

Abstract
Meningitis-causing Escherichia coli K1 internalization of the blood-brain barrier is required for penetration into the brain, but the host-microbial interactions involved in E. coli entry of the blood-brain barrier remain incompletely understood. We show here that a meningitis-causing E. coli K1 strain RS218 activates Rac1 (GTP-Rac1) of human brain microvascular endothelial cells (HBMEC) in a time-dependent manner. Both activation and bacterial invasion were significantly inhibited in the presence of a Rac1 inhibitor. We further showed that the guanine nucleotide exchange factor Vav2, not β-Pix, was involved in E. coli K1-mediated Rac1 activation. Since activated STAT3 is known to bind GTP-Rac1, the relationship between STAT3 and Rac1 was examined in E. coli K1 invasion of HBMEC. Downregulation of STAT3 resulted in significantly decreased E. coli invasion compared to control HBMEC, as well as a corresponding decrease in GTP-Rac1, suggesting that Rac1 activation in response to E. coli is under the control of STAT3. More importantly, two E. coli determinants contributing to HBMEC invasion, IbeA and OmpA, were shown to affect both Rac1 activation and their association with STAT3. These findings demonstrate for the first time that specific E. coli determinants regulate a novel mechanism of STAT3 cross talk with Rac1 in E. coli K1 invasion of HBMEC.
INTRODUCTION
Gram-negative bacillary meningitis continues to be an important cause of mortality and morbidity in neonates (4, 8, 14–16, 28, 29). A major contributing factor to such mortality and morbidity is our limited understanding of the pathogenesis of this disease (14, 15).
Escherichia coli is the most common Gram-negative bacteria causing neonatal meningitis (14, 15). The disease develops following bacterial adhesion to and invasion of the blood-brain barrier that is composed of human brain microvascular endothelial cells (HBMEC). A number of E. coli virulence factors such as FimH, outer membrane protein A (OmpA), IbeA, and cytotoxic necrotizing factor (CNF1) have been identified to contribute to HBMEC adhesion and invasion (9–15, 31, 32) and their contributions to E. coli meningitis have been described in a recent review (15).
The adhesion of E. coli virulence factors to membrane receptors of host cells triggers a cascade of host cell signal transduction pathways that result in host cell actin cytoskeleton rearrangements required for bacterial invasion of HBMEC (15). Regulation of the rearrangement of host actin cytoskeleton by E. coli K1 has been shown to be a complex multifactorial process, involving host actin binding proteins and signaling molecules, as well as specific microbial determinants (15). However, the underlying host-microbial interactions in the bacterial invasion of HBMEC remain incompletely understood.
The Rho family GTPases, including RhoA, Rac1, and Cdc42, have been identified as molecular switches that regulate actin cytoskeleton organization (7, 22–24). We have previously shown that RhoA and Rac1 contribute to type III group B streptococcus (GBS) invasion of HBMEC (25). Since GBS and E. coli represent the two most common bacteria causing neonatal meningitis (15), we examined whether E. coli exploits RhoA and Rac1 for invasion of HBMEC. Our previous studies showed that FimH and CNF1 of E. coli K1 activate RhoA (10, 12). However, there is no information on the role played by host Rac1 or its activation by specific virulence factors of meningitis-causing E. coli K1 for the invasion of HBMEC.
The signal transducers and activators of transcription (STATs) are identified as latent cytoplasmic transcription factors that are activated by cytokines and growth factors (34). The STAT family consists of seven members in mammals, with STAT3 being the most pleiotropic in mediating biological functions (5). STAT3 has been shown to bind GTP-Rac1 (27), as well as play a role in E. coli K1 invasion of HBMEC (19). The present study provides evidence for the role of Rac1 in E. coli K1 invasion of HBMEC, the bacterial virulence factors contributing to activation of Rac1, as well as the involvement of STAT3 in Rac1 activation in HBMEC.
MATERIALS AND METHODS
Reagents.
Rac1 inhibitor (NSC23766) was provided by Yi Zheng at Children's Hospital Medical Center in Cincinnati, OH, and 17-allylamino demethoxygeldanamycin (17-AAG) and G418 were from Sigma (St. Louis, MO). Antibodies to Rac1, STAT3, and pSTAT3 were obtained from Cell Signaling Technologies (Danvers, MA). Fugene-6 transfection reagent was from Roche Applied Sciences (Indianapolis, IN). Glutathione beads were acquired from Pierce (Rockford, IL).
Bacterial strains and plasmids.
E. coli K1 strain RS218 was the cerebrospinal fluid isolate from a neonate with meningitis. Mutants of strain RS218 with either cnf1, fimH, ibeA, or ompA deleted were constructed as described previously (9–13, 31, 32). The bacteria were cultured overnight at 37°C in the presence of antibiotics and then used for HBMEC adhesion and invasion, as well as for host cell signaling studies. N17Rac1 (the plasmid downregulating Rac1) was kindly provided by Alan Hall (University College of London, London, United Kingdom) (23), STAT3β plasmids (3) were obtained from Hua Yu (City of Hope, CA), the PAK1 plasmid encoding the PBD domain capable of binding activated Rac1 (1) was from Keith Burridge (University of North Carolina, Chapel Hill, NC), while downregulating plasmids of either β-Pix (26) and Vav2 (17) were from L. Romer (Johns Hopkins University, Baltimore, MD) and Y. Takai (University of Osaka, Osaka, Japan), respectively.
Treatment of HBMEC with inhibitors.
HBMEC were isolated and characterized from small fragments of cerebral cortex from individuals who have undergone surgical resections for treatment of seizure disorders, as previously described (30). For treatment with 17-AAG, HBMEC monolayer was treated with either 1 μM 17-AAG or the vehicle control (dimethyl sulfoxide) 1 day prior to the experiments. Initial pilot experiments showed that an 18-h treatment of HBMEC with 1 μM 17-AAG did not affect cell morphology (as observed by microscopy, as well as using Live/Dead cell stain [Molecular Probes]). After overnight treatment with 17-AAG, HBMEC were used for bacterial adhesion and invasion assays, as well as cell signaling studies. For the treatment of HBMEC with Rac1 inhibitor, NSC23766 was solubilized in water and used at a concentration of 100 μM.
Transfection of HBMEC with plasmids.
Transfection with plasmids was done using Fugene-6 (Roche) according to the manufacturer's instructions. Briefly, cells were grown to a 50% confluence in 100-mm dishes 1 day prior to transfection. A reagent to plasmid ratio of 3:1 was mixed in serum-free medium, and the complexes interacted with the cells. Two days later, the cells were analyzed by fluorescence microscopy for expression of green fluorescent protein (GFP) present in STAT3β plasmids. Transfection with N17Rac1 plasmid was analyzed by the inability to activate Rac1. β-Pix and Vav2 transfectants (cultured in G418 and neomycin, respectively) were analyzed by Western blot analysis using either Flag or c-myc antibodies, respectively. Transfected cells were used for bacterial adhesion and invasion assays, as well as for cell signaling analysis.
E. coli adhesion and invasion assays in HBMEC.
HBMEC were cultured as a monolayer either in 24-well tissue culture plates or 100-mm dishes at 37°C in 5% CO2 in RPMI with 20% fetal bovine serum, along with minimal essential vitamins, glutamine, sodium pyruvate, penicillin, and streptomycin. Bacterial invasion assays were performed in accordance with the well-established gentamicin protection assay (10, 11, 31, 32). Briefly, HBMEC were incubated with either wild-type RS218 or its mutants at a multiplicity of infection of 1:100 for 90 min, after which the cells were washed with RPMI 1640. The cells were further incubated with medium containing gentamicin (100 μg/ml) for 60 min to kill extracellular bacteria, after which the cells were lysed, and the internalized bacteria were enumerated on blood agar plates. The results were calculated as the percent invasion [i.e., (the number of intracellular bacteria recovered/the number of bacteria inoculated) × 100] and were expressed as the relative invasion (invasion as a percentage of E. coli K1 strain RS218 in HBMEC transfected with vector control or in the presence of vehicle control). For the HBMEC adhesion assays, the gentamicin step was omitted, and the total cell-associated bacteria was analyzed after the cells were lysed. The results were calculated as the percent adhesion [i.e., (the number of bacteria recovered/the number of bacteria inoculated) × 100] and were expressed as the relative adhesion (adhesion as a percentage of E. coli in HBMEC transfected with vector control or in the presence of vehicle control). Each set was run in triplicates.
Analysis of Rac1 activation.
HBMEC monolayers that were incubated with E. coli for various time points were washed quickly with ice-cold phosphate-buffered saline and then immediately lysed in assay buffer (50 mM Tris [pH 7.6], 150 mM NaCl, 1% Triton X-100, 0.5 mM MgCl2, protease inhibitor cocktail containing bestatin and pepstatin A [Sigma, MO], and 0.5 mM sodium vanadate) and briefly sonicated on ice. The lysates were centrifuged for 10 min at 4°C, and 400 μg of protein was incubated with 30 μg of glutathione-PBD (i.e., the Rac1-binding domain of P21-activated kinase) beads for 30 min to specifically pull down active Rac1 (GTP-Rac1). The PBD beads were quickly washed with the lysis buffer, boiled in SDS-PAGE sample buffer, and subjected to Western blot assay with anti-Rac1 antibody. The lysates were also examined for the total amount of Rac1, as described previously (25).
Analysis of interacting molecules by immunoprecipitation and Western blots.
Monolayers of HBMEC cultured in 100-mm dishes were incubated with bacteria for different time points and lysed in assay buffer as described above. Lysates were then analyzed for molecules of interest by immunoprecipitation, followed by Western blot assays with specific antibodies. Bands obtained were analyzed by ImageJ software (available at the National Institutes of Health [http://rsb.info.nih.gov/ij]) and graphically plotted in terms of fold change compared to controls.
Statistical analysis.
Differences in bacterial adhesion to and invasion of HBMEC were determined by using the Student t test. Standard deviations were calculated and are expressed as standard errors of means. A P value of <0.05 was considered significant.
RESULTS
Meningitis-causing E. coli K1 regulates host cell Rac1 activation for invasion of HBMEC.
Although earlier studies illustrate that host cell actin cytoskeletal rearrangements are required for meningitis-causing E. coli K1 entry into HBMEC (14, 15), the underlying mechanisms remain incompletely understood. Whereas RhoA, a member of the Rho family GTPases, has been shown to be involved in E. coli K1 invasion of HBMEC (10–13), the role of Rac1 in E. coli K1 entry of HBMEC is not known. To investigate the role of Rac1 in E. coli K1 invasion of HBMEC, the HBMEC monolayers were treated with various concentrations of the Rac1 inhibitor (NSC23766) and then examined for bacterial adhesion and invasion. While adhesion of E. coli to HBMEC was not affected by NSC23766, a dose-dependent inhibition of E. coli invasion was observed with a significant inhibition at 100 μM the inhibitor (Fig. 1A).
Fig 1.
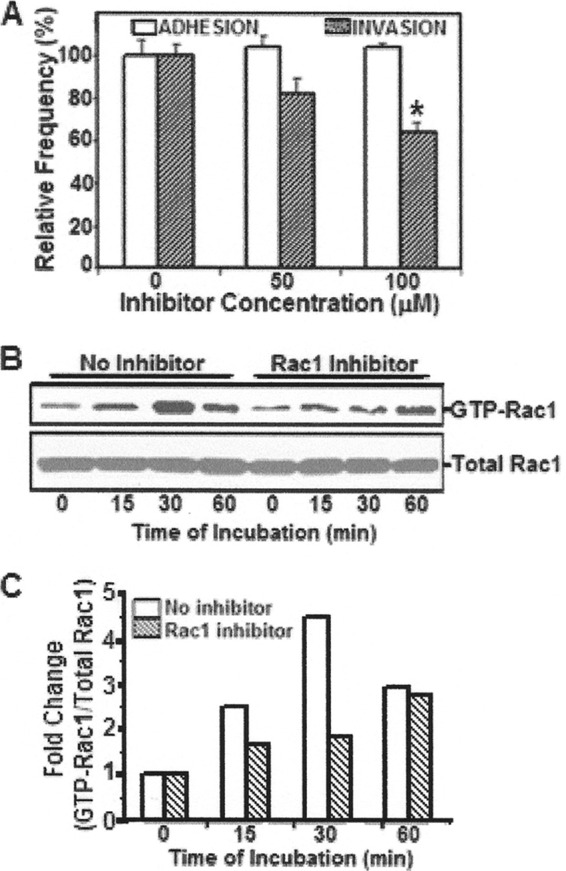
Examination of Rac1 activation in HBMEC incubated with E. coli K1 strain RS218. (A) To analyze the role of GTP-Rac1 in E. coli adhesion to and invasion of HBMEC, host cells were pretreated with either the Rac1 inhibitor (NSC23766) or vehicle control, incubated with the bacteria, and examined for bacterial adhesion and invasion. Values were graphically represented, with the results from control cells being taken as 100%. *, P < 0.05 compared to no inhibitor. (B) HBMEC treated with NSC23766 (100 μM) or vehicle control were incubated with E. coli for various time points and then lysed. Lysates were incubated with PBD-GST beads, boiled in SDS-PAGE buffer, and analyzed for activated Rac1 (GTP-Rac1) by Western blotting. Lysates were also examined for total Rac1. (C) Band intensities were estimated densitometrically by ImageJ software and expressed graphically as the ratio of GTP-Rac1 to total Rac1. The fold change was estimated with respect to time zero.
Since NSC23766 significantly inhibited E. coli invasion of HBMEC, the ability of the bacteria to activate Rac1 in HBMEC was examined. Briefly, HBMEC were treated with either NSC23766 or vehicle control, incubated with E. coli for various time points, and then lysed. Lysates were then mixed GST-PBD beads since PBD (PAK binding domain) specifically binds activated or GTP-Rac1 (see Materials and Methods). The beads were boiled and analyzed for Rac1 using Western blotting with Rac1 antibody. In addition, the lysates were examined for total Rac1 by Western blotting. As shown in Fig. 1B, a time-dependent activation of Rac1 occurred in response to E. coli K1 in control HBMEC, whereas HBMEC treated with NSC23766 (100 μM) exhibited minimal activation, corresponding to reduced bacterial invasion. Densitometric analysis, represented as a ratio of GTP-Rac1 to total Rac1, showed that the levels of GTP-Rac1 increase approximately 4- to 5-fold in control HBMEC compared to inhibitor-treated cells (Fig. 1C).
E. coli K1-dependent Rac1 activation requires the guanine exchange factor Vav2.
Since specific guanine nucleotide exchange factors (GEFs) involve Rac1 activation (GTP-Rac1) by promoting GDP to GTP exchange, we examined the two common GEFs, β-Pix and Vav2, for their involvement in Rac1 activation in response to E. coli in HBMEC. In brief, HBMEC were downregulated for either β-Pix or Vav2 by transfecting the host cells with either the wild-type plasmids or those expressing the nonfunctional molecule. Lysates were checked for expression of FLAG (tag present on β-Pix plasmids [Fig. 2A]) or c-myc (for Vav2[Fig. 2B]) by Western blot analysis. The transfected HBMEC were subsequently examined for E. coli adhesion and invasion, as well as Rac1 activation.
Fig 2.

Role of GEFs on Rac1 activation in response to E. coli K1 invasion. To identify the role of guanine nucleotide exchange factors (GEFs) in E. coli-dependent Rac1 activation, HBMEC were transfected with downregulating (DN) plasmids of the Rac1 GEFs β-Pix (A) and Vav2 (B) and analyzed for expression of either FLAG (β-Pix) or c-myc (Vav2) present in the constructs. Arrows denote the expression of either the wild-type or downregulating molecules. Next, HBMEC transfected with either β-Pix (C) or Vav2 (D) were examined for E. coli adhesion and invasion in comparison to control cells. Bacterial adhesion and invasion were graphically represented as the relative frequency (%), with values from control cells taken as 100%. *, P < 0.05 compared to wild-type transfected HBMEC. Cells transfected with either with β-Pix (E) or Vav2 (F) were grown in monolayers and incubated with E. coli for various time periods. Lysates were analyzed for either activated Rac1 (GTP-Rac1) using GST-PBD beads or total Rac1 by Western blot assays.
Although neither GEF affected E. coli adhesion to HBMEC, a significant decrease in bacterial invasion was observed in Vav2 downregulated cells compared to those transfected with wild-type Vav2 (Fig. 2D). Western blot analysis of GTP-Rac1 showed a corresponding decrease in GTP-Rac1 in Vav2-downregulated cells compared to the wild-type cells (Fig. 2F). In contrast, neither E. coli invasion nor Rac1 activation were affected by downregulation of β-Pix (Fig. 2C and E). These findings demonstrate that Rac1 activation in response to E. coli is dependent on Vav2.
STAT3 interaction with Rac1 in E. coli K1 invasion of HBMEC.
Earlier studies showed GTP-Rac1 to bind to phospho-STAT3 during the structural remodeling of actin cytoskeleton (27). Hence, the role of STAT3 and its association with GTP-Rac1 in E. coli K1 invasion of HBMEC was examined. Briefly, HBMEC were transfected and overexpressed for GFP-tagged STAT3β (a variant of STAT3 wherein a C-terminal truncation affects normal function) (21). The transfected cells (∼70% transfection frequency, as monitored for GFP by fluorescence microscopy) were examined for E. coli adhesion and invasion, as well as STAT3 phosphorylation and Rac1 activation.
Although no significant differences in E. coli adhesion were observed, E. coli invasion decreased significantly in HBMEC overexpressing STAT3β (Fig. 3A). Similar results were obtained when the HBMEC were transfected with STAT3 shRNA compared to control shRNA-transfected cells (data not shown).
Fig 3.
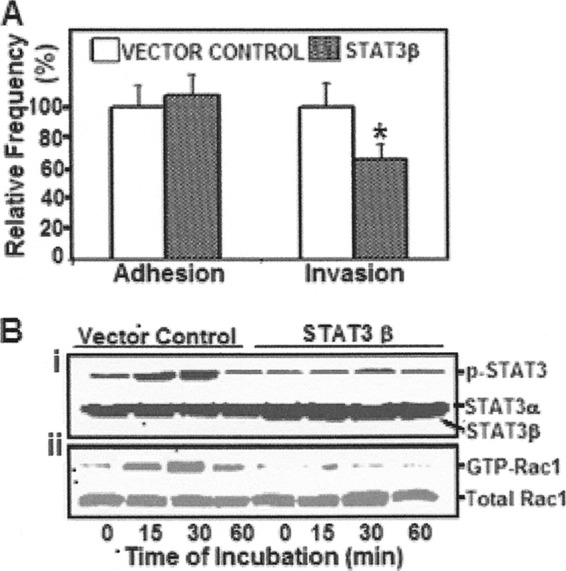
STAT3 regulation of GTP-Rac1 in E. coli invasion of HBMEC. (A) To examine the effect of STAT3 on E. coli adhesion and invasion of HBMEC, the host cells were transfected with STAT3β or the control vector and examined for E. coli adhesion and invasion. *, P < 0.05 compared to vector control. (B) To analyze the effect of STAT3 downregulation on the activation of Rac1, HBMEC transfected with either STAT3β or control vector were incubated with E. coli for various time periods. Cell lysates were then examined for either pSTAT3 (i) or Rac1 activation (ii) by Western blot analysis.
An examination of STAT3 phosphorylation by Western blotting showed a time-dependent increase in STAT3 activation in response to E. coli in HBMEC that were transfected with control vector. However, no such activation could be determined in cells expressing STATβ (Fig. 3Bi). Similarly, levels of GTP-Rac1 were increased when control HBMEC were incubated with E. coli for 15 or 30 min, but such increases in GTP-Rac1 were not observed in STAT3β-transfected cells (Fig. 3Bii). Total lysates contained similar amounts of either total Rac1 or total STAT3α, whereas strong expression of STAT3β was seen in HBMEC transfected with the dominant-negative plasmid (Fig. 3Bi). These findings suggest that the downregulation of STAT3 resulted in decreased E. coli invasion and decreased Rac1 activation in HBMEC.
17-AAG reduced E. coli K1 invasion of HBMEC and inhibited activation of STAT3 and Rac1.
17-AAG, an inhibitor of heat shock protein 90 (HSP90), has been shown to disrupt STAT3 activation and function (18). The inhibitor was also previously shown to inhibit E. coli invasion of HBMEC (20). To examine the effect of the inhibitor on STAT3 activation and association with Rac1-GTP in response to E coli K1, HBMEC with or without 17-AAG treatment were assessed for activation of STAT3 and Rac1 by Western blotting. As observed earlier (20), the inhibitor exhibited a significant decrease in E. coli invasion of HBMEC in comparison to vehicle control (Fig. 4A). While time-dependent increases in activation of STAT3 and GTP-Rac1 were observed in control cells (Fig. 4B), their activations were minimally affected in HBMEC that were pretreated with 17-AAG. Densitometric measurements revealed an ∼5-fold increase in STAT3 and Rac1 activation in control HBMEC compared to inhibitor-treated cells (Fig. 4C). Thus, the inhibition of STAT3 by 17-AAG, a molecule that probably affects HSP90 (a homolog of the OmpA receptor gp96, 11), resulted in decreased Rac1 activation and decreased E. coli invasion in HBMEC.
Fig 4.
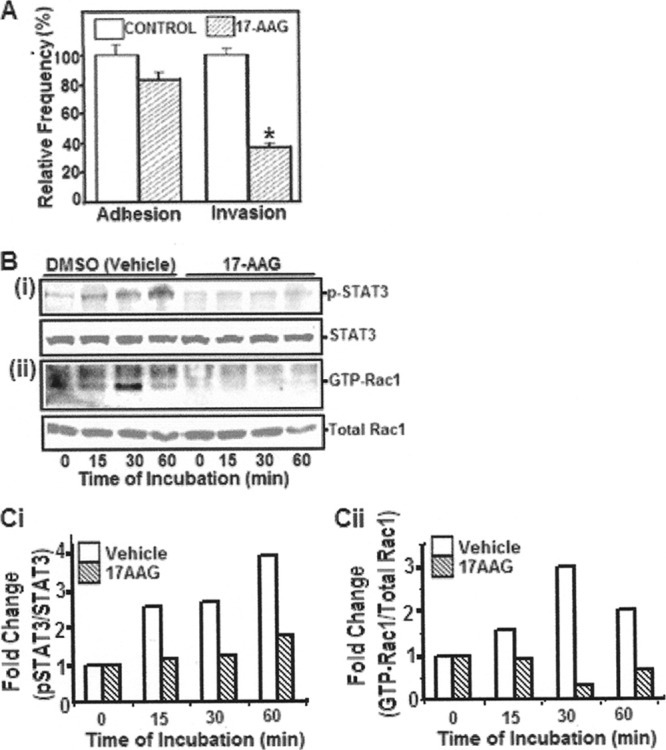
Effect of 17-AAG on STAT and Rac1 activation, as well as E. coli invasion of HBMEC. (A) To examine the effect of 17-AAG (a geldanomycin and HSP90/STAT3 inhibitor) on E. coli adhesion to and invasion of HBMEC, HBMEC were treated with the inhibitor or vehicle control and then examined for E. coli adhesion and invasion. *, P < 0.05 compared to vehicle control. (B and C) HBMEC treated with 17-AAG or vehicle control were incubated with E. coli for various time points, and the lysates were analyzed for either p-STAT3 (Bi) or Rac1 (Bii) activation by Western blotting. Band intensities were estimated densitometrically and are expressed as the fold increase in the pSTAT3/STAT3 (Ci) or GTP-Rac1/total Rac1 (Cii) ratio.
Taken together, these findings demonstrate that E. coli K1 triggers the activation of STAT3 and Rac1 to promote its invasion in HBMEC. Disruption of either STAT3 or Rac1 activation would adversely affect the ability of E. coli K1 to invade HBMEC.
Determination of specific virulence factors of E. coli K1 strain RS218 in activation of Rac1 and its association with STAT3.
Earlier studies showed that activated STAT3 bound to GTP-Rac1. We next incubated HBMEC with E. coli K1 for 30 min, and the lysates were examined for GTP-Rac1 by the PBD-GST pulldown assay. The PBD-GST beads were further examined for GTP-Rac1-bound STAT3. As observed earlier, incubation of HBMEC with E. coli K1 strain RS218 resulted in significant Rac1 activation and its association with STAT3 (Fig. 5A).
Fig 5.

Specific virulence factors of E. coli RS218 activate host cell Rac1. (A) To examine the association of STAT3 with Rac1 in response to RS218, HBMEC were incubated with the bacteria for 30 min, and lysates were examined for GTP-Rac1 using PBD-PAK1 beads. The beads were also examined for STAT3 in a Western blot assay. (B) To assess the contribution of the microbial factors to Rac1 activation, HBMEC were incubated with either wild-type RS218 or its mutants. Lysates were mixed with PBD-GST beads and examined for activated Rac1 (GTP-Rac1) and GTP-Rac1-associated STAT3. Total amounts of Rac1 in the lysates were also determined by Western blot analysis. The band intensities of Rac1, GTP-Rac1, and Rac1-bound STAT3 were measured by ImageJ software, and the results expressed as ratios of GTP-Rac1/total Rac1 or GTP-Rac1 bound STAT3/total Rac1 with the values of time zero as 1.
Our previous studies demonstrated the involvement of FimH and CNF1 in RhoA activation in HBMEC (10–13). We next examined the virulence factors that contribute to HBMEC adhesion and invasion for Rac1 activation. Briefly, HBMEC were incubated with either wild-type RS218 or its FimH, CNF1, IbeA, or OmpA mutants for up to 30 min. HBMEC lysates were then analyzed for GTP-Rac1.
Western blot analysis showed levels of GTP-Rac1 to be negligible in the lysates of HBMEC that were incubated with either IbeA or OmpA mutants compared to a time-dependent activation of Rac1 with wild-type bacteria (Fig. 5B). However, activation of Rac1 could be observed in HBMEC that were incubated with either FimH or CNF1 mutants.
Activated STAT3 has been shown to bind GTP-Rac1 and serves as a measure of the activated levels of the two molecules in HBMEC. Hence, the amount of STAT3 associated with GTP-Rac1 was further examined in the samples prepared above. HBMEC lysates that were incubated with wild-type RS218 showed higher amounts of GTP-Rac1 associated STAT3 (Fig. 5B). In line with Rac1 activation, the amounts of associated STAT3 were higher (1.3- to 1.8-fold increases) in the lysates of HBMEC incubated with either CNF1 or FimH mutants. However, the amounts of associated STAT3 were considerably less or negligible in HBMEC incubated with either IbeA or OmpA mutants. Taken together, these findings indicate that E. coli K1 exploits the STAT3-Rac1 pathway for successful entry into HBMEC by the aid of IbeA and OmpA.
DISCUSSION
The penetration of meningitis-causing pathogens into the brain is an essential step in the development of meningitis and has been the subject of intense study in recent years (15). Studies on E. coli K1 invasion of the blood-brain barrier, a prerequisite for penetration into the brain, have been rendered feasible due to the development of the in vitro blood-brain barrier model with HBMEC (14, 15, 30). Previous studies identified several microbial and host factors that contributed to E. coli binding to and invasion of HBMEC (14, 15), but the underlying host-microbial interactions remain incompletely understood.
We have shown that meningitis-causing E. coli K1 invasion of HBMEC requires the host actin cytoskeleton rearrangements. This is shown by the demonstration that cellular protrusions surround internalizing E. coli and internalized E. coli is found within membrane-bound vacuole (14, 15). The Rho family of small GTPases such as, RhoA, Rac1, and CDC42 have been shown to regulate host cytoskeleton organization (22–24), and various pathogens have been shown to exploit the Rho GTPases to aid their entry into the host cells (2, 6, 25). We have previously shown that RhoA activation occurs in response to meningitis-causing E. coli K1, and FimH and CNF1 contribute to RhoA-mediated E. coli invasion of HBMEC (11–13). Rac1 has also been shown to promote protrusions of the host cell membrane (7, 23, 24) and contribute to type III GBS invasion of HBMEC (25), but its role in E. coli K1 invasion of HBMEC is unclear. In the present study, the contribution of Rac1 to E. coli K1 invasion of HBMEC was demonstrated by our findings that Rac1 activation occurred in response to E. coli K1, as well as bacterial invasion, was significantly decreased by pharmacological inhibition and downmodulation of Rac1. Interestingly, however, Rac1 activation was observed in HBMEC that were incubated with either FimH or CNF1 mutants (though some decrease was observed with CNF1 mutant at 30 min, likely due to degradation of activated Rac1), but Rac1 activation was negligible with IbeA and OmpA mutants, suggesting the involvement of IbeA and OmpA in activating Rac1 in HBMEC. Taken together, FimH and CNF1 are likely to contribute to HBMEC invasion via RhoA activation, whereas IbeA and OmpA contribute to HBMEC invasion via Rac1 activation.
The role of STAT3 as a transcriptional factor on activation and homodimerization is well understood, but very little is known about its cytoplasmic functions. STAT3 has been shown to heterodimerize with other molecules. Simon et al. (27) showed that activated STAT3 binds GTP-Rac1. Our demonstrations of STAT3 association with GTP-Rac1 during E. coli K1 invasion of HBMEC, as well as reduced STAT3 interaction with GTP-Rac1 on incubation with IbeA and OmpA mutants defective in HBMEC invasion, clearly defines a specific role for STAT3-Rac1 interaction in E. coli K1 invasion of HBMEC.
The regulation of Rac1 by STAT3 was shown in mouse embryonic fibroblasts to involve the GEF β-Pix (33). Our present findings, however, did not implicate the role of β-Pix in either E. coli K1 invasion of HBMEC or E. coli K1-induced activation of Rac1. Rather, the activation of Rac1 in response to E. coli K1 was observed to involve the GEF Vav2. This is the first demonstration of implicating Vav2, not β-Pix, in E. coli K1 invasion of HBMEC. The regulation of Rac1 activation by STAT3 via Vav2 in E. coli K1 invasion of HBMEC, however, remains unclear and requires further investigation.
Although a number of bacterial virulence factors have been shown to contribute to E. coli adhesion to and invasion of HBMEC, it was interesting that only mutants deleted of either IbeA or OmpA were defective in Rac1 activation, as well as its association with STAT3. The data with 17-AAG (an inhibitor of HSP90 of which the OmpA receptor gp96 is a homolog [11]) led us to speculate that the OmpA-gp96 interaction is involved in activating Rac1 and STAT3. We hypothesize that both IbeA and OmpA act in tandem to activate the host cell molecules for efficient E. coli K1 invasion of HBMEC. We have previously shown that FimH and CNF1 are involved in RhoA activation (10–13), but our present studies showed the role of IbeA and OmpA in Rac1 activation of HBMEC. Our previous studies using single- and double-deletion mutants revealed that FimH and OmpA, as well as CNF1 and OmpA, double-deletion mutants exhibited significantly greater reduction in HBMEC invasion compared to single-deletion mutants (11, 32). On the other hand, HBMEC invasion by IbeA and OmpA double-deletion mutants was similar to that of single-deletion mutants (13). Based on these findings, we speculate that the contributions of IbeA and OmpA to activation of STA3-Rac1 are likely to be redundant and that deletion of both factors does not lead to additional decrease in HBMEC invasion. In contrast, FimH and CNF1 differ in host signaling mechanisms compared to those of OmpA, and deletions of either FimH or CNF1 and OmpA exhibited additional decreases in HBMEC invasion compared to single deletion mutants. These findings, therefore, lend further support the evidence demonstrated presented here that Rac1 activation contributes to E. coli K1 invasion of HBMEC and that its contribution is independent from that of RhoA. Our hypothesis on the involvement of IbeA and OmpA in activation of STAT3 and Rac1 of HBMEC, in addition to the activation of RhoA by FimH and CNF1, to aid in E. coli internalization is depicted in Fig. 6. Additional studies are needed to elucidate how the different bacterial factors regulate RhoA and Rac1 to aid in E. coli K1 invasion of HBMEC.
Fig 6.
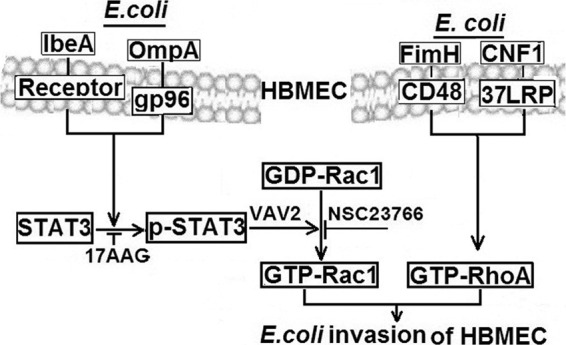
Diagrammatic representation of STAT3 regulation of Rac1 in E. coli K1 invasion of HBMEC. Whereas earlier studies showed that FimH and CNF1 activate RhoA for E. coli internalization of HBMEC (12, 13), we hypothesize, based on the information obtained in the present study, that OmpA and IbeA trigger the phosphorylation of STAT3, which aids in converting GDP-Rac1 to GTP-Rac1 (through an as-yet-unknown mechanism involving the GEF, Vav2). Inhibitors or downregulators of either STAT3 or Rac1 decrease E. coli invasion of HBMEC. This study shows for the first time a molecular mechanism of E. coli regulation of Rac1 activation via STAT3 for E. coli internalization into HBMEC.
In summary, we demonstrated here for the first time how meningitis-causing E. coli K1 regulates STAT3 cross talk with Rac1 for efficient invasion of HBMEC. In addition, our findings reveal the novel contribution of the bacterial determinants of IbeA and OmpA to activation of host cell molecules Rac1 and STAT3 in E. coli K1 invasion of HBMEC.
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Health grants NS26310 and AI84984.
We are grateful to Alan Hall (University College of London), Hua Yu (City of Hope, CA), Keith Burridge (North Carolina), L. Romer (Johns Hopkins University, Baltimore, MD), and Y. Takai (University of Osaka, Osaka, Japan) for providing plasmids.
The authors declare that they have no conflict of interest.
Footnotes
Published ahead of print 26 March 2012
REFERENCES
- 1. Bagrodia S, Derigard B, Davis RJ, Cerione RARA. 1995. Cdc42 and PAK mediated signaling leads to Jun kinase and p38 mitogen activated protein kinase activation. J. Biol. Chem. 270:27995–27998 [DOI] [PubMed] [Google Scholar]
- 2. Brandt S, et al. 2007. Use of a novel coinfection system reveals a role for Rac1, H-Ras, and CrkII phosphorylation in Helicobacter pylori-induced host cell actin cytoskeletal rearrangements. FEMS Immunol. Med. Microbiol. 50:190–205 [DOI] [PubMed] [Google Scholar]
- 3. Catlett-Falcone R, et al. 1999. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 10:105–115 [DOI] [PubMed] [Google Scholar]
- 4. Chang CJ, et al. 2004. Bacterial meningitis in infants: the epidemiology, clinical features, and prognostic factors. Brain Dev. 26:168–175 [DOI] [PubMed] [Google Scholar]
- 5. Darnell JE., Jr 1997. STATs and gene regulation. Science 277:1630–1635 [DOI] [PubMed] [Google Scholar]
- 6. Friebel A, et al. 2001. SopE and SopE2 from Salmonella typhimurium activate different sets of RhoGTPases of the host. Cell 36:34035–34040 [DOI] [PubMed] [Google Scholar]
- 7. Grabham PW, Reznik B, Goldberg DJ. 2003. Microtubule and Rac 1-dependent F-actin in growth cones. J. Cell Sci. 116:3739–3748 [DOI] [PubMed] [Google Scholar]
- 8. Holt DE, Halket S, DE Louvois J, Harvey D. 2001. Neonatal meningitis in England and Wales: 10 years on. Arch. Dis. Child. Fetal. Neonatal. Ed. 84:F85–F89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang SH, et al. 1995. Escherichia coli invasion of brain microvascular endothelial cells in vitro and in vivo: molecular cloning and characterization of invasion gene ibe10. Infect. Immun. 63:4470–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Khan NA, et al. 2002. Cytotoxic necrotizing factor-1 contributes to Escherichia coli K1 invasion of the central nervous system. J. Biol. Chem. 277:15607–15612 [DOI] [PubMed] [Google Scholar]
- 11. Khan NA, et al. 2003. Outer membrane protein A and cytotoxic necrotizing factor-1 use diverse signaling mechanisms for Escherichia coli K1 invasion of human brain microvascular endothelial cells. Microb. Pathog. 35:35–42 [DOI] [PubMed] [Google Scholar]
- 12. Khan NA, Kim Y, Shin S, Kim KS. 2007. FimH-mediated Escherichia coli K1 invasion of human brain microvascular endothelial cells. Cell Microbiol. 9:169–178 [DOI] [PubMed] [Google Scholar]
- 13. Kim KS. 2002. Strategy of Escherichia coli for crossing the blood-brain barrier. J. Infect. Dis. 186:S220–224 [DOI] [PubMed] [Google Scholar]
- 14. Kim KS. 2003. Neurological diseases: pathogenesis of bacterial meningitis: from bacteremia to neuronal injury. Nat. Rev. Neurosci. 4:376–385 [DOI] [PubMed] [Google Scholar]
- 15. Kim KS. 2008. Mechanisms of microbial traversal of the blood-brain barrier. Rev. Nat. Rev. Microbiol. 6:625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klinger G, Chin CN, Beyene J, Perlman M. 2000. Predicting the outcome of neonatal bacterial meningitis. Pediatrics 106:477–482 [DOI] [PubMed] [Google Scholar]
- 17. Kodama A, et al. 2000. The involvement of an SHP-2-Rho small G protein pathway in hepatocyte growth factor/scatter factor-induced cell scattering. Mol. Biol. Cell 11:2565–2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lang SA, et al. 2007. Targeting heat shock protein 90 in pancreatic cancer impairs insulin-like growth factor-I receptor signaling, disrupts an interleukin-6/signal-transducer and activator of transcription 3/hypoxia-inducible factor-1α autocrine loop, and reduces orthotopic tumor growth. Clin. Cancer Res. 13:6459–6468 [DOI] [PubMed] [Google Scholar]
- 19. Maruvada R, Argon Y, Prasadarao NV. 2008. Escherichia coli interaction with human brain microvascular endothelial cells induces signal transducer and activator of transcription 3 association with the C-terminal domain of Ec-gp96, the outer membrane protein A receptor for invasion. Cell. Microbiol. 10:2326–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maruvada R, Kim KS. 2011. Extracellular loops of the Escherichia coli outer membrane protein A contribute to the pathogenesis of meningitis. J. Infect. Dis. 203:131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Niu G, et al. 2001. Overexpression of a dominant-negative signal transducer and activator of transcription 3 variant in tumor cells leads to production of soluble factors that induce apoptosis and cell cycle. Cancer Res. 61:3276–3280 [PubMed] [Google Scholar]
- 22. Nobes CD, Hall A. 1995. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81:53–62 [DOI] [PubMed] [Google Scholar]
- 23. Ridley AJ, Paterson HF, Johnston CL, Dickmann D, Hall A. 1992. The small GTP-binding protein Rac regulates growth factor-induced membrane ruffling. Cell 70:401–410 [DOI] [PubMed] [Google Scholar]
- 24. Ridley AJ. 2006. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell. Biol;. 16:522–529 [DOI] [PubMed] [Google Scholar]
- 25. Shin S, Kim KS. 2006. RhoA and Rac1 contribute to type III group B streptococcal invasion of human brain microvascular endothelial cells. Biochem. Biophys. Res. Commun. 345:538–542 [DOI] [PubMed] [Google Scholar]
- 26. Shin EY, et al. 2004. Basic fibroblast growth factor stimulates activation of Rac1 through a p85 βPIX phosphorylation-dependent pathway. J. Biol. Chem. 279:1994–2004 [DOI] [PubMed] [Google Scholar]
- 27. Simon AR, et al. 2000. Regulation of STAT3 by Direct Binding to the Rac1 GTPase. Science 290:144–147 [DOI] [PubMed] [Google Scholar]
- 28. Smith PB, et al. 2006. A comparison of neonatal Gram-negative rod and Gram-positive cocci meningitis. J. Perinatol. 26:111–114 [DOI] [PubMed] [Google Scholar]
- 29. Stevens JP, et al. 2003. Long-term outcome of neonatal meningitis. Arch. Dis. Child. Fetal. Neonatal. ed. 88:F179–F184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stins MF, Gilles F, Kim KS. 1997. Selective expression of adhesion molecules on human brain microvascular endothelial cells. J. Neuroimmunol. 76:81–90 [DOI] [PubMed] [Google Scholar]
- 31. Teng CH, et al. 2005. Escherichia coli K1 RS218 interacts with human brain microvascular endothelial cells via type 1 fimbriae. Infect. Immun. 73:2923–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Teng CH, et al. 2006. Effects of OmpA deletion on expression of type 1 fimbriae in Escherichia coli K1 strain RS218 and on association of E. coli with human brain microvascular endothelial cells. Infect. Immun. 74:5609–5616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Teng TS, Lin B, Manser E, Ng DCH, Cao X. 2009. Stat3 promotes directional cell migration by regulating Rac1 activity via its activator βPIX. J. Cell Sci. 122:4150–4159 [DOI] [PubMed] [Google Scholar]
- 34. Zhong Z, Wen Z, Darnel JE., Jr 1994. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264:95–98 [DOI] [PubMed] [Google Scholar]