressed by S. dysenteriae serotype 1 (23, 53). All Stxs have an AB5 structure (11, 12). Stxs which cause disease in humans bind to the membrane glycolipid receptor globotriaosylceramide (Gb3) through interaction with the pentameric ring formed by Stx B subunits (33). Following internalization, the toxins undergo retrograde transport, trafficking within the cell first through early endosomes and then through the trans-Golgi network and Golgi apparatus to reach the endoplasmic reticulum (ER) (37, 50). The ER is the site of A-subunit cleavage, yielding the enzymatically active A1 fragment, which is subsequently retrotranslocated into the cytosol (14, 54). Stx A1 fragments cleave a single adenine residue from the 28S rRNA, leading to protein synthesis inhibition (9).
In contrast to studies using highly toxin-sensitive cells, such as Vero or HeLa cells, treatment of primary human monocytes or macrophage-like cell lines with Stxs did not result in rapid cytotoxicity. Rather, the cells initially responded to the toxins by upregulating expression of cytokines and chemokines (17, 48). These host response factors, in turn, may play a role in the pathogenesis of hemorrhagic colitis and HUS. Interleukin-1β (IL-1β) and tumor necrosis factor alpha (TNF-α) upregulate Gb3 synthesis in endothelial cells in vitro, rendering them more susceptible to Stx killing (61, 63). Stx-mediated secretion of neutrophil chemoattractants may lead to neutrophil infiltration within the lamina propria and fecal leukocytosis characteristic of hemorrhagic colitis (51). Monocyte infiltration into the kidney has been described in a murine model of HUS, and the macrophage chemoattractant monocyte chemoattractant protein 1 (MCP-1) has been detected in urine from HUS patients (26, 62). Finally, the activation of neutrophils by the chemokines secreted by macrophages in response to Stxs may explain the observation that neutrophils isolated from HUS patients showed increased adherence and destruction of an endothelial cell monolayer compared to neutrophils from healthy donors (11).
Agents which modify the 3′ end of the 28S rRNA trigger the initiation of signaling through mitogen-activated protein kinase (MAPK) cascades, a process termed the ribotoxic stress response (20). Stxs activate this response, leading to Jun N-terminal protein kinase (JNK), p38, and extracellular signal-regulated kinase (ERK) MAPK signaling pathway activation in human epithelial, endothelial, and myeloid cells. Downstream substrates of MAPKs include transcription factors such as AP-1, NF-κB, and early growth response factor 1 (Egr-1), which, in turn, may regulate cytokine and chemokine expression (4, 6, 17, 18, 32, 49). Therefore, we have investigated the role of the ribotoxic stress response in the regulation of cytokine/chemokine expression elicited by Stx1 treatment of human macrophage-like THP-1 cells. Our previous microarray analysis of the global transcriptional response of THP-1 cells to Stx1 showed that expression of mRNA of a subset of dual-specificity phosphatases (DUSPs) was upregulated (32). DUSPs, also known as MAP kinase phosphatases, are enzymes that dephosphorylate and inactivate MAPK isoforms in mammalian cells (43). Thus, we determined if the prototypical member of the dual-specificity phosphatase family, DUSP1, which is capable of dephosphorylating all three MAPKs with different substrate preferences, played a role in the regulation of MAPK activation elicited by Stx1. Utilizing pharmacological and small interfering RNA (siRNA) inhibition of DUSP1 expression, we show that DUSP1 plays a role in the regulation of MAPK activation and that MAPKs differentially regulate cytokine/chemokine and DUSP1 expression in Stx1-treated THP-1 cells.
MATERIALS AND METHODS
Reagents.
Antibodies directed against phospho-JNK1/2 (Thr183/Tyr185), phospho-p38 (Thr180/Tyr182), phospho-ERK1/2 (Thr202/Tyr204), and actin were obtained from Cell Signaling Technology (Beverly, MA). Antibodies against DUSP1, JNK1/2, p38, and ERK1/2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). JNK1/2 stress-activated protein kinase inhibitor SP600125, p38 MAPK inhibitor SB203580, ERK1/2 inhibitor PD98059, and DUSP1 inhibitor triptolide were purchased from Calbiochem (San Diego, CA). Except where noted, all other reagents were purchased from Sigma Chemical Co. (St. Louis, MO).
Toxin.
Stx1 was purified from cell lysates prepared from E. coli DH5α(pCKS112) by sequential ion-exchange and chromatofocusing chromatography (56). Purity of toxin preparations was assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with silver staining and Western blot analysis using anti-Stx1 antibodies. Toxin preparations contained <0.1 ng endotoxin per ml as determined by the Limulus amoebocyte lysate assay (Associates of Cape Cod, Falmouth, ME).
Macrophage differentiation and stimulation.
The human myelogenous leukemia cell line THP-1 (60) was obtained from the American Type Culture Collection, Manassas, VA. The cells were maintained in RPMI 1640 (Gibco-BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; HyClone Laboratories, Logan, UT), penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C in 5% CO2 in a humidified incubator. The mature macrophage-like state was induced by treating THP-1 cells (1 × 106 cells/ml) for 48 h with phorbol 12-myristate 13-acetate (PMA) at 50 ng/ml. Plastic-adherent cells were washed twice with cold, sterile Dulbecco's phosphate-buffered saline (PBS) and incubated with fresh RPMI 1640 lacking PMA but containing 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml). The medium was then changed every 24 h for 3 additional days. Experiments were performed on the 4th day after removal of PMA. To isolate total RNA, differentiated THP-1 cells (5 × 106 cells/ml) were washed once with cold PBS and fresh RPMI 1640 medium containing 10% FBS, with no antibiotics added prior to stimulation with Stx1 (400 ng/ml) for various times. We have previously demonstrated that this toxin dose produces maximal cytokine protein secretion in differentiated THP-1 cells in vitro (48). To prepare cellular lysates, THP-1 cells (5 × 106 cells/ml) were serum starved by growth in RPMI 1640 medium containing 0.5% FBS for 18 h to reduce background kinase signaling. Cells were washed as described above and stimulated with Stx1 (400 ng/ml) in RPMI 1640 plus 0.5% FBS for various times. For MAPK and DUSP1 inhibitor studies, cells were treated with the ERK1/2 inhibitor (PD98059; 50 μM), JNK1/2 stress-activated protein kinase inhibitor (SP600125, 50 μM), p38 MAPK inhibitor (SB203580, 20 μM), or the DUSP1 inhibitor triptolide (0.05 μM) for 1 h prior to and throughout treatment with Stx1 (400 ng/ml) in RPMI 1640 plus 10% FBS for various times.
siRNA transfection.
THP-1 cells (5 × 106 cells) were differentiated as described above. Two hours before transfection, medium was replaced with Dulbecco's modified Eagle medium plus 10% FBS. Complexes of transfection reagent and DUSP1- and early growth response factor 1 (Egr-1)-specific and nonspecific siRNAs were prepared according to the manufacturer's instructions (TransIT-TKO transfection reagent; Mirus Bio Corporation, Madison, WI). After addition of the siRNA complexes, cells were incubated for 48 h at 37°C in 5% CO2. Before Stx1 stimulation, viable cells were washed with 1× PBS, and RPMI plus 10% FBS was added. Cells were treated with Stx1 for 24 h. Supernatants were collected for measurement of soluble cytokines by enzyme-linked immunosorbent assay (ELISA), and total RNA was isolated. To assess transfection efficiency, quantitative reverse transcription-PCR (qRT-PCR) was performed using primers for DUSP1, Egr-1, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase).
qRT-PCR.
Total RNA was isolated using a TRIzol Plus kit (Invitrogen, Carlsbad, CA) with an RNase-free DNase (Invitrogen) treatment per the manufacturer's instructions. RNA was reverse transcribed to cDNA using a High-Capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA), and real-time PCR was performed on the resulting cDNAs using SYBR green I double-stranded DNA binding dye (Applied Biosystems). Real-time PCR primer sequences specific for DUSP1, Egr-1, GAPDH, growth-regulated protein β (GRO-β), IL-1β, IL-8, macrophage inflammatory protein 1α (MIP-1α), macrophage inflammatory 1β (MIP-1β), and TNF-α are shown in Table 1 (17, 25, 35, 66). The real-time PCRs were carried out with 100 nM concentrations (each) of forward and reverse primers in a final volume of 25 μl. To control for the presence of contaminating DNA in the real-time PCRs, reverse transcriptase-negative reaction mixtures were included. Nontemplate controls were run to test for DNA-contaminated primers. Real-time reactions were run and analyzed using a StepOne Plus Real-Time PCR system (Applied Biosystems). Dissociation curves for PCR samples were made to guarantee amplification of the correct genes. The amount of mRNA fold induction was determined from threshold cycle (ΔCT) values normalized for GAPDH expression and then normalized to the value derived from cells at time zero prior to medium change or treatment. Statistical analyses of real-time PCR data were performed using ΔCT values.
Table 1.
Real-time PCR primersa
| Targetb | Orientationc | Primer sequence (5′–3′) |
|---|---|---|
| DUSP1 (35) | F | GGCCCCGAGAACAGACAAA |
| R | GTGCCCACTTCCATGACCAT | |
| Egr-1 (25) | F | GCCTGCGACATCTGTGGAA |
| R | GCCGCAAGTGGATCTTGGTA | |
| GAPDH (17) | F | CAACGGATTTGGTCGTATTGG |
| R | GGCAACAATATCCACTTTACCAGAGT | |
| GRO-β (17) | F | CTGCCCTTACAGGAACAGAAGAG |
| R | CAAACACATTAGGCGCAATCC | |
| IL-1β (35) | F | TCCCCAGCCCTTTTGTTGA |
| R | TTAGAACCAAATGTGGCCGTG | |
| IL-8 (17) | F | AAGGAACCATCTCACTGTGTGTAAAC |
| R | ATCAGGAAGGCTGCCAAGAG | |
| MIP-1α (17) | F | TTGTGATTGTTTGCTCTGAGAGTTC |
| R | CGGTCGTCACCAGACACACT | |
| MIP-1β (17) | F | CCCTGGCCTTTCCTTTCAGT |
| R | AGCTTCCTCGCGGTGTAAGA | |
| TNF-α (66) | F | CCAGGCAGTCAGATCATCTTCTC |
| R | AGCTGGTTATCTCTCAGCTCCAC |
Primers were synthesized by Integral DNA Technologies (Coralville, IA).
Numbers in parentheses are references for each primer set.
F, forward; R, reverse.
Preparation of cellular lysates.
Cells were harvested and lysed at 4°C in modified radioimmunoprecipitation assay buffer (1.0% Nonidet P-40, 1.0% Na-deoxycholate, 150 mM NaCl, 50 mM Tris-HCl [pH 7.5], 0.25 mM Na-pyrophosphate, 2 mM each sodium vanadate and sodium fluoride, 10 μg/ml aprotinin, 1.0 μg/ml leupeptin and pepstatin, and 200 mM phenylmethylsulfonyl fluoride). Extracts were collected and cleared by centrifugation at 15,000 × g for 10 min. Cleared extracts were stored at −80°C until further use for Western blot analysis.
Western blot analysis.
Cell extracts prepared from stimulated THP-1 cells were used for determination of protein concentration using a DC protein assay kit (Bio-Rad, Hercules, CA). Equal amounts of protein (70 μg per gel lane) were separated by SDS-PAGE using 4 to 15% acrylamide gels, transferred to nitrocellulose membranes, which were blocked with 5% nonfat milk prepared in Tris-buffered saline (TBS)–Tween 20 (200 mM Tris [pH 7.6], 1.38 M NaCl, 0.1% Tween 20), and incubated overnight at 4°C with various primary antibodies in 5% bovine serum albumin made with TBS–0.1% Tween 20. Membranes were then incubated with secondary antibody (anti-rabbit or anti-mouse immunoglobulin G coupled to horseradish peroxidase [HRP]) for 1 h at room temperature. Bands were visualized using a Luminata Forte Western HRP substrate (Millipore, Billerica, MA). The intensities of protein bands captured on autoradiography film were quantitated using Image J software (NIH, Bethesda, MD). Fold induction was calculated as stimulated protein band intensity values divided by unstimulated control protein band intensity values after normalizing for loading controls.
Cytokine and chemokine ELISAs.
Quantitation of secreted IL-1β, IL-8, MIP-1β, and TNF-α protein concentrations was performed by the use of corresponding Quantikine colorimetric sandwich ELISA kits (R&D Systems, Minneapolis, MN). Cellular debris was removed from the supernatants of treated cells by centrifugation. Dilutions of supernatants from untreated control cells and cells treated with Stx1, Stx1 plus ERK1/2 inhibitor, Stx1 plus JNK1/2 inhibitor, Stx1 plus p38 MAPK inhibitor, Stx1 plus triptolide, Stx1 plus DUSP1 siRNA, Stx1 plus Egr-1 siRNA, and Stx1 plus scrambled siRNA were made when necessary, and kit-specified volumes of each sample were added in duplicate to the ELISA plate wells. The manufacturer's protocol was followed, and A450 and A570 were measured by a microtiter plate reader (MR5000; Dynatech Laboratories, Chantilly, VA). Protein concentrations were calculated on the basis of standard curves. The assay sensitivities were as follows: IL-1β, <1 pg/ml; IL-8, 4 pg/ml; MIP-1β, 10 pg/ml; and TNF-α, 1.6 pg/ml.
Statistical analysis.
Western blot data were analyzed using a simple t test or one-way analysis of variance (ANOVA) with Tukey's multiple-comparison posttest. Cytokine and chemokine mRNA expression kinetics were analyzed by one-way ANOVA with Dunnet's posttest. Secreted cytokine and chemokine production was analyzed utilizing one-way ANOVA with Bonferroni's posttest. qRT-PCR data of MAPK inhibitor- and siRNA-treated cells were analyzed by two-way ANOVA with the Bonferroni posttest using GraphPad Prism (version 5.00) for Windows (San Diego, CA).
RESULTS
Cytokine and chemokine expression kinetics induced by Stx1 treatment in differentiated THP-1 cells.
Previous studies showed that in response to Stx1 treatment, differentiated THP-1 cells produced cytokines and chemokines, including IL-1β, TNF-α, GRO-β, IL-8, MIP-1α, and MIP-1β (12, 17, 18). To further characterize the effect of Stx1 on the mRNA expression kinetics of these immunomodulating molecules, we carried out a time course study and evaluated their relative expression compared to that by untreated controls (Fig. 1). We noted two distinct expression patterns. IL-1β and IL-8 showed sustained expression throughout the time course studied, while the rest of the genes displayed a time-dependent transient expression, peaking ∼6 h after intoxication. IL-1β mRNA expression was elevated 2 h after intoxication (38-fold increase) and continued to increase throughout the time course, reaching a 348-fold increase by 24 h (P < 0.001). IL-8 mRNA expression at 2 h was increased 22-fold and also continued to increase throughout the time course, reaching a 176-fold increase by 24 h. MIP-1β mRNA expression was increased 141-fold by 2 h (P < 0.01), peaking at 6 h (209-fold; P < 0.001) and returning to near basal levels (13-fold increase) by 24 h. GRO-β mRNA expression increased by 21-fold by 2 h and peaked at 6 h (52-fold; P < 0.05), decreasing to a 35-fold increase at 24 h. MIP-1α mRNA expression was increased 20-fold at 2 h, peaking at 6 h (79-fold; P < 0.001) and decreasing to a 14-fold increase by 24 h. Finally, TNF-α mRNA expression was increased 54-fold at 2 h (P < 0.01), peaking at 6 h (84-fold; P < 0.001) and decreasing to a 11-fold increase by 24 h.
Fig 1.
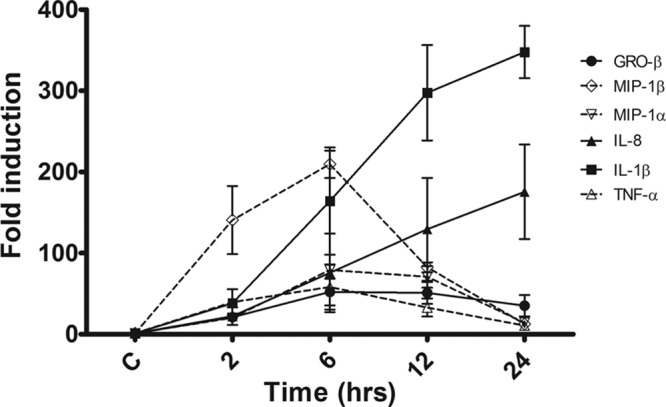
Cytokine and chemokine mRNA expression kinetics in macrophage-like THP-1 cells treated with Stx1. Cells were stimulated with Stx1 (400 ng/ml) for different times. Total RNA was isolated and treated with DNase, and cDNA was synthesized. Quantitative real-time PCR was performed with primers specific for GRO-β, IL-1β, IL-8, MIP-1α, MIP-1β, and TNF-α. The relative expression was calculated by using the ΔCT method. The data shown are the mean fold induction ± SEM of three independent experiments. C, untreated control.
Effect of MAPK inhibitors on Stx1-induced cytokine and chemokine mRNA expression.
Stx1 activates the ribotoxic stress response leading to signaling through the three major MAPK pathways (JNK, p38, and ERK) in macrophage-like THP-1 cells, and these kinases may play a role in the increased cytokine and chemokine expression elicited by Stx1. Previous studies in our laboratory showed that combinations of MAPK inhibitors negatively affected TNF-α, IL-1β, and IL-8 protein secretion (6, 13). To systematically elucidate the effect of each of these signaling cascades on the transcription of IL-1β, IL-8, MIP-1α, MIP-1β, GRO-β, and TNF-α, we treated macrophage-like THP-1 cells with Stx1 and ERK1/2 inhibitor (PD98059), JNK1/2 stress-activated protein kinase inhibitor (SP600125), or p38 MAPK inhibitor (SB203580) and measured the relative expression of these cytokine and chemokine genes compared to that with treatment with Stx1 alone. Treatment of cells with the MAPK inhibitors (p38, JNK, and ERK1/2) alone did not have an effect on cytokine or chemokine expression (data not shown). In cells treated with Stx1 plus ERK inhibitor, IL-1β mRNA expression was decreased to 41%, 30%, 18% (P < 0.05), and 13% (P < 0.05) of the levels induced by Stx1 alone after 2, 6, 12, and 24 h, respectively, showing evidence that ERK is a positive regulator of IL-1β expression. In contrast, treatment of cells with JNK and p38 inhibitors did not have a statistically significant effect on Stx1-induced IL-1β transcript levels (Fig. 2A). IL-8 expression was significantly downregulated in cells treated with Stx1 plus p38 inhibitor (61%, 52%, 32% [P < 0.05], and 20% [P < 0.01] of IL-8 transcript levels induced by Stx1 alone at 2, 6, 12, and 24 h, respectively). In comparison, treatment of cells with JNK and ERK inhibitors did not have a statistically significant effect on IL-8 mRNA expression (Fig. 2B). Both MIP-1α and MIP-1β showed similar patterns of MAPK-dependent regulation of expression. MIP-1α and MIP-1β showed a trend of downregulation throughout all the time points studied in response to treatment with Stx1 plus p38 inhibitor. MIP-1α and MIP-1β expression was not significantly affected by JNK or ERK inhibitor treatment, supporting the concept that JNK and ERK do not play a major regulatory role in MIP-1α or MIP-1β expression (Fig. 2C and D). GRO-β expression was decreased significantly compared to that with treatment with Stx1 alone in the presence of ERK and p38 inhibitors, suggesting that both these kinases play a positive regulatory role in GRO-β expression in Stx1-treated cells. In cells treated with Stx1 plus JNK inhibitor, GRO-β expression did not change significantly from that elicited by Stx1 treatment alone, suggesting that JNK does not play a major role in the regulation of this chemokine (Fig. 2E). Finally, TNF-α expression was not significantly affected by any of the inhibitors; however, treatment with the ERK and JNK inhibitors showed a trend of downregulation, while at early time points, treatment with the p38 inhibitor showed an upward trend in TNF-α mRNA expression (Fig. 2F).
Fig 2.

Effect of MAPK inhibitors on Stx1-induced cytokine and chemokine mRNA expression in macrophage-like THP-1 cells. Cells were stimulated with Stx1 (400 ng/ml) in the presence or absence of inhibitors of ERK1/2 (PD98059, 50 μM; black bars), JNK1/2 (SP600125, 50 μM; white bars), or p38 (SB203580, 20 μM; hatched bars) for different times. Dotted lines represent cytokine/chemokine mRNA expression in response to treatment with Stx1 alone (100%). Total RNA was isolated and treated with DNase, and cDNA was synthesized. Quantitative real-time PCR was performed with primers specific for IL-1β (A), IL-8 (B), MIP-1α (C), MIP-1β (D), GRO-β (E), TNF-α (F), and GAPDH. The relative expression was calculated by using the ΔCT method. The data shown are the mean fold induction ± SEM of three independent experiments normalized for Stx1 for the corresponding time points (Stx1 = 100%). Statistical significance was calculated using two-way ANOVA (P value versus Stx1, *, <0.05; **, <0.01).
Effect of MAPK inhibitors on cytokine and chemokine secretion.
To assess the effect of the ERK, JNK, and p38 MAPK signaling cascades on the translation of cytokine and chemokine genes, we quantified the secretion of immunoreactive IL-1β, IL-8, MIP-1α, MIP-1β, and TNF-α in cell culture supernatants from cells that were treated for 24 h with Stx1 and inhibitors specific for the MAPKs mentioned above (Fig. 3). In response to Stx1 plus ERK inhibitor treatment, IL-1β secretion decreased compared to that with Stx1 alone (47 ± 17 versus 20 ± 7 pg/ml). In cells treated with Stx1 plus JNK inhibitor compared to Stx1 alone, IL-1β secretion decreased slightly, from 47 ± 17 pg/ml to 39 ± 11 pg/ml. Finally, in cells exposed to Stx1 plus p38 inhibitor compared to Stx1, IL-1β secretion increased from 47 ± 17 pg/ml to 74 ± 18 pg/ml (Fig. 3A). Treatment of macrophage-like THP-1 cells with Stx1 induced a robust IL-8 response (14,005 ± 1,936 pg/ml) that was significantly reduced by treatment with all the MAPK inhibitors (Stx1 plus ERK inhibitor, 3,212 ± 894 pg/ml, P < 0.001; Stx1 plus JNK inhibitor, 5,516 ± 1,180 pg/ml, P < 0.001; Stx1 plus p38 inhibitor, 5,368 ± 987 pg/ml, P < 0.001) (Fig. 3B). MIP-1β secretion in response to Stx1 alone versus Stx1 plus ERK inhibitor was similar (2,928 ± 846 versus 3,285 ± 694 pg/ml). In contrast, MIP-1β secretion was diminished by JNK inhibition (2,928 ± 846 versus 1,478 ± 64 pg/ml) and p38 inhibition (2,928 ± 846 pg/ml versus 2,402 ± 323 pg/ml) (Fig. 3C). In the presence of ERK and JNK inhibitors, TNF-α secretion was reduced (37 ± 9 pg/ml to 23 ± 3 pg/ml and 20 ± 2 pg/ml, respectively). However, TNF-α secretion in cells treated with Stx1 plus p38 increased from 37 ± 9 pg/ml to 75 ± 16 pg/ml (Fig. 3D). Thus, inhibition of p38 MAPK activity in Stx1-treated cells increased soluble TNF-α levels, but the change was not significant. Collectively, these data suggest that cytokine/chemokine gene expression is differentially regulated by the ribotoxic stress response at transcriptional and posttranscriptional levels.
Fig 3.

Effect of MAPK inhibitors on Stx1-induced cytokine and chemokine secretion in macrophage-like THP-1 cells. Cells were stimulated with Stx1 (400 ng/ml) in the presence or absence of inhibitors of ERK1/2 (PD98059, 50 μM), JNK1/2 (SP600125, 50 μM), or p38 (SB203580, 20 μM) for 24 h. Supernatants were collected, and soluble IL-1β (A), IL-8 (B), MIP-1β (C), and TNF-α (D) levels were measured in supernatants collected from cells using a sandwich ELISA. Standards provided with the kits were used to calculate soluble cytokine/chemokine protein amounts. Values are given as pg/ml and expressed as the mean ± standard error of three independent experiments. Statistical significance was calculated using one-way ANOVA (P value versus control, #, <0.05; ##, <0.01; ###, <0.001; P value versus Stx1, *, <0.05; **, <0.01; ***, <0.001).
Stx1 induces upregulation of dual-specificity phosphatase 1 in macrophage-like THP-1 cells.
Our previous studies showed that in response to Stx1 treatment, macrophage-like THP-1 cells express select members of the dual-specificity phosphatase family (DUSP1, -5, and -10) (32). These enzymes dephosphorylate threonine and tyrosine residues to inactivate MAPK isoforms in mammalian cells. DUSP1, the prototypical member of the DUSP family, is capable of dephosphorylating all the MAPKs that we studied, with a substrate preference of p38 > JNK > ERK (43). To confirm that this phosphatase was expressed at the protein level, we stimulated differentiated THP-1 cells with Stx1 for various times and analyzed DUSP1 expression by Western blotting (Fig. 4). DUSP1 expression was detected as early as 1 h after intoxication (5.7-fold increase); increased expression became statistically significant at 2 h (8.8-fold, P < 0.05), peaking at 4 h (9-fold, P < 0.05); and DUSP1 was continually expressed throughout the rest of the time course, with a 7-fold increase in expression still detected 24 h after intoxication (P < 0.01).
Fig 4.

DUSP1 protein expression elicited by Stx1 treatment of macrophage-like THP-1 cells. Cells were stimulated with Stx1 (400 ng/ml) for various times. (A) Whole-cell lysates (70 μg) were subjected to SDS–4 to 15% PAGE and probed with polyclonal antibodies against DUSP1 and actin (loading control). The blot shown is representative of three independent experiments. (B) Mean fold change ± SEM calculated from densitometric scanning of three individual experiments. Statistical significance was calculated using a t test (P values, *, <0.05; **, <0.01). WB, Western blot.
Effect of triptolide-mediated DUSP1 inhibition on MAPK phosphorylation induced by Stx1.
Triptolide, a diterpenoid isolated from the Chinese herb Tripterygium wilfordii hook f, has been shown to inhibit DUSP1 induction in various cell types treated with different stimuli (5, 46, 64). Therefore, we investigated the effect of triptolide and Stx1 treatments on MAPK activation. We hypothesized that inhibition of DUSP1 would result in increased ERK, JNK, and p38 phosphorylation following Stx1 treatment (Fig. 5). ERK phosphorylation in the presence of Stx1 was increased 8.8-fold, 14.2-fold, and 17.8-fold compared to that for untreated cells after 6, 12, and 24 h, respectively. When cells were treated with Stx1 plus triptolide, ERK phosphorylation compared to that after treatment with toxin alone was not significantly altered at 6 h (7.8-fold increase) but was increased (29.6-fold) at 12 h and significantly increased (39.6-fold) at 24 h. We examined p38 phosphorylation in cells treated with Stx1 for 6, 12, and 24 h and found that it was increased by 4.7-fold, 10.9-fold, and 31.7-fold, respectively, compared to that in untreated cells. Phosphorylation of p38 in cells treated with Stx1 plus triptolide increased slightly at 6 and 12 h (8.4-fold and 14.9-fold, respectively) and increased significantly at 24 h (70.8-fold). Finally, we measured JNK phosphorylation levels in cells treated with Stx1 compared to untreated controls and found that JNK phosphorylation was increased to 2.3-fold, 4.0-fold, and 2.4-fold after 6, 12, and 24 h of treatment, respectively. JNK phosphorylation in response to Stx1 plus triptolide treatment did not change significantly from that in Stx1-treated cells (2.7-fold and 2.2-fold at 6 and 12 h, respectively) but was dramatically reduced after 24 h of triptolide treatment (0.5-fold), although this change was not statistically significant. Cell viability assays were performed to assess the effect of triptolide on THP-1 cells, and we found that treatment with triptolide alone did not affect cell viability at the concentrations used in this study (data not shown). Treatment with triptolide alone for the duration of the time course did not induce ERK, JNK, or p38 phosphorylation (data not shown). These data confirm the hypothesis that DUSP1 inhibition by triptolide enhances Stx1-induced phosphorylation of ERK and p38. While JNK phosphorylation did not appear to be regulated by this treatment at early time points, JNK phosphorylation appeared to be downregulated by triptolide treatment at 24 h (Fig. 5).
Fig 5.

MAPK phosphorylation in macrophage-like THP-1 cells treated with Stx1 and triptolide. Cells were stimulated with Stx1 (400 ng/ml) in the presence or absence of 0.05 μM triptolide for various time points. Whole-cell lysates (70 μg) were subjected to SDS–4 to 15% PAGE and probed with polyclonal antibodies against phospho-ERK, phospho-JNK, phospho-p38, ERK, JNK, and p38 MAPKs. C, untreated controls. The blots shown are representative of three to four independent experiments. The bar graphs represent mean fold change ± SEM calculated from densitometric scanning of three to four individual experiments for phospho-ERK (p-ERK), phospho-JNK (p-JNK), and phospho-p38 (p-p38) levels normalized to total MAPKs. Statistical significance was calculated using one-way ANOVA (**, P <0.01).
Stx1-induced cytokine and chemokine production is reduced in triptolide-treated cells.
Triptolide is known to have anti-inflammatory effects and is capable of decreasing cytokine and chemokine secretion in response to various stimuli in different cell types (34). However, since we observed increased ERK and p38 phosphorylation in cells treated with Stx1 plus triptolide for 24 h, we predicted that this observation would correlate with increased secretion of IL-1β, IL-8, MIP-1β, and TNF-α. Therefore, we measured soluble cytokine and chemokine levels in supernatants isolated from THP-1 cells treated with Stx1 in the presence and absence of triptolide (Fig. 6). IL-1β secretion in response to Stx1 increased significantly compared to that by untreated cells (43 ± 6 pg/ml versus 6 ± 2 pg/ml) and decreased to 25 ± 4 pg/ml in cells treated with Stx1 plus triptolide. IL-8 secretion in untreated cells was 26 pg/ml, increased significantly to 8,720 ± 1,980 pg/ml in response to Stx1, and decreased significantly to 1,330 ± 270 pg/ml in cells treated with Stx1 plus triptolide. The secretion of MIP-1β increased significantly in cells stimulated with Stx1 compared to untreated controls (3,557 ± 1,322 versus 52 ± 19 pg/ml) and decreased to 800 ± 239 pg/ml in cells treated with Stx1 plus triptolide. TNF-α secretion in Stx1-treated cells was significantly increased compared to that in untreated cells. TNF-α secretion decreased very slightly in cells treated with Stx1 plus triptolide compared to Stx1 alone (43 ± 7 versus 52 ± 8 pg/ml). Therefore, it appears that the increased MAPK phosphorylation induced by Stx1 plus triptolide does not correlate with increased cytokine/chemokine secretion. Alternatively, treatment of THP-1 cells with Stx1 and triptolide for 24 h caused a downregulation of JNK (Fig. 5) that caused downregulation of IL-8 and MIP-1β (Fig. 3B and C). We speculate that inhibition of DUSP1 activity does not overcome downstream anti-inflammatory mechanisms induced by triptolide, which may be regulated independently of DUSP1 activity.
Fig 6.

Soluble cytokine and chemokine production in macrophage-like THP-1 cells treated with Stx1 and triptolide. Cells were stimulated with Stx1 (400 ng/ml) in the presence or absence of triptolide (0.05 μM) for 24 h. IL-1β (A), IL-8 (B), MIP-1β (C), and TNF-α (D) levels were measured in supernatants collected from cells using a sandwich ELISA. Standards provided with the kits were used to calculate soluble cytokine/chemokine protein amounts. Values are given as pg/ml and expressed as the mean ± standard error of three independent experiments. Statistical significance was calculated using one-way ANOVA (P value versus control, #, <0.05; ##, <0.01; P value versus Stx1, **, <0.01).
Secretion of cytokines and chemokines in Stx1-treated cells expressing reduced DUSP1 and Egr-1 mRNA levels.
To further characterize the role of DUSP1 on the regulation of MAPK-mediated cytokine and chemokine expression elicited by Stx1, we transiently knocked down DUSP1 mRNA expression utilizing siRNA transfection. To confirm the knockdown, DUSP1 mRNA was measured in cells treated for 24 h with Stx1, Stx1 plus DUSP1 siRNA, and Stx1 plus an unspecific scrambled (sc) siRNA. DUSP1 mRNA expression was reduced by 48% in cells transfected with the DUSP1 siRNA compared to Stx1-treated cells, while transfection of the scrambled siRNA did not affect the levels of DUSP1 elicited by Stx1 (Fig. 7). We predicted that decreased DUSP1 expression would correlate with increased secretion of proinflammatory mediators. In contrast to the prediction, we found that reduced DUSP1 expression did not significantly alter the IL-1β, IL-8, MIP-β, and TNF-α secretion elicited by Stx1 (data not shown). We previously showed that increased expression of the transcription factor Egr-1 is an ERK-mediated response in THP-1 macrophage-like cells treated with Stx1, and Egr-1 is known to positively regulate expression of genes encoding various inflammatory mediators, including cytokines and chemokines (27, 32). Therefore, we measured the secretion of IL-1β, IL-8, MIP-β, and TNF-α in Stx1-treated cells transfected with an Egr-1 siRNA. Using qRT-PCR, we determined that Egr-1 mRNA expression was decreased by 39% in cells treated with Stx1 and Egr-1 siRNA compared to Stx1 alone and that treatment with an unspecific scrambled siRNA did not affect Egr-1 transcript levels in response to Stx1 (data not shown). We hypothesized that reduced levels of Egr-1 would result in decreased IL-1β, MIP-1β, and TNF-α secretion since it is known that the genes corresponding to these inflammatory mediators are targets of Egr-1 (44). However, diminution of Egr-1 mRNA expression did not have a significant effect on IL-1β, MIP-1β, and TNF-α secretion (data not shown). The level of reduction of DUSP1 and Egr-1 mRNA expression may have been inadequate to detect differences in cytokine and chemokine expression in response to Stx1 treatment, and other isoforms of DUSPs and Egrs may compensate for the loss of DUSP1 and Egr-1. Alternatively, other regulatory factors may be involved in the expression of cytokines and chemokines in response to toxin treatment.
Fig 7.
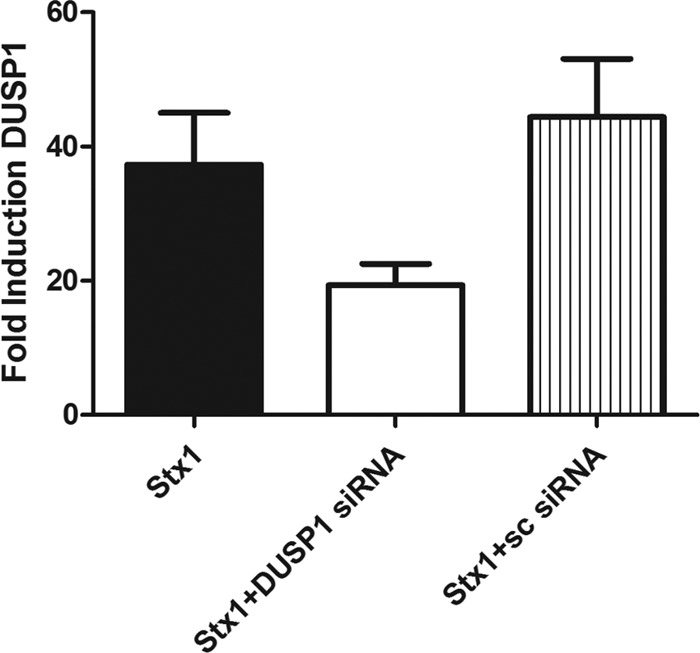
Confirmation of DUSP1 siRNA knockdown. Macrophage-like THP-1 cells were treated with DUSP1 siRNA or scrambled (sc) and subsequently stimulated with Stx1 (400 ng/ml) for 24 h. Total RNA was isolated and treated with DNase, and cDNA was synthesized. Quantitative real-time PCR was performed with primers specific for DUSP1 and GAPDH. The relative expression was calculated by using the ΔCT method. The data shown are the mean fold induction ± standard error of three independent experiments.
Transcriptional regulation of DUSP1 by the ERK, JNK, and p38 MAPKs.
Dual-specificity phosphatases are subject to tight regulation and are expressed in very low levels under basal conditions, and MAPKs can control induction of gene expression and posttranslational modification of the DUSPs (2, 3). To examine the role of ERK, JNK, and p38 MAPKs in DUSP1 mRNA induction by Stx1, we used the pharmacological inhibitors for these pathways that have been described above (Fig. 8). ERK and JNK inhibitors had no significant effect on DUSP1 mRNA levels compared to that for Stx1-treated cells. In contrast, cells stimulated with Stx1 in the presence of p38 inhibitor showed almost complete ablation of the DUSP1 transcript at all time points, and this change was statistically significant at 12 and 24 h (P < 0.001 and P < 0.01, respectively). These results lead us to believe that at the transcriptional level, p38 is the major MAPK that regulates DUSP1 expression in response to Stx1.
Fig 8.
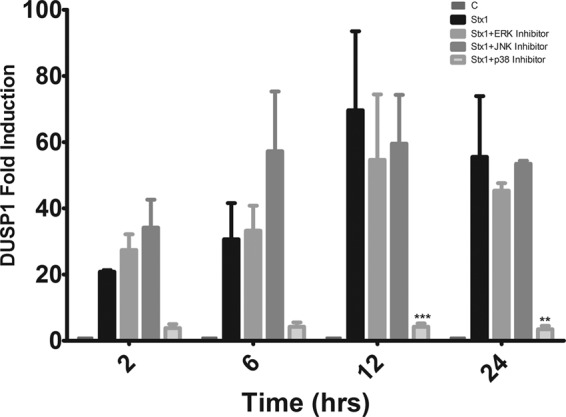
Stx1-induced DUSP1 mRNA expression in macrophage-like THP-1 cells treated with MAPK inhibitors. Cells were stimulated with Stx1 (400 ng/ml) in the presence or absence of ERK1/2 (PD98059, 50 μM), JNK1/2 (SP600125, 50 μM), or p38 (SB203580, 20 μM) inhibitors for different times. Total RNA was isolated and treated with DNase, and cDNA was synthesized. Quantitative real-time PCR was performed with primers specific for DUSP1 and GAPDH. The relative expression was calculated by using the ΔCT method. The data shown are the mean fold induction ± SEM from three to four independent experiments. Statistical significance was calculated using two-way ANOVA (P values versus Stx1, **, <0.01; ***, <0.001).
DISCUSSION
The capacity of Stxs to mediate protein synthesis inhibition is well characterized. However, recent studies suggest that the toxins are multifunctional proteins. The toxins are capable of inducing apoptosis or autophagy, and toxin B subunits lacking enzymatic activity have been shown to induce apoptosis in some cell types (16, 31, 37). Thus, the induction of programmed cell death may be dissociated from protein synthesis inhibition. Primary human monocytes and macrophage-like cell lines are relatively resistant to the cytotoxic action of Stxs (4, 16) yet respond to the toxins by producing soluble products that regulate the innate immune response, including prostaglandins, cytokines, and chemokines (12, 13, 17, 18, 32, 42, 48, 57). How toxins capable of protein synthesis inhibition mediate increased protein expression has been a subject of intense scrutiny, and it is now known that Stxs activate the transcription factors AP-1, NF-κB, and Egr-1 (22, 32, 49). Finally, the depurination reaction catalyzed by Stx A1 fragments following retrotranslocation from the ER lumen into the cytoplasm activates the ribotoxic stress response (4, 6, 52).
The term ribotoxic stress response describes the reaction of cells to ribosome-inactivating proteins which alter the peptidyltransferase center (domains V and VI) of the 28S rRNA ribosomal subunit leading to variable activation of ERK, JNK, and p38, depending on the cell type and toxicant (19–21). Although the precise mechanisms linking Stx-mediated ribosomal depurination with activation of MAPKs remain to be fully characterized, it is known that Stx A1 fragments and the ribosome-inactivating protein ricin A chain interact with acidic ribosomal phosphoproteins that form the ribosomal stalk (8, 40). This interaction appears to be necessary for the depurination reaction mediated by both Stx A1 fragments and ricin A chain. Gray et al. (15) showed that double-stranded RNA-activated protein kinase (PKR) inhibitors significantly reduced the capacity of Stx1 to induce IL-8 expression by human U-937 monocytic cells. PKR is a serine/threonine kinase which binds to and is activated by damaged ribosomes. Thus, the interaction of Stxs with the ribosomal stalk and the subsequent depurination reaction may induce sufficient alterations in ribosomal conformation to allow PKR and other kinases which recognize altered ribosomal structure to bind and be activated (1). Activated PKR phosphorylates and inactivates eukaryotic translation initiation factor-2α (eIF-2α), which downregulates global translation, although expression of proteins involved in host cell stress responses is maintained and/or upregulated. Other downstream substrates of PKR include IκB, leading to NF-κB activation, and the MAP kinase kinase kinase (MAP3K) apoptosis signal-regulating kinase-1 (ASK-1). ASK-1 may subsequently activate MAP kinase kinases (MAP2Ks) MKK3/6 and MKK4/7, which in turn activate p38 MAPK and JNK activation, respectively (reviewed in reference 55). Finally, inhibition of the MAP3K zipper sterile α-motif kinase (ZAK) was shown to block Stx2- and ricin-induced activation of JNK and p38 MAPKs in Hct8 and Vero cells, implicating this MAP3K in Stx activation of the ribotoxic stress response (24).
We have shown that Stxs activate the ribotoxic stress response in THP-1 macrophage-like cells. Furthermore, the MAPKs regulate the expression and secretion of the proinflammatory cytokines IL-1β and TNF-α and the CXC chemokine IL-8 (6, 13). However, macroarray analyses of global changes in cytokine and chemokine gene expression in THP-1 cells treated with Stx1 showed that additional chemokine genes are expressed in cells exposed to Stx1 (17). Therefore, we systematically explored the individual roles of the ERK, JNK, and p38 pathways at both the transcriptional (mRNA expression) and translational (protein secretion) levels for the molecules mentioned above, in addition to the CXC chemokine GRO-β and the CC chemokines MIP-1α and MIP-1β. We found two distinct patterns of expression: GRO-β, MIP-1α, MIP-1β, and TNF-α peak transcript levels were detected approximately 6 h after toxin exposure and then decreased. IL-1β and IL-8 mRNA expression continually increased throughout the time course of the experiments. These results correlate with previous observations in THP-1 cells, although the kinetics of GRO-β, MIP-1α, and MIP-1β mRNA expression had not been examined in detail (17, 18). While MAPKs may be involved in transcriptional regulation of cytokine/chemokine gene expression by activation of transcription factors, the kinases may also regulate expression at the posttranscriptional level by stabilization of mRNA and phosphorylation of eIF4E and 4E-BP (4E-binding protein), leading to increased translation initiation (6). Therefore, it was important to investigate differential regulation of cytokine/chemokine expression at the mRNA and protein levels.
IL-1β mRNA was significantly reduced when ERK was inhibited, while JNK and p38 MAPKs did not appear to play an important role in expression. The pattern of decreased IL-1β expression in the presence of the ERK inhibitor was recapitulated when IL-1β protein levels were measured in cell supernatants. As we have previously noted (18), there was no direct correlation between Stx1-induced IL-1β transcript and protein expression; we detected a >300-fold increase in IL-1β mRNA, while protein levels were increased only ∼10-fold. The precise mechanisms responsible for the differential expression of IL-1β transcript and protein remain to be characterized, although it is known that IL-1β protein undergoes extensive posttranslational processing prior to secretion (65).
Neutrophil infiltration into the lamina propria and fecal leukocytosis is seen in the colitis caused by STEC (51). IL-8 and GRO-α, neutrophil chemoattractants, are produced by intestinal epithelial cells in vitro in response to Stx treatment and may aid in neutrophil recruitment (59). IL-8 has also been found in the urine of HUS patients, suggesting the presence of neutrophils in the kidney during extraintestinal disease (11). Stx-mediated IL-8 secretion was reduced when p38 was inhibited in human intestinal cells (58), and we found that p38 inhibition significantly reduced IL-8 mRNA expression and secretion, providing further evidence that p38 plays an important role in IL-8 production elicited by Stxs. Individual inhibition of ERK and JNK did not alter IL-8 transcripts levels but significantly reduced Stx1-mediated IL-8 secretion to a similar extent as p38 MAPK inhibition, suggesting that all three pathways are important in the regulation of IL-8 protein production. This finding correlates with our previous study that showed that treating THP-1 cells with all three inhibitors reduced IL-8 secretion by 84% (6). When ERK and p38 were inhibited, significantly decreased GRO-β mRNA levels were observed, suggesting that both kinase pathways positively regulate transcription of this chemokine.
MIP-1α and MIP-1β are monocyte chemoattractants that can activate cells by increasing intracellular Ca2+ concentrations, which may lead to the release of arachidonic acid (41). Biopsy specimens from patients with glomerulonephritis showed increased expression of both MIPs and a monocytic infiltrate (41). In a murine model of Stx-mediated renal damage, Keepers et al. (26) showed a monocytic infiltrate in the kidneys of mice administered Stx2 which corresponded with increased renal expression of the monocytic chemoattractants MIP-1α, MIP-1β, and RANTES. MIP-1α and MIP-1β are expressed and secreted in THP-1 cells that have been exposed to Stx1 (17), but the role of MAPKs in their regulation has not been explored. We show here that MIP-1α transcript levels were reduced in the presence of p38 inhibitor, while ERK and JNK inhibition did not significantly alter mRNA levels. As was the case with MIP-1α mRNA expression, inhibition of the p38 MAPK pathway decreased MIP-1β mRNA expression and JNK inhibition increased mRNA levels at the 24-h time point. However, we noted a trend of increased MIP-1β mRNA expression in THP-1 cells treated with Stx1 plus ERK inhibitor.
TNF-α and IL-1β have been shown to upregulate the expression of Gb3, the Stx receptor, on vascular endothelial cell membranes (61, 63). TNF-α is expressed and secreted by macrophages in response to Stx exposure (48), and combinations of MAPK inhibitors partially abrogate TNF-α secretion in THP-1 cells (6, 13, 47). Cameron et al. (4) showed that Stx1- and Stx2-mediated TNF-α secretion was almost completely abolished by inhibiting p38 and partially diminished by inhibiting ERK and JNK in primary human peripheral blood monocytes. In accordance with these studies, we show that inhibition of ERK and JNK partially decreased TNF-α mRNA levels at early time points; however, p38 inhibition did not affect TNF-α mRNA levels in THP-1 cells treated with Stx1. Decreased levels of soluble TNF-α detected in cell supernatants correlated with transcript levels in ERK- and JNK-inhibited cells. However, we noted an increase in soluble TNF-α produced by cells treated with Stx1 plus p38 inhibitors, and the increase did attain statistical significance compared to the result for cells treated with Stx1 only.
Stx1 treatment is known to elicit DUSP1 mRNA expression, a negative regulator of all the MAPK pathways discussed above, in macrophages, intestinal epithelial cells, and endothelial cells (29, 32, 39). We confirmed DUSP1 expression at the protein level and found that it is produced in a time-dependent manner and that it remains upregulated throughout the studied time course. To our knowledge, this is the first time that DUSP1 protein expression elicited by Stx1 exposure has been shown. We hypothesized that the inhibition of DUSP1 would result in increased MAPK phosphorylation following activation of the ribotoxic stress response by Stx1. Therefore, THP-1 cells were treated with Stx1 plus triptolide, a pharmacological inhibitor of DUSP1 that is also known to dampen proinflammatory responses (30). Our hypothesis was confirmed for ERK and p38, which showed increased phosphorylation levels in the presence of Stx1 plus triptolide compared to Stx1 alone. JNK phosphorylation levels were not significantly different, suggesting that DUSP1 does not play an important role in JNK regulation in response to Stx1 in this cell type. Zhao et al. (67) showed that in murine alveolar macrophages treated with lipopolysaccharide (LPS) and triptolide, dephosphorylation of JNK and p38 was delayed but there was little effect on ERK. However, it is also known that in murine RAW264.7 macrophages, ERK dephosphorylation was modestly delayed by triptolide treatment, while phosphorylation of JNK and p38 was significantly delayed by triptolide treatment. Intervertebral disc cells stimulated with IL-1β and triptolide showed increased phosphorylation of ERK and p38 but not JNK (5, 28). Thus, patterns of DUSP1 regulation of MAPK may be stimulus and cell type specific. It will be interesting to determine if MAPK phosphorylation patterns differ in other cell types treated with Stxs and triptolide.
Microarray analysis of murine macrophages exposed to LPS plus triptolide showed that many of the genes upregulated by LPS, genes that are known to regulate immune function and host defense, were downregulated by triptolide, including the genes encoding TNF-α and IL-1β. This downregulation correlated with reduced cytokine secretion. For example, LPS-induced IL-8 secretion was decreased in corneal fibroblasts treated with triptolide (36). In keeping with these studies, we show here that even though ERK and p38 phosphorylation was increased by triptolide, we still saw a dampening of the inflammatory response in THP-1 cells treated with both Stx1 and triptolide, as evidenced by decreased cytokine and chemokine secretion. These data suggest that other downstream effects of triptolide, such as NF-κB activation, are sufficient to suppress the inflammatory response elicited by Stx1 (36, 38).
Delayed JNK and p38 MAPK dephosphorylation and a subsequent increase in TNF-α production were observed in bone marrow-derived macrophages isolated from DUSP1-knockout mice (7). To establish that DUSP1-mediated alterations in the phosphorylation status of MAPKs are involved in the regulation of cytokine and chemokine production by Stxs, we knocked down DUSP1 by transfecting THP-1 cells with a DUSP1-specific siRNA and achieved a 48% knockdown at the mRNA level. However, we did not see significant changes in cytokine and chemokine production compared to that by cells treated with Stx1 alone. Other members of the dual-specificity family are also expressed by THP-1 cells in response to Stx1 treatment and may be able to compensate for the partial loss of DUSP1. Alternatively, complete abrogation of DUSP1 expression may be necessary to observe a significant effect on cytokine production if reduced levels of DUSP1 are capable of MAPK dephosphorylation (32). We also transfected cells with an Egr-1 siRNA and achieved a partial knockdown of Egr-1 mRNA. Egr-1 is a zinc finger transcription factor that belongs to a group of early response genes. It has over 300 target genes; among these are several inflammation-related genes, such as those for TNF-α, IL-1β, IL-6, MCP-1, MIP-1α, MIP-1β, tissue factor, plasminogen activator inhibitor 1, intercellular adhesion molecule 1, CD44, and transforming growth factor β. We previously observed that ERK inhibition significantly decreased Stx-mediated Egr-1 and TNF-α expression (27, 32, 44). We did not observe significant changes in TNF-α, IL-8, and MIP-1β secretion in THP-1 cells treated with Stx1 plus Egr-1 siRNA compared to cells treated with Stx1 alone. This may be due to the fact that we achieved only partial loss of Egr-1 expression or that other transcription factors may compensate for Egr-1 loss. Finally, we wanted to explore the role of MAPK on transcriptional regulation of the dusp1 gene. In murine macrophages, LPS-induced DUSP1 expression was primarily regulated by ERK, while p38 played only a minor role (5). In contrast to these findings, we found that Stx1-induced DUSP1 expression was primarily mediated by p38 MAPK, while ERK and JNK had little effect. These data reinforce the concept that the consequences of activation of the ribotoxic stress response are stimulus and cell type specific.
In summary, we show that Stx1 activates the ribotoxic stress response in THP-1 macrophage-like cells, as evidenced by activation of the ERK, p38, and JNK MAPKs. ERK and p38 activation is negatively regulated by DUSP1, while DUSP1 mRNA expression is regulated by p38 MAPKs, creating an autoregulatory loop (Fig. 9). Thus, the signal for deactivation of the ribotoxic stress response may be a part of the activation cascade. ERK has positive regulatory effects on IL-1β, IL-8, GRO-β, and TNF-α expression. JNK positively regulates IL-8, MIP-1α, MIP-β, and TNF-α. Finally, p38 MAPKs have a positive effect on IL-8, MIP-1α, MIP-1β, GRO-β, and TNF-α expression. Therefore, cytokine and chemokine production are differentially regulated by MAPKs. The data suggest that Stxs are capable of eliciting both pro- and anti-inflammatory responses in human macrophage-like cells and the balance between the two responses may be important for either the resolution or progression of disease. Further study of the innate immune response to Stxs will be necessary to develop therapeutic agents regulating the pro- and anti-inflammatory effects of Stxs.
Fig 9.
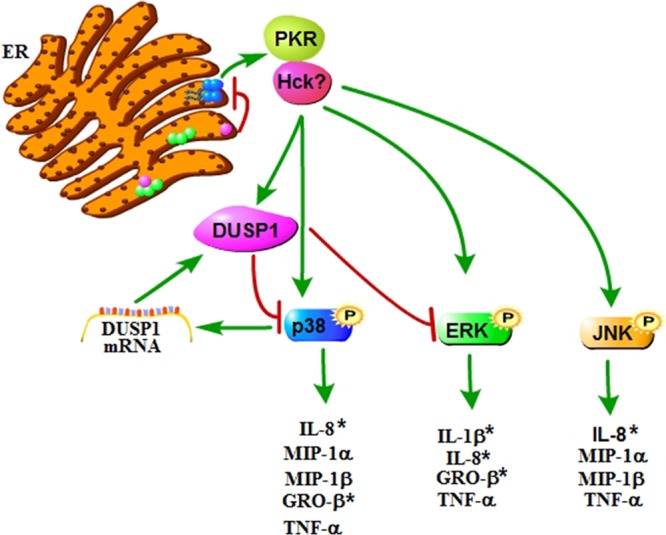
Model of the regulation of proinflammatory cytokine and chemokine production by MAPKs in THP-1 cells. After retrotranslocation of the Stx A1 fragment (purple circle) across the ER membrane, depurination of a single adenine in the 28S rRNA ribosomal subunit (blue circles) may lead to a conformational change that allows PKR and possibly other kinases to bind the ribosome. ERK, p38, and JNK MAPKs are activated, resulting in differential regulation of cytokine and chemokine expression. ERK and p38 activation is negatively regulated by DUSP1, while DUSP1 mRNA expression is regulated by p38 MAPKs, creating an autoregulatory loop. Asterisks denote statistical significance. (Adapted from reference 55 with permission of the publisher, John Wiley & Sons.) The figure was made using the Pathway Builder (version 2.0) software.
ACKNOWLEDGMENT
This work was supported by U.S. Public Health Service grants 2RO1 AI34530-10.
Footnotes
Published ahead of print 19 March 2012
REFERENCES
- 1. Bae H, et al. 2010. Hematopoietic cell kinase associates with the 40S ribosomal subunit and mediates the ribotoxic stress response to deoxynivalenol in mononuclear phagocytes. Toxicol. Sci. 115:444–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bermudez O, Pages G, Gimond C. 2010. The dual-specificity MAP kinase phosphatases: critical roles in development and cancer. Am. J. Physiol. Cell Physiol. 299:C189–C202 [DOI] [PubMed] [Google Scholar]
- 3. Britson JS, Barton F, Balko JM, Black EP. 2009. Deregulation of DUSP activity in EGFR-mutant lung cancer cell lines contributes to sustained ERK1/2 signaling. Biochem. Biophys. Res. Commun. 390:849–854 [DOI] [PubMed] [Google Scholar]
- 4. Cameron P, Smith SJ, Giembycz MA, Rotondo D, Plevin R. 2003. Verotoxin activates mitogen-activated protein kinase in human peripheral blood monocytes: role in apoptosis and proinflammatory cytokine release. Br. J. Pharmacol. 140:1320–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen P, et al. 2002. Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J. Immunol. 169:6408–6416 [DOI] [PubMed] [Google Scholar]
- 6. Cherla RP, Lee SY, Mees PL, Tesh VL. 2006. Shiga toxin 1-induced cytokine production is mediated by MAP kinase pathways and translation initiation factor eIF4E in the macrophage-like THP-1 cell line. J. Leukoc. Biol. 79:397–407 [DOI] [PubMed] [Google Scholar]
- 7. Chi H, et al. 2006. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc. Natl. Acad. Sci. U. S. A. 103:2274–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chiou JC, Li XP, Remacha M, Ballesta JP, Tumer NE. 2008. The ribosomal stalk is required for ribosome binding, depurination of the rRNA and cytotoxicity of ricin A chain in Saccharomyces cerevisiae. Mol. Microbiol. 70:1441–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Endo Y, et al. 1988. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 171:45–50 [DOI] [PubMed] [Google Scholar]
- 10. Ferens WA, Hovde CJ. 2011. Escherichia coli O157:H7: animal reservoir and sources of human infection. Foodborne Pathog. Dis. 8:465–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Forsyth KD, Simpson AC, Fitzpatrick MM, Barratt TM, Levinsky RJ. 1989. Neutrophil-mediated endothelial injury in haemolytic uraemic syndrome. Lancet ii:411–414 [DOI] [PubMed] [Google Scholar]
- 12. Foster GH, Armstrong CS, Sakiri R, Tesh VL. 2000. Shiga toxin-induced tumor necrosis factor alpha expression: requirement for toxin enzymatic activity and monocyte protein kinase C and protein tyrosine kinases. Infect. Immun. 68:5183–5189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Foster GH, Tesh VL. 2002. Shiga toxin 1-induced activation of c-Jun NH(2)-terminal kinase and p38 in the human monocytic cell line THP-1: possible involvement in the production of TNF-alpha. J. Leukoc. Biol. 71:107–114 [PubMed] [Google Scholar]
- 14. Garred O, van Deurs B, Sandvig K. 1995. Furin-induced cleavage and activation of Shiga toxin. J. Biol. Chem. 270:10817–10821 [DOI] [PubMed] [Google Scholar]
- 15. Gray JS, Bae HK, Li JC, Lau AS, Pestka JJ. 2008. Double-stranded RNA-activated protein kinase mediates induction of interleukin-8 expression by deoxynivalenol, Shiga toxin 1, and ricin in monocytes. Toxicol. Sci. 105:322–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harrison LM, et al. 2005. Comparative evaluation of apoptosis induced by Shiga toxin 1 and/or lipopolysaccharides in human monocytic and macrophage-like cells. Microb. Pathog. 38:63–76 [DOI] [PubMed] [Google Scholar]
- 17. Harrison LM, van den Hoogen C, van Haaften WC, Tesh VL. 2005. Chemokine expression in the monocytic cell line THP-1 in response to purified Shiga toxin 1 and/or lipopolysaccharides. Infect. Immun. 73:403–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harrison LM, van Haaften WC, Tesh VL. 2004. Regulation of proinflammatory cytokine expression by Shiga toxin 1 and/or lipopolysaccharides in the human monocytic cell line THP-1. Infect. Immun. 72:2618–2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iordanov MS, et al. 2002. The UV (ribotoxic) stress response of human keratinocytes involves the unexpected uncoupling of the Ras-extracellular signal-regulated kinase signaling cascade from the activated epidermal growth factor receptor. Mol. Cell. Biol. 22:5380–5394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iordanov MS, et al. 1997. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol. 17:3373–3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iordanov MS, et al. 1998. Ultraviolet radiation triggers the ribotoxic stress response in mammalian cells. J. Biol. Chem. 273:15794–15803 [DOI] [PubMed] [Google Scholar]
- 22. Ishii H, Takada K, Higuchi T, Sugiyama J. 2000. Verotoxin-1 induces tissue factor expression in human umbilical vein endothelial cells through activation of NF-kappaB/Rel and AP-1. Thromb. Haemost. 84:712–721 [PubMed] [Google Scholar]
- 23. Jackson MP, O'Brien AD, Holmes RK, Newland JW. 1987. Nucleotide sequence analysis and comparison of the structural genes for Shiga-like toxin I and Shiga-like toxin II encoded by bacteriophages from Escherichia coli. FEMS Microbiol. Lett. 44:109–114 [DOI] [PubMed] [Google Scholar]
- 24. Jandhyala DM, Ahluwalia A, Obrig T, Thorpe CM. 2008. ZAK: a MAP3Kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell. Microbiol. 10:1468–1477 [DOI] [PubMed] [Google Scholar]
- 25. Jean S, et al. 2001. The expression of genes induced in melanocytes by exposure to 365-nm UVA: study by cDNA arrays and real-time quantitative RT-PCR. Biochim. Biophys. Acta 1522:89–96 [DOI] [PubMed] [Google Scholar]
- 26. Keepers TR, Gross LK, Obrig TG. 2007. Monocyte chemoattractant protein 1, macrophage inflammatory protein 1 alpha, and RANTES recruit macrophages to the kidney in a mouse model of hemolytic-uremic syndrome. Infect. Immun. 75:1229–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khachigian LM, Collins T. 1998. Early growth response factor 1: a pleiotropic mediator of inducible gene expression. J. Mol. Med. (Berl.) 76:613–616 [DOI] [PubMed] [Google Scholar]
- 28. Klawitter M, et al. 26 July 2011. Triptolide exhibits anti-inflammatory, anti-catabolic as well as anabolic effects and suppresses TLR expression and MAPK activity in IL-1beta treated human intervertebral disc cells. Eur. Spine J. doi:10.1007/s00586-01101919y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kojima S, et al. 2000. mkp-1 encoding mitogen-activated protein kinase phosphatase 1, a verotoxin 1 responsive gene, detected by differential display reverse transcription-PCR in Caco-2 cells. Infect. Immun. 68:2791–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee KY, Chang W, Qiu D, Kao PN, Rosen GD. 1999. PG490 (triptolide) cooperates with tumor necrosis factor-alpha to induce apoptosis in tumor cells. J. Biol. Chem. 274:13451–13455 [DOI] [PubMed] [Google Scholar]
- 31. Lee MS, et al. 2011. Shiga toxins induce autophagy leading to differential signalling pathways in toxin-sensitive and toxin-resistant human cells. Cell. Microbiol. 13:1479–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leyva-Illades D, Cherla RP, Galindo CL, Chopra AK, Tesh VL. 2010. Global transcriptional response of macrophage-like THP-1 cells to Shiga toxin type 1. Infect. Immun. 78:2454–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lingwood CA, Binnington B, Manis A, Branch DR. 2010. Globotriaosyl ceramide receptor function—where membrane structure and pathology intersect. FEBS Lett. 584:1879–1886 [DOI] [PubMed] [Google Scholar]
- 34. Liu Q. 2011. Triptolide and its expanding multiple pharmacological functions. Int. Immunopharmacol. 11:377–383 [DOI] [PubMed] [Google Scholar]
- 35. Locati M, et al. 2002. Analysis of the gene expression profile activated by the CC chemokine ligand 5/RANTES and by lipopolysaccharide in human monocytes. J. Immunol. 168:3557–3562 [DOI] [PubMed] [Google Scholar]
- 36. Lu Y, et al. 2006. Inhibition by triptolide of chemokine, proinflammatory cytokine, and adhesion molecule expression induced by lipopolysaccharide in corneal fibroblasts. Invest. Ophthalmol. Vis. Sci. 47:3796–3800 [DOI] [PubMed] [Google Scholar]
- 37. Mangeney M, et al. 1993. Apoptosis induced in Burkitt's lymphoma cells via Gb3/CD77, a glycolipid antigen. Cancer Res. 53:5314–5319 [PubMed] [Google Scholar]
- 38. Matta R, et al. 2009. Triptolide induces anti-inflammatory cellular responses. Am. J. Transl. Res. 1:267–282 [PMC free article] [PubMed] [Google Scholar]
- 39. Matussek A, et al. 2003. Molecular and functional analysis of Shiga toxin-induced response patterns in human vascular endothelial cells. Blood 102:1323–1332 [DOI] [PubMed] [Google Scholar]
- 40. McCluskey AJ, et al. 2008. The catalytic subunit of Shiga-like toxin 1 interacts with ribosomal stalk proteins and is inhibited by their conserved C-terminal domain. J. Mol. Biol. 378:375–386 [DOI] [PubMed] [Google Scholar]
- 41. Menten P, Wuyts A, Van Damme J. 2002. Macrophage inflammatory protein-1. Cytokine Growth Factor Rev. 13:455–481 [DOI] [PubMed] [Google Scholar]
- 42. O'Brien AD, et al. 1992. Shiga toxin: biochemistry, genetics, mode of action, and role in pathogenesis. Curr. Top. Microbiol. Immunol. 180:65–94 [DOI] [PubMed] [Google Scholar]
- 43. Owens DM, Keyse SM. 2007. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 26:3203–3213 [DOI] [PubMed] [Google Scholar]
- 44. Pawlinski R, et al. 2003. Regulation of tissue factor and inflammatory mediators by Egr-1 in a mouse endotoxemia model. Blood 101:3940–3947 [DOI] [PubMed] [Google Scholar]
- 45. Proulx F, Tesh VL. 2007. Renal diseases in the pediatric intensive care unit: thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenia purpura, p 1189–1203 In Wheeler DS, Wong HR, Shanley TP. (ed), Pediatric care medicine: basic science and clinical evidence. Springer-Verlag, London, United Kingdom [Google Scholar]
- 46. Purwana IN, Kanasaki H, Oride A, Miyazaki K. 2010. Induction of dual specificity phosphatase 1 (DUSP1) by gonadotropin-releasing hormone (GnRH) and the role for gonadotropin subunit gene expression in mouse pituitary gonadotroph L beta T2 cells. Biol. Reprod. 82:352–362 [DOI] [PubMed] [Google Scholar]
- 47. Ramegowda B, Samuel JE, Tesh VL. 1999. Interaction of Shiga toxins with human brain microvascular endothelial cells: cytokines as sensitizing agents. J. Infect. Dis. 180:1205–1213 [DOI] [PubMed] [Google Scholar]
- 48. Ramegowda B, Tesh VL. 1996. Differentiation-associated toxin receptor modulation, cytokine production, and sensitivity to Shiga-like toxins in human monocytes and monocytic cell lines. Infect. Immun. 64:1173–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sakiri R, Ramegowda B, Tesh VL. 1998. Shiga toxin type 1 activates tumor necrosis factor-alpha gene transcription and nuclear translocation of the transcriptional activators nuclear factor-kappaB and activator protein-1. Blood 92:558–566 [PubMed] [Google Scholar]
- 50. Sandvig K, et al. 1992. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 358:510–512 [DOI] [PubMed] [Google Scholar]
- 51. Slutsker L, et al. 1997. Escherichia coli O157:H7 diarrhea in the United States: clinical and epidemiologic features. Ann. Intern. Med. 126:505–513 [DOI] [PubMed] [Google Scholar]
- 52. Smith WE, et al. 2003. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 71:1497–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Strockbine NA, et al. 1986. Two toxin-converting phages from Escherichia coli O157:H7 strain 933 encode antigenically distinct toxins with similar biologic activities. Infect. Immun. 53:135–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tam PJ, Lingwood CA. 2007. Membrane cytosolic translocation of verotoxin A1 subunit in target cells. Microbiology 153:2700–2710 [DOI] [PubMed] [Google Scholar]
- 55. Tesh VL. 2012. Activation of cell stress response pathways by Shiga toxins. Cell. Microbiol. 14:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tesh VL, et al. 1993. Comparison of the relative toxicities of Shiga-like toxins type I and II for mice. Infect. Immun. 61:3392–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tesh VL, Ramegowda B, Samuel JE. 1994. Purified Shiga-like toxins induce expression of proinflammatory cytokines from murine peritoneal macrophages. Infect. Immun. 62:5085–5094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thorpe CM, et al. 1999. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect. Immun. 67:5985–5993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thorpe CM, Smith WE, Hurley BP, Acheson DW. 2001. Shiga toxins induce, superinduce, and stabilize a variety of C-X-C chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infect. Immun. 69:6140–6147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tsuchiya S, et al. 1980. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 26:171–176 [DOI] [PubMed] [Google Scholar]
- 61. van de Kar NC, Monnens LA, Karmali MA, van Hinsbergh VW. 1992. Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: implications for the pathogenesis of the hemolytic uremic syndrome. Blood 80:2755–2764 [PubMed] [Google Scholar]
- 62. van Setten PA, et al. 1998. Monocyte chemoattractant protein-1 and interleukin-8 levels in urine and serum of patents with hemolytic uremic syndrome. Pediatr. Res. 43:759–767 [DOI] [PubMed] [Google Scholar]
- 63. van Setten PA, et al. 1997. Effects of TNF alpha on verocytotoxin cytotoxicity in purified human glomerular microvascular endothelial cells. Kidney Int. 51:1245–1256 [DOI] [PubMed] [Google Scholar]
- 64. Wang J, Zhou JY, Zhang L, Wu GS. 2009. Involvement of MKP-1 and Bcl-2 in acquired cisplatin resistance in ovarian cancer cells. Cell Cycle 8:3191–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Weber A, Wasiliew P, Kracht M. 2010. Interleukin-1beta (IL-1beta) processing pathway. Sci. Signal. 3:cm2. [DOI] [PubMed] [Google Scholar]
- 66. Zhang L, et al. 2003. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 348:203–213 [DOI] [PubMed] [Google Scholar]
- 67. Zhao Q, et al. 2005. The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. J. Biol. Chem. 280:8101–8108 [DOI] [PubMed] [Google Scholar]