

Abstract
Listeriolysin O (LLO) is a pore-forming toxin of the cholesterol-dependent cytolysin (CDC) family and a primary virulence factor of the intracellular pathogen Listeria monocytogenes. LLO mediates rupture of phagosomal membranes, thereby releasing bacteria into the growth-permissive host cell cytosol. Several unique features of LLO allow its activity to be precisely regulated in order to facilitate phagosomal escape, intracellular growth, and cell-to-cell spread. To improve our understanding of the multifaceted contribution of LLO to the pathogenesis of L. monocytogenes, we developed a screen that combined saturation mutagenesis and signature tags, termed in vivo analysis by saturation mutagenesis and signature tags (IVASS). We generated a library of LLO mutant strains, each harboring a single amino acid substitution and a signature tag, by using the previously described pPL2 integration vector. The signature tags acted as molecular barcodes, enabling high-throughput, parallel analysis of 40 mutants in a single animal and identification of attenuated mutants by negative selection. Using the IVASS technique we were able to screen over 90% of the 505 amino acids present in LLO and identified 60 attenuated mutants. Of these, 39 LLO residues were previously uncharacterized and potentially revealed novel functions of the toxin during infection. The mutants that were subsequently analyzed in vivo each conferred a 2- to 4-orders of magnitude loss in virulence compared to wild type, thereby validating the screening methods. Phenotypic analysis of the LLO mutant library using common in vitro techniques suggested that the functional contributions of some residues could only have been revealed through in vivo analysis.
INTRODUCTION
Among the biggest challenges facing investigators of microbial pathogenesis is evaluation of the roles of pathogenic determinants in vivo. A number of strategies have been developed to identify determinants that are essential for in vivo growth (6, 24, 40, 54, 82). Once a potential determinant of pathogenesis has been identified, its biological activity can be analyzed in vitro. But ultimately, a complete understanding of the role of a protein, domain, or single amino acid residue must include in vivo analysis. For this report, we developed a rapid and unbiased strategy, termed IVASS (in vivo analysis by saturation mutagenesis and signature tags), for individually mutating every single residue of listeriolysin O (LLO), an essential virulence factor of Listeria monocytogenes. We used this library to evaluate the effects of every mutant on the virulence of L. monocytogenes in vivo using a murine model.
L. monocytogenes is a Gram-positive, facultative, food-borne pathogen capable of intracellular growth in a wide variety of cell types (83). L. monocytogenes is the causative agent of listeriosis, a severe systemic illness that affects immunocompromised individuals and pregnant women (66). The intracellular life cycle of L. monocytogenes is well characterized and begins with cell entry by phagocytosis of bacteria and compartmentalization into a single-membrane phagosome. Bacteria rapidly escape from this primary vacuole and are released into the cytosol. The growth-permissive cytosol allows bacteria to proliferate and to induce genetic programs necessary for spreading into neighboring cells by entering within a double-membrane vacuole (12, 79). Escape from this secondary vacuole releases L. monocytogenes into the cytosol of the new host cell, and the life cycle begins anew. The ability of L. monocytogenes to replicate in the host cytosol and spread from cell to cell protects the bacterium from the extracellular defenses of the host (19, 58).
Escape from a phagosome is primarily mediated by the cytolysin LLO, making this toxin a crucial determinant for intracellular growth as well as cell-to-cell spread (16, 27, 41, 75). LLO is a member of the cholesterol-dependent cytolysin (CDC) family, comprising toxins from 28 species of pathogenic Gram-positive bacteria (25), including perfringolysin O (PFO), streptolysin O (SLO), anthrolysin O (ALO), and intermedilysin (ILY). The CDCs act as potent virulence factors by assembling into large, homo-oligomeric pores upon encountering a mammalian cell (reviewed in references 18, 25, and 80). These toxins share a high degree of sequence identity (40 to 70%) and adopt the same four-domain secondary structure (4, 56, 65). The structural similarity of the CDCs suggests that these toxins also exhibit similar pore-forming activities. Detailed structure-function analyses have suggested a general three-step model for CDC pore formation whereby secreted monomers bind to cholesterol-containing membranes, oligomerize, and then undergo profound structural changes in order to insert into membranes and form pores (18, 25, 81).
The pore-forming activity of LLO is essential for the virulence of L. monocytogenes in the murine model of listeriosis. Strains deficient in LLO production or activity remain trapped within a phagosome and are attenuated for virulence by 5 orders of magnitude (9, 29, 59). However, LLO activity must be restricted to the acidic phagosomal compartment in order to prevent premature lysis of the host cell and disruption of the protective intracellular niche. Mutants incapable of restricting LLO activity to the vacuole produce host cell toxicity and are attenuated for virulence (13, 19, 20, 34, 35). Thus, proper temporal and spatial regulation of LLO is crucial both for facilitating phagosomal escape and for preventing disruption of the protective cytosolic niche.
L. monocytogenes is the only intracellular pathogen that contains a CDC and, consequently, LLO possesses several novel features that are essential for intracellular growth. In addition to the canonical CDC four-domain secondary structure, LLO possesses an N-terminal amino acid recognized by the eukaryotic N-end rule degradation pathway, a 26-amino-acid PEST-like sequence (P, Pro; E, Glu; S, Ser; T, Thr) that regulates the toxin's translation, and an “acidic triad” that produces the toxin's optimal pH, all of which may be essential for preventing toxicity to the cell (13, 70, 71). The toxin also encodes a consensus nuclear localization signal (NLS), but whether or not the toxin translocates into the host nucleus has not been determined.
It is important that LLO elicits a myriad of host responses. It can trigger host signaling in a pore-independent manner (17), induce apoptosis (7, 22), stimulate mitogen-activated protein kinases (78), modulate histone modifications (23), and enhance bacterial uptake (14, 84). Furthermore, LLO paradoxically acts both as a potent T-cell-reactive antigen and as a suppressor of antigen presentation (2, 8). How LLO elicits such diverse cellular responses remains poorly understood.
While detailed in vitro analyses have provided valuable information about the mechanisms of CDC pore formation, these methods are inadequate for evaluation of the sophisticated ways in which these toxins interact with the host. To improve our understanding of the multifaceted contribution of LLO to the pathogenesis of L. monocytogenes, we developed a novel in vivo screen, termed IVASS. This screen combines saturation mutagenesis and a signature-tag system (24, 42). Saturation mutagenesis has been shown to be a powerful and extremely accurate tool for the unbiased determination of the functional residues of proteins (11, 38, 43, 47, 48). While the high level of resolution afforded by saturation mutagenesis is quite powerful for analysis of protein function, the technical challenges of analyzing many mutants in parallel have largely restricted these studies to in vitro analyses. The signature-tag method was developed as an in vivo alternative to the slow and arduous analysis of individual mutants (24). Various 40-bp tags serve as molecular barcodes to enable high-throughput screening of many mutants in a single animal. Avirulent mutants are identified by negative selection. Variations of this approach have been applied in both Gram-positive and Gram-negative bacteria (10, 21, 28, 32, 42, 45, 74). Combining these two techniques enabled us to analyze LLO function in vivo at the resolution of a single amino acid. Importantly, this approach allowed us to identify features of LLO that contribute to unexpected functions of this toxin during a mammalian infection.
By using the IVASS system, we were able to screen over 90% of the amino acids present in LLO and identify 60 attenuated LLO mutants. Mutants from all four domains of the protein were isolated. They included 39 previously unidentified functional residues. We hypothesized that this methodology would reveal novel residues whose function could be revealed only by in vivo analysis. Indeed, in vitro screening of the LLO mutant library for hemolytic activity on sheep blood agar identified mutants with zones of hemolysis indistinguishable from that of wild-type LLO. Yet, when these LLO mutants were tested in vivo, they were found to be attenuated. These findings underscore the power of in vivo analyses for thoroughly evaluating the roles of pathogenic determinants. To our knowledge, this is the first study that has combined alanine scanning mutagenesis with signature tagging for detailed, high-throughput analysis of a particular virulence factor.
MATERIALS AND METHODS
Bacterial strains and generation of signature-tagged pPL2 plasmids.
All L. monocytogenes strains used in this study were derived from strain 10403S (3). Escherichia coli strains XL1-Blue and SM10 were used for cloning. Forty unique 40-bp signature tags (ST) (see Table S3 in the supplemental material) were inserted into the L. monocytogenes site-specific integration vector pPL2 (33, 37, 44) by using QuikChange site-directed mutagenesis (Stratagene) according to the manufacturer's protocol. Once produced, the hly gene encoding LLO was cloned into the multiple-cloning site of each ST-pPL2 plasmid to produce plasmids ST-pJM1.1 to ST-pJM1.40. Signature-tagged plasmids were transformed into E. coli XL1-Blue cells by using standard techniques.
Generation of the signature-tag LLO library.
Each of the 40 ST-pJM1 plasmids were isolated from E. coli XL1-Blue by using the QIAprep Spin miniprep kit (Qiagen) and used iteratively as templates in mutagenic PCRs. The individual alanine substitutions were introduced onto the ST-pJM1 template by using QuikChange site-directed mutagenesis (Stratagene) (see Table S4 in the supplemental material). Mutations were confirmed by sequencing. ST-pJM1 plasmids with the desired mutations were isolated and transformed into the E. coli SM10 donor strain (Fig. 1). To generate L. monocytogenes strains carrying the ST-pJM1 mutant plasmids, the SM10 donor strains were mixed with the L. monocytogenes recipient strain DP-2161 (Δhly) by streaking equal amounts together on a brain heart infusion (BHI) agar plate. After incubation at 37°C for 2 h, bacteria were removed from the plate and dilutions were plated onto BHI agar supplemented with chloramphenicol (7.5 μg/ml) and streptomycin (200 μg/ml) and incubated at 37°C for 2 days. Individual colonies were picked and screened for plasmid integration within the chromosome as previously described (33).
Fig 1.

Generation of the signature-tag LLO library. The library of signature-tagged LLO saturation mutagenesis plasmids (ST-pJM1) was constructed using the backbone of the site-specific integration vector pPL2 (33). Each plasmid in the library harbors a unique combination of an ST (49) and hly mutant gene harboring a single amino acid substitution (LLOala). The signature-tagged LLO mutant constructs were individually transformed into E. coli SM10 cells and then introduced into a Δhly L. monocytogenes strain by conjugative transfer. The resulting strains each harbor an ST-LLO mutant integrated at the tRNAArg chromosomal locus. These strains were organized into pools of 40 individually tagged strains.
Animal infections.
All animals were housed and handled in accordance with federal and institutional guidelines. The animal use committees at the University of California, Berkeley, approved the animal protocol describing our studies. For the screen, mixtures of ST L. monocytogenes (ST-Lm) strains were prepared for intravenous inoculation as follows: overnight cultures of 40 ST-Lm strains each carrying a different signature tag (36 ST-Lmmutant strains, 2 ST-LmΔhly strains, and 2 ST-Lmwild-type strains) were mixed in equal amounts, diluted, and grown in BHI broth at 37°C until mid-log phase was reached. Aliquots were stored at −80°C. On the day of infection, one aliquot was thawed, diluted in BHI broth, and grown to mid-log phase at 37°C with constant shaking (250 rpm). Three 6- to 8-week-old female CD1 mice (Charles River Laboratories) were injected intravenously with a total of 105 bacteria in phosphate-buffered saline for each injection round containing a pool of 40 mutants and controls (Fig. 2). A portion of the inoculum was retained, diluted in BHI broth, and grown overnight at 37°C for the generation of the input bacterial pool. At 48 h postinfection, the animals were killed by carbon dioxide asphyxiation followed by cervical dislocation, and the spleens and livers were removed and homogenized in 0.2% NP-40. Dilutions were plated onto Luria-Bertani agar for quantitative CFU/ml determination. The remainder of the homogenate for each organ was added to BHI broth and grown overnight at 37°C with constant shaking. The following day, the cultures were diluted 1:100 in BHI broth and grown again overnight at 37°C to generate the output bacterial pools. The second round of dilution was carried out to ensure removal of contaminating organ DNA.
Fig 2.

Selection of signature-tagged mutations. Input pools of 40 signature-tagged strains were used to inoculate three mice. Forty-eight hours postinfection, bacteria were recovered from the livers and spleens, and aliquots were split to enumerate the bacterial load and to generate output pools of genomic DNA. The tags present in the DNA pools were identified by short-cycle standard PCR and compared to the pool of input DNA prepared from the original inocula. Each PCR mixture contained 1 of 40 ST-specific primers and a primer common to all the mutant strains. Mutants whose tags were absent or present at low levels were considered attenuated.
For individual infections, five 6- to 8-week-old female CD1 mice (Charles River Laboratories) were injected intravenously with one 50% lethal dose (1 × 105 CFU) of ST-Lmwild-type, ST-LmΔhly, or one of the ST-Lmmutant strains, and the spleens and livers were harvested 48 h later as previously described (60).
DNA purification and PCR.
Chromosomal DNA was purified from bacterial cultures from the inoculum (input pool) and organs (output pools) by using the Epicentre Biotechnologies Gram-positive DNA purification kit, replacing lysozyme with mutanolysin (5 U/μl; Sigma). Bacterial genomic DNA from the input and output pools was used to compare the presence of the signature tags pre- and postinfection in a 23-cycle end point PCR. PCR mixtures (20-μl volumes) contained 10 μM ST-specific primer (see Table S5 in the supplemental material), 10 μM reverse primer (pPL2-359R [see Table S5 in the supplemental material]), 40 ng isolated chromosomal DNA, Pfx50 polymerase (Invitrogen), 1× Pfx50 buffer, 1 mM deoxynucleoside triphosphates, and nuclease-free water. All cycling conditions were 94°C for 2 min, 23 cycles of 94°C for 1 min, 60°C for 2 min, and 72°C for 3 min. The entire reaction was run on a 1% agarose gel stained with ethidium bromide for visualization of PCR products.
Quantitative PCR.
All quantitative PCRs (qPCRs) were performed in 20-μl volumes in a Stratagene Mx3000P qPCR machine and analyzed using MxPro software (Stratagene). Reaction mixtures contained 10 μl Platinum SYBR green 2× reaction mix (Invitrogen), 10 μM ST-specific primer (see Table S5 in the supplemental material), 10 μM reverse primer (pPL2-359R [see Table S5]), 40 ng chromosomal DNA, and nuclease-free water. Bacterial genomic DNA from the input pool was used to generate standard curves for each ST in each experiment. Standard curve reaction mixtures were identical to the sample reaction mixtures except for the amount of chromosomal DNA (200 ng, 20 ng, 2 ng, or 0.2 ng per reaction mixture). All cycling conditions were 50°C for 2 min, 95°C for 2 min, and 40 cycles of 95°C for 15 s and 55°C for 1 min, followed by 95°C for 1 min, 55°C for 30 s, and 95°C for 30 s.
Western blotting.
To detect secreted LLO, proteins from the supernatants of mid-log-phase cultures were precipitated with 10% trichloroacetic acid (TCA). Samples were fractionated by SDS-PAGE and transferred to a polyvinylidene difluoride membrane for immunoblotting with polyclonal antibodies raised against either LLO or phosphatidylinositol-specific phospholipase C (PI-PLC; a kind gift from H. Goldfine), followed by infrared fluorescence detection with the Odyssey imaging system (LI-COR Biosciences).
Blood plate analysis for hemolytic phenotypes.
Bacteria were grown in BHI overnight at 37°C. The mutant strains were struck on Luria-Bertani (LB)-G1P agar (LB medium containing 25 mM glucose-1-phosphate, 5% activated charcoal, and 50 mM MOPS [morpholinepropanesulfonic acid], pH 7.4) supplemented with 5% defibrinated sheep blood (HemoStat Laboratories, Dixon, CA). Plates were incubated at 37°C for 2 days and then for 3 to 9 days at 4°C before being scanned by eye.
L2 plaque assay.
The plaque assay was performed as previously described by Sun et al. (77). L2 mouse fibroblasts were propagated in Dulbecco modified Eagle medium with high glucose (Gibco/Invitrogen) supplemented with 10% fetal bovine serum (GemCell), 1% glutamine, and 1% sodium pyruvate at 37°C with 5% CO2. Plaques formed in monolayers of L2 cells were visualized by staining the cells with neutral red (N6264; Sigma-Aldrich) 3 days postinfection. Plaque size was determined for at least 20 plaques from three independent experiments by using ImageJ software (http://rsbweb.nih.gov/ij/). The mean plaque size was compared to the mean plaque size of the JM-1 control wild-type strain.
RESULTS
Generation of signature-tagged LLO alanine mutant library.
The library of individual signature-tagged LLO alanine mutant strains was generated in a multistep process. First, the 40 STs (see Table S3 in the supplemental material) were individually inserted into the site-specific integration vector pPL2 previously described by Lauer et al. (33). Next, the hly gene encoding LLO was cloned into the multiple-cloning site of each ST-pPL2 plasmid to produce plasmids ST-pJM1.1 to ST-pJM1.40. Finally, these vectors were used as templates for the systematic substitution of each amino acid of LLO by site-directed mutagenesis. The resulting plasmids, each harboring a single amino acid substitution within the hly gene encoding LLO and a unique signature tag, were individually transformed into E. coli SM10 and integrated into the chromosome of the L. monocytogenes recipient strain DP-2161 (Δhly) by conjugative transfer as previously described (33) (Fig. 1). This method was used to generate 481 (95%) of the possible 505 mutant L. monocytogenes strains. The signal sequence comprises the first 24 amino acids of LLO and was excluded from these analyses since this portion of the protein is cleaved prior to secretion.
Evaluation of the signature tag system in vivo.
To evaluate the efficacy of the signature tag system and to ensure that the signature tags alone had no intrinsic effect on virulence, mixed pools of ST-Lmwild-type and ST-LmΔhly bacteria were introduced into mice. Two pools were generated, each containing 20 ST-Lmwild-type and 20 ST-LmΔhly tags. The pools differed only in the 20 STs used to tag the wild-type and Δhly strains. Each pool was used to inoculate three mice in two independent experiments. After 48 h, the bacteria were recovered from the spleens and livers. The Δhly strain was attenuated by nearly 4 orders of magnitude compared to wild type (15, 29, 57, 59). Quantitative PCR analysis of DNA prepared from recovered bacteria confirmed that each signature tag in the collection was reproducibly detectable from the pool of wild-type strains and that the ability to detect these tags was significantly reduced in the pool of Δhly strains (data not shown). The dynamic range afforded by these methods was greater than 3 orders of magnitude. These data validated the methodology and suggested that any false positives that arose from use of the library were not due to inherent defects in the tagging system.
To rule out the possibility that this system introduced unintended physiological side effects, the growth rates of each LLO mutant in the library were compared to the parental wild-type and Δhly strains. Growth curves were prepared for individual strains grown in rich medium (BHI) (data not shown). None of the mutants tested exhibited a significant change in growth rate. This result is consistent with previous evaluations of the pPL2 integration vector (33) and with those characterizing other LLO point mutations, where no change in growth rate has been reported (20, 70).
IVASS identification of LLO residues that exhibited attenuated virulence.
To screen for LLO mutations that conferred attenuated virulence, 13 input pools, each composed of 40 different signature-tagged strains, including 36 LLO mutants and two different signature-tagged wild-type and Δhly control strains (105 organisms in total) were injected intravenously into three mice each (Fig. 2). Forty-eight hours postinfection, the bacteria from the livers and spleens were recovered and plated to enumerate the bacterial loads. The remaining bacteria from each organ homogenate were cultured to generate output pools from which to extract genomic DNA. The tags present in the DNA pools were identified by short-cycle standard PCR and compared to the pool of input DNA prepared from the original inocula.
In total, 60 mutants were detected in the inocula but not in the bacteria recovered from animals and were therefore candidates for being attenuated. The positions of these residues within LLO are depicted in Fig. 3A. Of these, 21 mutations corresponded to previously characterized functional residues of the toxin (thus demonstrating the validity of the screen), and the remainder of the mutations mapped to previously uncharacterized portions of LLO. The screen identified mutants in each of the four structural domains of LLO.
Fig 3.
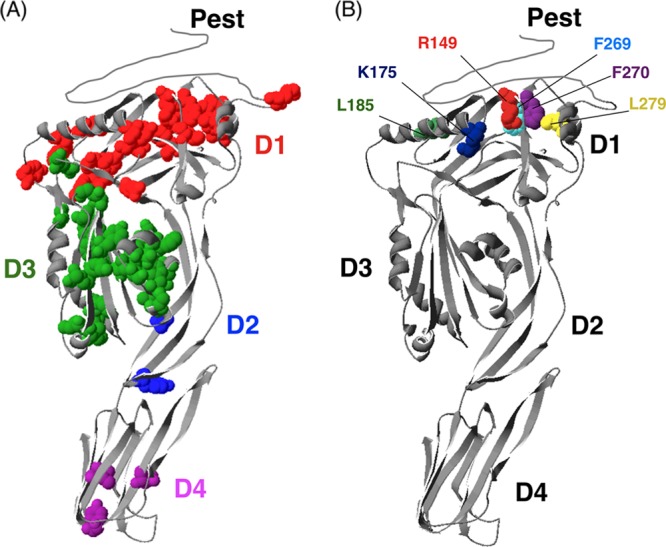
Modified three-dimensional ribbon structure of the LLO toxin (based on the crystal structure of perfringolysin O [PFO] [65]). (A) The original structural model was modified to include the LLO-specific PEST domain. Avirulent mutants identified in the screen are highlighted and colored by domain. (B) The domain 1 mutants characterized in this study are indicated in different colors to highlight their locations.
Characterization of selected mutants identified by IVASS.
Due to a large number of attenuated mutants identified in the screening process, it was not practical to characterize each mutant strain. Attenuated mutants from domain 1 were selected, since this domain is the least well described (Fig. 3B). Analysis of CDC pore structures predicts a role for domain 1 in oligomerization; however, the function of this portion of the toxin remains to be fully evaluated. This study allowed for near-saturation analysis of this domain in vivo (91% of residues were analyzed), and we identified 21 domain 1 substitutions that were attenuated in vivo. None of these had been previously characterized in L. monocytogenes.
Validation that mutants identified by IVASS are attenuated in vivo.
To confirm the in vivo defects of the mutants identified by IVASS, six domain 1 mutant strains were used to infect five mice each in two independent experiments. These mutants were selected because they produce wild-type levels of LLO protein and are therefore likely to be defective in function rather than expression or stability of the protein. Livers and spleens from these animals were harvested 48 h postinfection and analyzed for bacterial load (Fig. 4). In agreement with previously published results, the Δhly mutant was significantly attenuated compared to the wild-type strain (59). Each of the mutants tested exhibited attenuated virulence by several logs compared to the wild type. To address the possibility of false negatives generated by IVASS, five mutants that were not identified by the screen, yet which were quite hypohemolytic, were individually evaluated as described above. Each of the mutants tested retained virulence and was present in the spleens (5 of 5 mutants) or livers (4 of 5) of mice at levels that were significantly different from that of the Δhly strain (data not shown). Taken together, these data suggest that the screening methodology was able to accurately identify residues whose function is required in vivo, while limiting the number of false negatives.
Fig 4.

In vivo confirmation of attenuated mutants identified by IVASS. Forty-eight hours after the infection of five mice with 10 mutant strains each, we measured the total bacterial load from the spleens and livers. The median values for each mutant are represented as horizontal bars. Statistical significance was evaluated using a Mann-Whitney test for each mutant versus the wild-type strain. Data from the spleen and liver were all significant and gave P values of <0.01 and <0.05, respectively. Data are representative of two independent experiments with similar results.
Hemolytic activity.
The ability to form pores in membranes is a hallmark of CDC function and depends on monomer production, activity, and secretion. To evaluate IVASS against a commonly used in vitro assay of CDC function, we monitored hemolysis of red blood cells in a blood plate assay. Here, hemolytic activity of each mutant strain within the signature-tagged library was evaluated on sheep blood agar. The size and opacity of the zone of hemolysis around each colony were scored by visual inspection and categorized as hypohemolytic, hyperhemolytic, or as having wild-type hemolysis, as previously described (86). Of the 60 mutants identified by IVASS, 8 were hyperhemolytic, 3 were ahemolytic, and 48 exhibited low hemolytic activity (Table 1). Interestingly, under normal plating conditions the attenuated mutants I204A and A374G displayed zones of clearance indistinguishable from the wild-type strain (see Fig. S1 in the supplemental material). In addition, the blood plate analysis revealed differences in hemolytic activity for many of the mutant strains that retained virulence in vivo. While blood plate analyses are obviously an important first step for evaluating CDC function, these data suggest that this assay is unable to identify mutants whose in vivo function may affect virulence in ways not detectable by a blood plate screen.
Table 1.
Attenuated mutants identified by IVASS
| PEST |
Domain 1 |
Domain 2 |
Domain 3 |
Domain 4 N-terminal region |
|||||
|---|---|---|---|---|---|---|---|---|---|
| LLO mutanta | Blood plate phenotypeb | LLO mutant | Blood plate phenotype | LLO mutant | Blood plate phenotype | LLO mutant | Blood plate phenotype | LLO mutant | Blood plate phenotype |
| S44A | + | I61A | − | Y98A | − | I204A | −c | I421A | + |
| P45A | + | D62A | − | Y406A | o | Y206A | − | Y427A | o |
| A47G | + | I65A | − | T409A | − | Q216A | − | N432A | o |
| P49A | + | N119A | − | K220A | − | ||||
| K50A | + | Y126A | − | F221A | − | ||||
| P52A | + | L145A | − | G222A | − | ||||
| K55A | − | R149A | − | F225A | − | ||||
| D157A | − | N229A | − | ||||||
| K175A | − | V234A | − | ||||||
| L185A | − | F236A | − | ||||||
| W189A | − | G237A | − | ||||||
| Y197A | − | I239A | − | ||||||
| Y255A | − | V248A | − | ||||||
| Y256A | − | F251A | − | ||||||
| N261A | − | Q253A | − | ||||||
| F269A | − | Y303A | − | ||||||
| F270A | − | A318G | − | ||||||
| L279A | − | F319A | − | ||||||
| Y292A | − | I337A | − | ||||||
| Y298A | − | I338A | − | ||||||
| G299A | − | K344A | − | ||||||
| G349A | o | ||||||||
| G350A | − | ||||||||
| I359A | + | ||||||||
| A374G | WT | ||||||||
| R378A | − | ||||||||
Avirulent mutants were not detected in the livers and spleens of mice 48 h postinfection.
Qualitative hemolysis activity was tested by streaking the individual mutant strains on sheep blood agar plates and comparing zones of hemolysis with those of wild-type bacteria. The activities are reported as follows: +, hyperhemolytic; −, hypohemolytic; o, ahemolytic. WT, wild type.
Phenotype after extended incubation (8 to 9 days).
Listeriolysin O mutant protein expression.
Listeriolysin O expression is regulated through several novel mechanisms at the transcriptional, translational, and posttranslational levels (68). To determine whether the mutants identified by IVASS were attenuated as a result of altered expression of LLO, we analyzed secreted polypeptides from these strains by quantitative Western blotting (Fig. 5). The S44A mutant had previously been isolated from the PEST region of the LLO protein and was used as a positive control for overproduction of the polypeptide (13). Six of the 12 mutants examined produced low levels of secreted LLO protein (30 to 70% of wild-type LLO), an observation that suggests that the loss of virulence in these mutants may be attributable to lower protein expression or stability. The remaining mutants exhibited wild-type levels of LLO protein expression and therefore are likely to represent residues important for LLO function.
Fig 5.
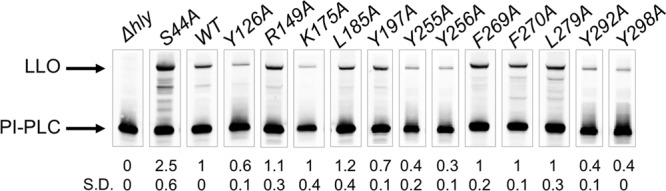
Western blot analysis of secreted LLO. Protein bands were quantified using LI-COR Odyssey software and are presented as the band intensity relative to the wild-type LLO band after normalization to the internal control, PI-PLC. Protein levels represent the means of at least three experiments. SD, standard deviation.
LLO mutants exhibit reduced plaque size.
LLO is essential for facilitating escape from the primary and spreading vacuoles (68). To determine the capacity of the LLO mutants to facilitate phagosomal escape and cell-to-cell spread, we analyzed strains for their capacity to form plaques in monolayers of mouse L2 cell fibroblasts. The size and shape of plaques depend on the ability of the bacterium to infect a cell, escape from the primary vacuole, replicate, spread to neighboring cells, and efficiently escape from the secondary spreading vacuole. In all cases, the mutant strains produced small plaques, ranging from undetectable to 70% of the wild-type plaque size (Table 2). These results suggest that the mutants isolated with IVASS were attenuated in vivo by conferred defects in intracellular LLO activity or by improper regulation.
Table 2.
Plaque sizes of LLO domain 1 mutants
| Mutated residuea | Plaque sizeb |
|---|---|
| Y126A | 65.0 ± 5.4** |
| R149A | 14.8 ± 2.8*** |
| K175A | 44.1 ± 1.1*** |
| L185A | 47.5 ± 1.5*** |
| Y197A | 18.1 ± 3.0* |
| Y255A | 66.7 ± 4.7** |
| Y256A | 8.1 ± 0.3**** |
| F269A | 33.7 ± 4.3** |
| F270A | 47.8 ± 4.0** |
| L279A | 44.3 ± 4.2** |
| Y292A | 49.8 ± 5.9** |
| Y298A | 21.6 ± 8.1** |
Each mutant strain was analyzed in at least three experiments.
Diameters of bacterial plaques is expressed as a percentage of the diameter of wild-type plaques and includes the standard deviation. Student's t test was used to analyze statistical significance versus wild-type bacteria.
, P < 0.05;
, P < 0.01;
, P < 0.001;
, P < 0.0001.
DISCUSSION
The work described here is the first comprehensive analysis of a single virulence factor in vivo in an animal model. These methods allowed for analysis of over 90% of the amino acids of LLO and identified 60 mutations within the toxin that conferred a loss of virulence in the murine model of listeriosis. Because of the high degree of sequence similarity shared by the CDCs, we predicted that the screen would isolate mutations in residues whose contribution to function had previously been identified. We further hypothesized that the screen would reveal novel residues with the potential to contribute to previously unknown functions of LLO. Indeed, 21 of the 60 attenuated mutants are previously described residues important for CDC function, and 39 mutants were previously uncharacterized. Of these uncharacterized mutants, the function of 22 residues can be inferred from their location within or near previously characterized features of the toxin. Therefore, these mutants comprise novel residues that contribute to known functions of the toxin. The remaining 17 mutants potentially constitute an exciting class of mutants that may facilitate hitherto-undiscovered activities of this toxin or simply new regions of the protein that contribute to known functions. Interestingly, a third class of mutants was identified that comprised residues whose mutation did not obviously alter in vitro hemolysis but were nonetheless attenuated in vivo. Below, we discuss these classes of mutants within each domain and speculate on their importance for LLO function.
Mutations in the PEST region.
The PEST-like region of LLO is unique within the CDCs and is important for facilitating the intracellular life cycle of L. monocytogenes. The primary nucleotide sequence within this region negatively regulates translation of the hly transcript through an unknown mechanism (67). The role of the PEST-like region is dependent on the primary nucleotide sequence, since mutants harboring codon substitutions encoding silent amino acid changes overproduce LLO protein, are cytotoxic to host cells, and exhibit a loss of virulence (13, 34, 35, 67, 69). In this study, seven codon substitutions within the PEST region (S44A, P45A, A47A, P49A, K50A, P52A, and K55A) conferred a loss of virulence. Of these, the contributions of the RNA sequences encoding P45, A47, and K55 had not previously been identified. Taken together, these data expand the sequences identified within the hly RNA sequence that facilitate the ability of this region to regulate its own translation.
To determine whether these mutants exhibited phenotypes similar to those previously described for PEST sequence mutants, we compared the hemolytic activities and LLO protein expression levels of these mutants. Each of the PEST mutants identified in the screen, with the exception of K55A, conferred a hyperhemolytic phenotype on blood agar and was expressed at levels higher than that of the wild-type protein (data not shown). These data are in agreement with previous reports that mutations within this domain result in increased protein expression and loss of virulence (13, 67, 69). Conversely, the K55A mutant was expressed at low levels and conferred a hypohemolytic phenotype. Additional analysis will be required to determine what distinguishes the K55A substitution from other mutations in this region. It is possible that the alanine substitution at the K55 transcript results in an RNA secondary structure that impairs its normal translation. These data suggest that changes to the RNA sequence at positions P45 and A47 have functions similar to those of previously characterized PEST mutants and thus expand the key sequences identified within this domain. Future studies will be required to shed light on the mechanisms underpinning PEST function.
Domain 1.
This study allowed for near-saturation analysis of domain 1 in vivo (91% of residues were analyzed). In total, 21 domain 1 substitutions were attenuated in vivo. Of these, the counterparts of several residues have been identified and described for other CDCs (1, 51, 52, 76, 85). However, the importance of these residues in LLO has not yet been described. On the basis of the structure of PFO, Rossjohn et al. (64) suggested that this domain functions in oligomerization, but very few studies have directly tested this hypothesis.
Truncation analysis of recombinant SLO by Yamamoto et al. identified a region spanning 4 amino acids (SLOE108-D111) within domain 1 that were critical to hemolytic activity in vitro. In that study, analysis of an intact LLO also identified this region as functionally important in vivo and resolved the residues responsible for this effect to two key amino acids, I61 and D62 (SLOI109 and SLON110). In addition, Yamamoto et al. identified a double mutant in SLO harboring substitutions at D324E and L325P that showed a significant reduction in hemolytic activity. Mutation of the corresponding leucine residue in LLO (L279) was identified in our screen, while the D324 counterpart, LLOQ278A, remained virulent. These studies suggest that the leucine residue in both toxins is important for both hemolytic activity and virulence and highlight the high degree of resolution conferred by the alanine scanning method.
The hypothesized role for domain 1 in oligomerization remains understudied. The potential role of these newly identified domain 1 mutants in oligomerization can now be further analyzed through detailed in vitro analyses. It will be interesting to determine whether these residues contribute to pore formation or another, novel function of LLO.
Domain 2.
Within domain 2, we were able to screen 47 of the 51 total residues and found that only three mutants were attenuated (Fig. 3A). The level of tolerance for mutations in this portion of the protein suggests that vertical collapse mediated by this domain is refractory to small changes in the primary sequence. Indeed, reports of mutations with phenotypes in this domain are limited (36).
LLO contains a consensus bipartite NLS within domain 2 that has been hypothesized to target the toxin to the host nucleus during intracellular growth. Because the nucleus has very little membrane cholesterol, LLO could be sequestered here as an additional mechanism for preventing dissolution of the host cytosolic membrane during the bacterium's cytosolic replicative phase. If the NLS of LLO constitutes a novel regulatory region of the protein toxin, then we would expect that mutation of this sequence would affect virulence. Previous work has shown that single amino acid changes to eukaryotic NLS consensus sequences can alter nuclear targeting (62). However, none of the six alanine NLS substitutions tested conferred a virulence defect. These data suggested that if LLO is targeted to the host nucleus, single alanine substitutions do not affect LLO's role in virulence to a measurable extent in CD1 mice.
Domain 3.
Domain 3 contains the transmembrane hairpins responsible for insertion of the prepore complex into a membrane and formation of the mature pore (72, 73). Within domain 3, 119 (90%) of the total 132 residues were analyzed in this study. Of these, 26—nearly one-fifth—were attenuated. These data highlight the importance of this domain for cytolysin function. Tightly coordinated structural rearrangements within domain 3 regulate the timing of oligomerization and membrane insertion (18). Two conserved glycines act as a hinge to allow a portion of domain 3 to swivel and expose the underlying polypeptide for intermolecular interaction. Mutation of these glycines allows monomers to bind to membranes but abrogates their ability to associate and form pores (61). The screen identified both of these residues in LLO (G349 and G350) and supported the proposed importance of these residues. In addition, this study identified I204 and Y206 upstream of the first transmembrane hairpin, which are essential for the conversion of the prepore complex into a membrane-inserted pore in PFO. Mutation of the PFOY181 counterpart (LLOY206) prevents prepore complexes from converting into an inserted pore complex (26). Mutation of the PFO counterpart to LLOI204, PFOT179, confers a defect in pore formation in vitro (61). The crystal structure of PFO predicts that this residue lies at the interface between two PFO monomers of the prepore and may be important for oligomerization (64). Data from this study highlight the importance of this residue for virulence and suggest that the function of this residue may be conserved across CDCs.
Hydrophobic loops of domain 4.
During the CDC monomer interaction with a membrane surface, the conserved tryptophan-rich undecapeptide loop and three hydrophobic loops of domain 4 insert into the membrane and anchor the CDC monomer in an upright position. The C-terminal portion of domain 4 harboring the undecapeptide region and the cholesterol recognition loop 1 has been extensively studied (reviewed in reference 18). We therefore focused our analysis on the first 50 amino acids of this domain, containing hydrophobic loops 2 and 3. We tested 37 amino acids in this region and found three mutants that were attenuated for virulence. Although none of these mutants has been previously described, each of these residues (I421, Y427, and N432) are next to or within hydrophobic loop 2, a fact that is suggestive of a role for these residues in membrane insertion as previously hypothesized (61). Interestingly, despite the high degree of conservation of seven residues within loop 2, only one of the analyzed mutants, Y427, was attenuated for virulence in vivo. Finally, the identification of the two residues flanking loop 2 (I421 and Y427) suggests that the regions outside the loop may have underappreciated interactions with the membrane.
Comparison of in vivo and in vitro phenotypes.
The fundamental pore-forming activity of CDCs can easily be visualized by hemolysis of erythrocytes on blood agar plates. The feasibility of scoring large numbers of mutants makes this an attractive assay for identifying mutants deficient in CDC function or other bacterial genes that contribute to hemolytic phenotypes (86). While the advantages of blood agar plate analysis are obvious, this assay alone was insufficient to accurately identify mutants whose functions were important in vivo. Under normal plating conditions, two mutant strains from domain 3, I204A and A374G, gave zones of clearance indistinguishable from that of the wild-type strain. These mutants had not been identified prior to this work, and their isolation underscores the power of in vivo analysis for fully evaluating CDC function. Although zones of clearance are normally evaluated 3 to 4 days after plating, further incubation (8 to 9 days total) revealed altered activity for the I204A but not the A374G strain. These data suggest that I204A activity differs from that of the wild-type toxin in a way that would have been missed under normal in vitro assay conditions.
Evaluation of I204A and A374G in a plaque assay revealed that the A374G mutant produces small plaques (53% of wild type), while the I204A mutant does not form plaques (data not shown). To date, all of the small plaque mutants identified have invariably exhibited in vivo defects (77), a fact that highlights the predictive power of this assay. Taken together, these data suggest that a combination of screening techniques would be required to identify novel mutants like I204A and A374G. Residue A374 lies outside characterized motifs within domain 3, and it is therefore difficult to speculate on its function. Since hemolytic activity is not obviously altered in this mutant, we propose that this residue contributes a novel function for the toxin during infection.
The hemolytic activity of individual mutants can be semiquantitatively measured through a variety of methods, including relative release of hemoglobin from erythrocytes incubated with purified toxin proteins (39, 50, 55, 59, 73). These assays have been used to characterize residues that contribute to the CDC pore-forming mechanism. However, comparison between data from this study and from previous studies has led to some interesting observations that could not have been predicted by analysis of hemolytic activity alone. For example, previous cysteine scan analyses demonstrated wild-type hemolytic activity for many of the substituted residues within the transmembrane hairpin 1 (TMH1) of recombinant PFO (73). However, the LLO counterparts of several of these (6 of 16) were attenuated in vivo. It is not clear whether these data represent discrepancies in methodologies, differences in the toxins, or the requirement for these residues to function only in vivo. Further analysis of these TMH residues is needed to establish the reason behind these observed differences.
Future prospects.
The collection of LLO mutants generated here constitutes a powerful resource for detailed analysis of known CDC functions and the discovery of novel ones. In addition, this new resource can be used to address other questions concerning LLO regulation, the role of LLO in the intracellular niche, and how the sequential mechanisms of pore formation affect different aspects of immunity. For example, how do mutants of particular domains specifically affect host cell signaling? Listeriolysin O has been shown to be essential for triggering a wide variety of host immune responses, including apoptosis (7, 22), NF-κB activation (31, 63), autophagy (46), stimulation of both proinflammatory and gamma-interferon-inducing cytokines (30, 53), and acquired immunity (2, 5). The contribution of LLO to these responses is often determined through the use of a Δhly strain. However, it is not clear to what extent the toxin itself or its activities contribute to detection by the host immune system. Listeriolysin O is likely to trigger host responses through a variety of means, including detection of the toxin by host receptors, pore-dependent disruption of membranes, translocation of effector molecules, and perturbation of other host processes. The collection of LLO mutants generated in this study is a rich resource for probing the contribution of LLO to triggering host responses. The mutants described here can be used to functionally uncouple host detection of LLO from the toxin's activity. For example, the mutant library can be used to probe for how mutants deficient in particular steps of pore formation and the intracellular life cycle are sensed by the host.
IVASS identified 21 unique residues that did not map to previously characterized functional domains of LLO and therefore are promising candidates for novel in vivo functions of LLO. We believe that the IVASS approach, coupled with subsequent in vitro analysis of known CDC functions, holds promise for identifying and characterizing novel roles for other members of the CDC family. The feasibility of high-throughput scanning has already been demonstrated for other proteins, such as the protective antigen (PA) moiety of anthrax toxin (48). Application of the IVASS system to these existing libraries will allow for rapid and efficient in vivo analysis of these proteins and further expand our understanding of how important virulence factors, such as anthrax toxin, function in vivo.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Madhani for signature tag sequences, H. Goldfine for providing the PI-PLC antibody, V. Van Voorhis, S. Mochegova, T. Burke, and B. Kline for their help with experiments, N. Meyer-Morse, J. D. Sauer, and K. Archer for their generous assistance with animal work, and J. D. Sauer and B. Kline for critical reading of the manuscript.
This research was supported by National Institutes of Health grants PO1 A1063302 and R01 AI27655 (to D.A.P.), F32 AI072988 (to J.A.M.-W.), and 5T32CA009179-34 (to S.L.M.).
D. A. Portnoy consults with and has a financial interest in Aduro BioTech, a company that may stand to benefit from the results of this research.
Footnotes
Published ahead of print 26 March 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Abdel Ghani EM, et al. 1999. Streptolysin O: inhibition of the conformational change during membrane binding of the monomer prevents oligomerization and pore formation. Biochemistry 38:15204–15211 [DOI] [PubMed] [Google Scholar]
- 2. Beattie IA, Swaminathan B, Ziegler HK. 1990. Cloning and characterization of T-cell-reactive protein antigens from Listeria monocytogenes. Infect. Immun. 58:2792–2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bishop DK, Hinrichs DJ. 1987. Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J. Immunol. 139:2005–2009 [PubMed] [Google Scholar]
- 4. Bourdeau RW, et al. 2009. Cellular functions and X-ray structure of anthrolysin O, a cholesterol-dependent cytolysin secreted by Bacillus anthracis. J. Biol. Chem. 284:14645–14656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bouwer HG, Barry RA, Hinrichs DJ. 1997. Acquired immunity to an intracellular pathogen: immunologic recognition of L. monocytogenes-infected cells. Immunol. Rev. 158:137–146 [DOI] [PubMed] [Google Scholar]
- 6. Camilli A, Mekalanos JJ. 1995. Use of recombinase gene fusions to identify Vibrio cholerae genes induced during infection. Mol. Microbiol. 18:671–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carrero JA, Calderon B, Unanue ER. 2004. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J. Exp. Med. 200:535–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cluff CW, Ziegler HK. 1987. Inhibition of macrophage-mediated antigen presentation by hemolysin-producing Listeria monocytogenes. J. Immunol. 139:3808–3812 [PubMed] [Google Scholar]
- 9. Cossart P, et al. 1989. Listeriolysin O is essential for virulence of Listeria monocytogenes: direct evidence obtained by gene complementation. Infect. Immun. 57:3629–3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coulter SN, et al. 1998. Staphylococcus aureus genetic loci impacting growth and survival in multiple infection environments. Mol. Microbiol. 30:393–404 [DOI] [PubMed] [Google Scholar]
- 11. Cunningham BC, Wells JA. 1989. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244:1081–1085 [DOI] [PubMed] [Google Scholar]
- 12. Dabiri GA, Sanger JM, Portnoy DA, Southwick FS. 1990. Listeria monocytogenes moves rapidly through the host-cell cytoplasm by inducing directional actin assembly. Proc. Natl. Acad. Sci. U. S. A. 87:6068–6072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Decatur AL, Portnoy DA. 2000. A PEST-like sequence in listeriolysin O essential for Listeria monocytogenes pathogenicity. Science 290:992–995 [DOI] [PubMed] [Google Scholar]
- 14. Dramsi S, Cossart P. 2003. Listeriolysin O-mediated calcium influx potentiates entry of Listeria monocytogenes into the human Hep-2 epithelial cell line. Infect. Immun. 71:3614–3618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gaillard JL, Berche P, Sansonetti P. 1986. Transposon mutagenesis as a tool to study the role of hemolysin in the virulence of Listeria monocytogenes. Infect. Immun. 52:50–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gedde MM, Higgins DE, Tilney LG, Portnoy DA. 2000. Role of listeriolysin O in cell-to-cell spread of Listeria monocytogenes. Infect. Immun. 68:999–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gekara NO, Jacobs T, Chakraborty T, Weiss S. 2005. The cholesterol-dependent cytolysin listeriolysin O aggregates rafts via oligomerization. Cell. Microbiol. 7:1345–1356 [DOI] [PubMed] [Google Scholar]
- 18. Gilbert RJ. 2010. Cholesterol-dependent cytolysins. Adv. Exp. Med. Biol. 677:56–66 [DOI] [PubMed] [Google Scholar]
- 19. Glomski IJ, Decatur AL, Portnoy DA. 2003. Listeria monocytogenes mutants that fail to compartmentalize listerolysin O activity are cytotoxic, avirulent, and unable to evade host extracellular defenses. Infect. Immun. 71:6754–6765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Glomski IJ, Gedde MM, Tsang AW, Swanson JA, Portnoy DA. 2002. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J. Cell Biol. 156:1029–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grant AJ, et al. 2008. Modelling within-host spatiotemporal dynamics of invasive bacterial disease. PLoS Biol. 6:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guzman CA, et al. 1996. Apoptosis of mouse dendritic cells is triggered by listeriolysin, the major virulence determinant of Listeria monocytogenes. Mol. Microbiol. 20:119–126 [DOI] [PubMed] [Google Scholar]
- 23. Hamon MA, et al. 2007. Histone modifications induced by a family of bacterial toxins. Proc. Natl. Acad. Sci. U. S. A. 104:13467–13472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hensel M, et al. 1995. Simultaneous identification of bacterial virulence genes by negative selection. Science 269:400–403 [DOI] [PubMed] [Google Scholar]
- 25. Heuck AP, Moe PC, Johnson BB. 2010. The cholesterol-dependent cytolysin family of gram-positive bacterial toxins. Subcell. Biochem. 51:551–577 [DOI] [PubMed] [Google Scholar]
- 26. Hotze EM, et al. 2002. Monomer-monomer interactions drive the prepore to pore conversion of a beta-barrel-forming cholesterol-dependent cytolysin. J. Biol. Chem. 277:11597–11605 [DOI] [PubMed] [Google Scholar]
- 27. Jones S, Preiter K, Portnoy DA. 1996. Conversion of an extracellular cytolysin into a phagosome-specific lysin which supports the growth of an intracellular pathogen. Mol. Microbiol. 21:1219–1225 [DOI] [PubMed] [Google Scholar]
- 28. Karlyshev AV, et al. 2001. Application of high-density array-based signature-tagged mutagenesis to discover novel Yersinia virulence-associated genes. Infect. Immun. 69:7810–7819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kathariou S, Metz P, Hof H, Goebel W. 1987. Tn916-induced mutations in the hemolysin determinant affecting virulence of Listeria monocytogenes. J. Bacteriol. 169:1291–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kayal S, Charbit A. 2006. Listeriolysin O: a key protein of Listeria monocytogenes with multiple functions. FEMS Microbiol. Rev. 30:514–529 [DOI] [PubMed] [Google Scholar]
- 31. Kayal S, et al. 1999. Listeriolysin O-dependent activation of endothelial cells during infection with Listeria monocytogenes: activation of NF-kappa B and upregulation of adhesion molecules and chemokines. Mol. Microbiol. 31:1709–1722 [DOI] [PubMed] [Google Scholar]
- 32. Lau GW, et al. 2001. A functional genomic analysis of type 3 Streptococcus pneumoniae virulence. Mol. Microbiol. 40:555–571 [DOI] [PubMed] [Google Scholar]
- 33. Lauer P, Chow MY, Loessner MJ, Portnoy DA, Calendar R. 2002. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J. Bacteriol. 184:4177–4186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lety MA, Frehel C, Berche P, Charbit A. 2002. Critical role of the N-terminal residues of listeriolysin O in phagosomal escape and virulence of Listeria monocytogenes. Mol. Microbiol. 46:367–379 [DOI] [PubMed] [Google Scholar]
- 35. Lety MA, et al. 2001. Identification of a PEST-like motif in listeriolysin O required for phagosomal escape and for virulence in Listeria monocytogenes. Mol. Microbiol. 39:1124–1139 [DOI] [PubMed] [Google Scholar]
- 36. Lety MA, Frehel C, Raynaud C, Dupuis M, Charbit A. 2006. Exploring the role of the CTL epitope region of listeriolysin O in the pathogenesis of Listeria monocytogenes. Microbiology 152:1287–1296 [DOI] [PubMed] [Google Scholar]
- 37. Liu OW, et al. 2008. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell 135:174–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Loeb DD, et al. 1989. Complete mutagenesis of the HIV-1 protease. Nature 340:397–400 [DOI] [PubMed] [Google Scholar]
- 39. Magassa N, Chandrasekaran S, Caparon MG. 2010. Streptococcus pyogenes cytolysin-mediated translocation does not require pore formation by streptolysin O. EMBO Rep. 11:400–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mahan MJ, Slauch JM, Mekalanos JJ. 1993. Selection of bacterial virulence genes that are specifically induced in host tissues. Science 259:686–688 [DOI] [PubMed] [Google Scholar]
- 41. Marquis H, Doshi V, Portnoy DA. 1995. The broad-range phospholipase C and a metalloprotease mediate listeriolysin O-independent escape of Listeria monocytogenes from a primary vacuole in human epithelial cells. Infect. Immun. 63:4531–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mazurkiewicz P, Tang CM, Boone C, Holden DW. 2006. Signature-tagged mutagenesis: barcoding mutants for genome-wide screens. Nat. Rev. Genet. 7:929–939 [DOI] [PubMed] [Google Scholar]
- 43. Melton-Witt JA, Bentsen LM, Tweten RK. 2006. Identification of functional domains of Clostridium septicum alpha toxin. Biochemistry 45:14347–14354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Melton-Witt JA, Rafelski SM, Portnoy DA, Bakardjiev AI. 2012. Oral infection with signature-tagged Listeria monocytogenes reveals organ-specific growth and dissemination routes in guinea pigs. Infect. Immun. 80:720–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Merrell DS, Hava DL, Camilli A. 2002. Identification of novel factors involved in colonization and acid tolerance of Vibrio cholerae. Mol. Microbiol. 43:1471–1491 [DOI] [PubMed] [Google Scholar]
- 46. Meyer-Morse N, et al. 2010. Listeriolysin O is necessary and sufficient to induce autophagy during Listeria monocytogenes infection. PLoS One 5:e8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Milla ME, Brown BM, Sauer RT. 1994. Protein stability effects of a complete set of alanine substitutions in Arc repressor. Nat. Struct. Biol. 1:518–523 [DOI] [PubMed] [Google Scholar]
- 48. Mourez M, et al. 2003. Mapping dominant-negative mutations of anthrax protective antigen by scanning mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 100:13803–13808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nelson RT, Hua J, Pryor B, Lodge JK. 2001. Identification of virulence mutants of the fungal pathogen Cryptococcus neoformans using signature-tagged mutagenesis. Genetics 157:935–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ohno-Iwashita Y, Iwamoto M, Mitsui K, Ando S, Nagai Y. 1988. Protease-nicked theta-toxin of Clostridium perfringens, a new membrane probe with no cytolytic effect, reveals two classes of cholesterol as toxin-binding sites on sheep erythrocytes. Eur. J. Biochem. 176:95–101 [DOI] [PubMed] [Google Scholar]
- 51. Palmer M, et al. 1998. Assembly mechanism of the oligomeric streptolysin O pore: the early membrane lesion is lined by a free edge of the lipid membrane and is extended gradually during oligomerization. EMBO J. 17:1598–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Palmer M, et al. 1996. Membrane-penetrating domain of streptolysin O identified by cysteine scanning mutagenesis. J. Biol. Chem. 271:26664–26667 [DOI] [PubMed] [Google Scholar]
- 53. Pamer EG. 2004. Immune responses to Listeria monocytogenes. Nat. Rev. Immunol. 4:812–823 [DOI] [PubMed] [Google Scholar]
- 54. Pascopella L, Collins FM, Martin JM, Jacobs WR, Jr, Bloom BR. 1993. Identification of a genomic fragment of Mycobacterium tuberculosis responsible for in vivo growth advantage. Infect. Agents Dis. 2:282–284 [PubMed] [Google Scholar]
- 55. Polekhina G, Giddings KS, Tweten RK, Parker MW. 2004. Crystallization and preliminary X-ray analysis of the human-specific toxin intermedilysin. Acta Crystallogr. D Biol. Crystallogr. 60:347–349 [DOI] [PubMed] [Google Scholar]
- 56. Polekhina G, Giddings KS, Tweten RK, Parker MW. 2005. Insights into the action of the superfamily of cholesterol-dependent cytolysins from studies of intermedilysin. Proc. Natl. Acad. Sci. U. S. A. 102:600–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Portnoy DA. 1992. Innate immunity to a facultative intracellular bacterial pathogen. Curr. Opin. Immunol. 4:20–24 [DOI] [PubMed] [Google Scholar]
- 58. Portnoy DA, Auerbuch V, Glomski IJ. 2002. The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity. J. Cell Biol. 158:409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Portnoy DA, Jacks PS, Hinrichs DJ. 1988. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 167:1459–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rae CS, Geissler A, Adamson PC, Portnoy DA. 2011. Mutations of the Listeria monocytogenes peptidoglycan N-deacetylase and O-acetylase result in enhanced lysozyme sensitivity, bacteriolysis, and hyperinduction of innate immune pathways. Infect. Immun. 79:3596–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ramachandran R, Tweten RK, Johnson AE. 2004. Membrane-dependent conformational changes initiate cholesterol-dependent cytolysin oligomerization and intersubunit beta-strand alignment. Nat. Struct. Mol. Biol. 11:697–705 [DOI] [PubMed] [Google Scholar]
- 62. Robbins J, Dilworth SM, Laskey RA, Dingwall C. 1991. Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: identification of a class of bipartite nuclear targeting sequence. Cell 64:615–623 [DOI] [PubMed] [Google Scholar]
- 63. Rose F, et al. 2001. Human endothelial cell activation and mediator release in response to Listeria monocytogenes virulence factors. Infect. Immun. 69:897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rossjohn J, Feil SC, McKinstry WJ, Tweten RK, Parker MW. 1997. Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell 89:685–692 [DOI] [PubMed] [Google Scholar]
- 65. Rossjohn J, et al. 2007. Structures of perfringolysin O suggest a pathway for activation of cholesterol-dependent cytolysins. J. Mol. Biol. 367:1227–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schlech WF., III 2000. Foodborne listeriosis. Clin. Infect. Dis. 31:770–775 [DOI] [PubMed] [Google Scholar]
- 67. Schnupf P, et al. 2006. Regulated translation of listeriolysin O controls virulence of Listeria monocytogenes. Mol. Microbiol. 61:999–1012 [DOI] [PubMed] [Google Scholar]
- 68. Schnupf P, Portnoy DA. 2007. Listeriolysin O: a phagosome-specific lysin. Microbes Infect. 9:1176–1187 [DOI] [PubMed] [Google Scholar]
- 69. Schnupf P, Portnoy DA, Decatur AL. 2006. Phosphorylation, ubiquitination and degradation of listeriolysin O in mammalian cells: role of the PEST-like sequence. Cell. Microbiol. 8:353–364 [DOI] [PubMed] [Google Scholar]
- 70. Schnupf P, Zhou J, Varshavsky A, Portnoy DA. 2007. Listeriolysin O secreted by Listeria monocytogenes into the host cell cytosol is degraded by the N-end rule pathway. Infect. Immun. 75:5135–5147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schuerch DW, Wilson-Kubalek EM, Tweten RK. 2005. Molecular basis of listeriolysin O pH dependence. Proc. Natl. Acad. Sci. U. S. A. 102:12537–12542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shatursky O, et al. 1999. The mechanism of membrane insertion for a cholesterol-dependent cytolysin: a novel paradigm for pore-forming toxins. Cell 99:293–299 [DOI] [PubMed] [Google Scholar]
- 73. Shepard LA, et al. 1998. Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: an alpha-helical to beta-sheet transition identified by fluorescence spectroscopy. Biochemistry 37:14563–14574 [DOI] [PubMed] [Google Scholar]
- 74. Shi L, Jung YJ, Tyagi S, Gennaro ML, North RJ. 2003. Expression of Th1-mediated immunity in mouse lungs induces a Mycobacterium tuberculosis transcription pattern characteristic of nonreplicating persistence. Proc. Natl. Acad. Sci. U. S. A. 100:241–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Smith GA, Portnoy DA, Theriot JA. 1995. Asymmetric distribution of the Listeria monocytogenes ActA protein is required and sufficient to direct actin-based motility. Mol. Microbiol. 17:945–951 [DOI] [PubMed] [Google Scholar]
- 76. Suarez-Alvarez B, Garcia-Suarez Mdel M, Mendez FJ, de los Toyos JR. 2003. Characterisation of mouse monoclonal antibodies for pneumolysin: fine epitope mapping and V gene usage. Immunol. Lett. 88:227–239 [DOI] [PubMed] [Google Scholar]
- 77. Sun AN, Camilli A, Portnoy DA. 1990. Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect. Immun. 58:3770–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tang P, Rosenshine I, Cossart P, Finlay BB. 1996. Listeriolysin O activates mitogen-activated protein kinase in eucaryotic cells. Infect. Immun. 64:2359–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tilney LG, Portnoy DA. 1989. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J. Cell Biol. 109:1597–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tweten RK. 2005. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 73:6199–6209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tweten RK, Parker MW, Johnson AE. 2001. The cholesterol-dependent cytolysins. Curr. Top. Microbiol. Immunol. 257:15–33 [DOI] [PubMed] [Google Scholar]
- 82. Valdivia RH, Falkow S. 1997. Probing bacterial gene expression within host cells. Trends Microbiol. 5:360–363 [DOI] [PubMed] [Google Scholar]
- 83. Vazquez-Boland JA, et al. 2001. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14:584–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wadsworth SJ, Goldfine H. 2002. Mobilization of protein kinase C in macrophages induced by Listeria monocytogenes affects its internalization and escape from the phagosome. Infect. Immun. 70:4650–4660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yamamoto I, Kimoto H, Taketo Y, Taketo A. 2001. Mutational and comparative analysis of streptolysin O, an oxygen-labile streptococcal hemolysin. Biosci. Biotechnol. Biochem. 65:2682–2689 [DOI] [PubMed] [Google Scholar]
- 86. Zemansky J, et al. 2009. Development of a mariner-based transposon and identification of Listeria monocytogenes determinants, including the peptidyl-prolyl isomerase PrsA2, that contribute to its hemolytic phenotype. J. Bacteriol. 191:3950–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.