

Abstract
The human pathogens enterohemorrhagic and enteropathogenic Escherichia coli (EHEC and EPEC), as well as the related mouse pathogen Citrobacter rodentium, utilize a type III secretion system (T3SS) to inject multiple effector proteins into host cells. The E. coli O157:H7 strain EDL933 carries two copies of non-locus of enterocyte effacement (LEE)-encoded protein H, designated NleH1 and NleH2, both of which bind to the human ribosomal protein S3 (RPS3), a subunit of NF-κB transcriptional complexes. In this study, we describe significant functional differences between NleH1 and NleH2 in their ability to regulate the host NF-κB pathway. We show that the EHEC and EPEC NleH effectors are functionally equivalent in their ability to affect RPS3 nuclear translocation. NleH1, but not NleH2, inhibited NF-κB activity without altering the kinetics of IκBα phosphorylation/degradation. We also determined that the class I PSD-95/Disc Large/ZO-1 (PDZ)-binding domain of NleH was important for its activity in the NF-κB pathway. In addition to binding RPS3, we found that NleH1 and NleH2 are able to bind to each other in vitro and in vivo, suggesting an additional mechanism by which the E. coli NleH effectors may regulate the extent and duration of NF-κB activation after their T3SS-dependent translocation. We also performed mouse infection experiments and established that mouse mortality and Citrobacter colonization were reduced in mice infected with ΔnleH. Complementing ΔnleH with NleH1 restored Citrobacter virulence and colonization to wild-type levels, whereas complementing with NleH2 reduced them. Taken together, our data show that NleH1 and NleH2 have pronounced functional differences in their ability to alter host transcriptional responses to bacterial infection.
INTRODUCTION
Enterohemorrhagic Escherichia coli (EHEC) and other Shiga-like toxin-producing E. coli (STEC) strains are transmitted to humans through consumption of fruit juice, raw/undercooked meat, and vegetables contaminated with manure. STEC causes diseases ranging from bloody diarrhea to severe kidney and neurological complications and is a leading cause of pediatric renal failure (hemolytic uremic syndrome [HUS]). There are no treatments of proven therapeutic value (25), and administering antibiotics is often contraindicated because they can enhance the progression of enteritis to HUS.
While E. coli O157:H7 is the most frequently isolated STEC serotype in North America, non-O157-STEC serotypes (e.g., O26, O103, and O111) also cause outbreaks of bloody diarrhea and HUS of comparable severity (14). The presence of a type III secretion system (T3SS) and specific virulence proteins (effectors) correlates with the ability of a STEC strain to cause severe disease and human outbreaks (2).
STEC effectors are translocated directly into intestinal epithelial cells through a T3SS (3). In attaching and effacing (A/E) pathogens, which also include enteropathogenic E. coli (EPEC) and Citrobacter rodentium, a natural pathogen of mice used for in vivo studies (4), the T3SS and several effectors are encoded by a pathogenicity island termed the locus of enterocyte effacement (LEE) (13). Many other non-LEE-encoded [Nle] effectors are encoded by other pathogenicity islands identified more recently (5).
Some effectors (e.g., NleB, NleC, NleD, NleE, and NleH) are key modulators of the innate immune system of intestinal epithelial cells, especially pathways regulated by the transcription factor NF-κB (6, 16, 17). For example, NleC is a protease that cleaves the NF-κB p65 subunit (1, 15, 18), as well as the p300 acetyltransferase (24). NleD cleaves the c-Jun N-terminal kinase (JNK) to prevent AP-1 activation (1). NleE inhibits both p65 nuclear translocation and the degradation of the inhibitory NF-κB chaperone IκBα (17) to block NF-κB activation, in conjunction with NleB (16, 17). A recent report also suggests that Tir, in addition to its role as the translocated intimin receptor, also functions to inhibit NF-κB activity by targeting tumor necrosis factor (TNF) receptor-associated factors (TRAFs) (22).
Activation of the inhibitor of κB kinase (IKK) complex during infection stimulates the NF-κB pathway by promoting IκBα degradation. After nuclear import, NF-κB binds to κB sites within target gene promoters and regulates transcription by recruiting coactivators/repressors (26). The ribosomal protein S3 (RPS3) guides NF-κB to specific κB sites (26). We previously showed that RPS3 is inducibly associated with and phosphorylated by IKKβ on serine 209 (S209) in concert with NF-κB pathway activation (27). RPS3 binds to the p65 NF-κB subunit and increases the affinity of NF-κB for a subset of target genes (26).
E. coli O157:H7 strain EDL933 carries the T3SS effectors NleH1 and NleH2, the amino acid sequences of which are 84% identical. We have shown that both NleH1 and NleH2 bind RPS3 (6). NleH1, but not NleH2, prevents RPS3 association with NF-κB in the nucleus by inhibiting IKKβ phosphorylation of RPS3 (27). However, others have suggested that both NleH1 and NleH2 can inhibit NF-κB through a mechanism involving attenuation of IκBα degradation, rather than by interacting with RPS3 (21).
Other studies have characterized NleH interactions with different host proteins. NleH1 also binds to the Bax inhibitor 1, inhibiting caspase-3 activation during EPEC infection (8, 20). NleH1 also binds the Na+/H+ exchanger regulatory factor 2 (NHERF2), a scaffold protein involved in tethering and recycling ion channels in polarized epithelia (12). Overexpressing NHERF2 diminishes the antiapoptotic activity of NleH1 (12). The interaction between NleH1 and NHERF2 has been suggested to function as a plasma membrane sorting site to control spatial and temporal NleH activity (12).
Several animal studies have been performed to elucidate NleH function in vivo, but with some conflicting results. The mouse pathogen C. rodentium carries only one copy of NleH (19), a protein that functions equivalently to EHEC NleH1 (6). In wild-type C57BL/6 mice, a ΔnleH C. rodentium mutant is cleared more rapidly than wild-type bacteria (9). At early but not late stages of infection, ΔnleH also displays reduced colonization of mouse colons (7). Unexpectedly, deleting nleH caused a reduced TNF response (9). Infecting streptomycin-treated mice with EPEC revealed that NleH inhibits proinflammatory cytokine expression and promotes EPEC colonization (21). Deleting both nleH1 and nleH2 from E. coli O157:H7 caused increased shedding compared with that of the parental strain in calves, yet had a reduced competitive advantage in mixed infections of lambs (9). Our earlier studies of E. coli O157:H7 in gnotobiotic piglets revealed a hypervirulent ΔnleH1 phenotype, but with reduced piglet colonization (6).
The goal of this study was to clarify the biochemical basis for the reported differences in NleH activities. In the course of doing so, we also determined that NleH1 and NleH2 are capable of interacting with each other and identified the NleH class I PSD-95/Disc Large/ZO-1 (PDZ)-binding domain as contributing to NF-κB pathway regulation.
MATERIALS AND METHODS
Ethics statement.
All animal experiments were performed according to Institutional Animal Care and Use Committee-approved protocols (Animal Welfare Assurance no. A3958-01).
Chemicals and antibodies.
All chemicals and antibodies were used according to manufacturers' recommendations. Antibodies were obtained from the following sources: poly(ADP-ribose) polymerase (PARP), BD Biosciences; IκBα and phospho-IκBα, Cell Signaling; phospho-RPS3, Primm Biotech; RPS3, Proteintech Group; tubulin, Santa Cruz Biotechnology; and actin, FLAG, glutathione S-transferase (GST), hemagglutinin (HA), and His, Sigma.
Bacterial strains, cell culture, and infection experiments.
The bacterial strains and plasmids used in this study are described in Table 1. Oligonucleotide sequences will be made available upon request. HeLa and HEK293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 4.5 g/liter glucose, l-glutamine, and sodium pyruvate, supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Cells were transfected using Lipofectamine 2000 (Invitrogen) or TransPass COS/293 (New England BioLabs). Bacteria were cultured in Luria-Bertani (LB) broth at 37°C for 18 h. Overnight LB cultures were subcultured 1:10 into DMEM, followed by a further incubation for 3 h at 37°C and 5% CO2. IPTG (isopropyl-β-d-thiogalactopyranoside) (1 mM) was added for the final 1 h of incubation when NleH was expressed from pFLAG-CTC. Cell culture medium was replaced with DMEM prior to infection, and bacteria were added at a multiplicity of infection of 25 to 50.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| Strains | ||
| E. coli EDL933 | Wild-type E. coli O157:H7 isolate | CDC |
| E. coli E2348/69 | Wild-type E. coli O127:H6 isolate | 11 |
| E. coli BL21(DE3) | E. coli F−ompT hsdSB(rB− mB−) gal dcm (DE3) | Novagen |
| BL21(DE3)/pNleH1-pET28a | His-NleH1 | 6 |
| BL21(DE3)/pNleH2-pET28a | His-NleH2 | 6 |
| BL21(DE3)/6-PFK-pET28a | His-6-PFK | This study |
| BL21(DE3)/RPS3-pET42a | GST-RPS3 | 27 |
| BL21(DE3)/GST-pET42a | GST | Novagen |
| BL21(DE3)/NleH1-pET42a | GST-NleH1 | This study |
| BL21(DE3)/NleH2-pET42a | GST-NleH2 | This study |
| C. rodentium DBS100 | Wild-type C. rodentium ATCC 51459 | 23 |
| C. rodentium ΔnleH | C. rodentium nleH mutant | J. Puente |
| C. rodentium ΔnleH/pNleH1-FLAG | C. rodentium ΔnleH expressing EHEC NleH1-FLAG | This study |
| C. rodentium ΔnleH/pNleH2-FLAG | C. rodentium ΔnleH expressing EHEC NleH2-FLAG | This study |
| C. rodentium ΔnleH/pNleH1(K159A)-FLAG | C. rodentium ΔnleH expressing EHEC NleH1(K159A)-FLAG | This study |
| C. rodentium ΔnleH/pNleH2(K169A)-FLAG | C. rodentium ΔnleH expressing EHEC NleH2(K169A)-FLAG | This study |
| Plasmids | ||
| κB (5X)-luc | Firefly luciferase driven by RPS/κB site | Promega |
| pTKRL-luc | Renilla luciferase | Promega |
| IKKβ | IKKb expression plasmid | 27 |
| pET28a | Bacterial hexahistidine fusion expression | Novagen |
| NleH1-pET28a | His-NleH1 | 6 |
| NleH2-pET28a | His-NleH2 | 6 |
| NleH1(K159A)-pET28a | His-NleH1 K159A mutant | 6 |
| NleH2(K169A)-pET28a | His-NleH2 K169A mutant | 6 |
| 6-PFK-pET28a | His-6-PFK | This study |
| HA | HA fusion expression for transfection | 6 |
| EHEC NleH1-HA | EHEC NleH1 fused to HA | 6 |
| EHEC NleH2-HA | EHEC NleH2 fused to HA | 6 |
| EPEC NleH1-HA | EPEC NleH1 fused to HA | This study |
| EPEC NleH2-HA | EPEC NleH2 fused to HA | This study |
| VN | Venus fluorescence protein (aaa 1–173) | 6 |
| VC | Venus fluorescence protein (aa 155–238) | 6 |
| VN-actin | Venus 1–173 fused to human actin | 6 |
| VC-actin | Venus 155–238 fused to human actin | 6 |
| VN-NleH1 | Venus 1–173 fused to EHEC NleH1 | 6 |
| VC-NleH1 | Venus 155–238 fused to EHEC NleH1 | 6 |
| VN-NleH2 | Venus 1–173 fused to EHEC NleH2 | 6 |
| VC-NleH2 | Venus 155–238 fused to EHEC NleH2 | 6 |
| pET42a | Bacterial GST fusion protein expression | Novagen |
| NleH1-pET42a | GST-NleH1 | This study |
| NleH2-pET42a | GST-NleH2 | This study |
| pFLAG-CTC | Bacterial FLAG fusion protein expression | Sigma |
| NleH1-pFLAG-CTC | NleH1-FLAG | 6 |
| NleH2-pFLAG-CTC | NleH2-FLAG | 6 |
| NleH1(K159A)-pFLAG-CTC | NleH1(K159A)-FLAG | 6 |
| NleH2(K169A)-pFLAG-CTC | NleH2(K169A)-FLAG | 6 |
| FLAG NleH1 | NleH1-FLAG fusion for transfection | This study |
| FLAG NleH2 | NleH2-FLAG fusion for transfection | This study |
| FLAG NleH1 ΔLSKI | NleH1 ΔLSKI-FLAG fusion for transfection | This study |
| FLAG NleH2 ΔLSKI | NleH2 ΔLSKI-FLAG fusion for transfection | This study |
aa, amino acids.
Protein purification.
NleH1 and NleH2 were cloned into either pET-28a (His) or pET-42a (GST) and expressed in E. coli BL21(DE3). Bacterial cultures were grown to an optical density at 600 nm (OD600) of 0.2 to 0.5, at which time IPTG was added (1 mM) and cells were grown for 2 additional hours. Cells were pelleted and lysed in either His lysis buffer or GST Bugbuster protein extraction reagent (Novagen). After centrifugation, the supernatants were applied to either PerfectPro Ni-NTA agarose or glutathione bead slurries and incubated at 4°C overnight. After the epitope-tagged proteins were washed and eluted, purity was analyzed using 12% SDS-PAGE.
Gel overlays.
GST-NleH1 and NleH2 (5 μg) were electrophoresed, transferred to nitrocellulose, and sequentially washed first in 50 mM Tris (pH 8.0)–20% isopropanol, then in 50 mM Tris (pH 8.0)–6 M guanidine HCl, and finally in 50 mM Tris (pH 8.0)–0.05% Tween 20. Membranes were blocked in Odyssey blocking buffer and then overlaid with 2.5 μg of purified His-RPS3. The transferred and overlaid proteins were then probed using appropriate primary and secondary antibodies. After rinsing in phosphate-buffered saline (PBS), blots were imaged using an Odyssey infrared imaging system.
ELISAs.
Immulon-2 96-well plates (Dynatech) were coated with 2 μg His-NleH1 or His-NleH2 and incubated at 37°C for 1 h. Plates were washed with 0.05% PBS-Tween and blocked in 5% (wt/vol) milk in PBS-Tween. After washing, the plates were overlaid with various amounts of GST-tagged proteins (RPS3, NleH1, and NleH2) and incubated at 37°C for 1 h. After incubating with primary and secondary antibodies, the plates were developed with 1-Step Ultra TMB-ELISA solution (Thermo Scientific) and then quenched with 2 M H2SO4. Absorbance at 450 nm was measured. Competitive enzyme-linked immunosorbent assays (ELISAs) utilized a 5-fold molar excess of His-NleH1 or titrations of either His-NleH1 or a nonspecific competitor, His-6-phosphofructokinase (His-6-PFK). Kd values were estimated, using Kaleidagraph, through curve fitting to the following equation, where θ is the fractional occupancy of the prebound NleH substrate: θ = ligand/(ligand +Kd).
Immunoblotting.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris [pH 8.0], 0.5% sodium deoxycholate, 0.1% SDS, 1% Nonidet P-40 [NP-40]), incubated on ice for 30 min, and centrifuged. Equal amounts of protein from the supernatants were separated by SDS-PAGE, transferred to nitrocellulose membranes, blocked in Odyssey blocking buffer (Li-Cor), and then probed with appropriate primary and secondary antibodies. After rinsing in PBS, blots were imaged using an Odyssey infrared imaging system.
Nuclear fractionation.
Cytosolic and nuclear protein extracts were prepared from 293T cells transfected with NleH plasmids, as previously described (6). TNF stimulation (20 ng/ml, 30 min) (Cell Signaling) was used to promote RPS3 translocation into the nucleus. Data were analyzed by Western blotting for nuclear RPS3. PARP was used to normalize the nuclear protein concentrations.
Luciferase assays.
293T cells were cotransfected with a firefly luciferase construct driven by a consensus κB site, together with the renilla luciferase pTKRL plasmid, and with NleH expression plasmids as indicated in the figure legends. Transfected cells were cultured for 36 h and then stimulated with TNF (20 ng/ml, 30 min). Cells were lysed with passive lysis buffer, and lysates were analyzed using the Dual-Luciferase kit (Promega), with firefly fluorescence units (FU) normalized to renilla FU. Luciferase assays were performed in triplicate with at least three independently transfected cell populations.
Mouse infections.
Four-to-six-week-old C3H/HeJ and C57BL/6 mice were kept in sterilized cages with filter tops, handled in tissue culture hoods, and fed autoclaved food and water under specific-pathogen-free conditions. Mice were infected by oral gavage with 5.0 × 109 CFU of overnight cultures of C. rodentium in 100 μl of LB broth. Mice were monitored twice daily for up to 1 week. At necropsy, the first 4 cm of each colon, beginning at the anal verge, was collected. Fecal pellets were removed before colonic tissue was weighed. For viable bacterial count studies, colons and fecal pellets were collected in ice-cold PBS. Colonic tissues were homogenized, serially diluted, and plated onto MacConkey agar. Bacterial colonies were enumerated the following day. Tissues were collected in PBS for viable bacterial count studies or transferred to TRIzol reagent (Gibco), frozen in liquid N2, and stored at −80°C until required for RNA analysis.
RT-PCR.
cDNA was prepared from 1 μg RNA using the Superscript First Strand system (Invitrogen) with oligo(dT) primer. Real-time PCR (RT-PCR) was performed in triplicate using a SYBR green PCR Master Mix (Ambion) in a Fast 7500 sequence detection system (Applied Biosystems). Relative transcription levels were calculated using the ΔΔCT method.
Statistical analyses.
RPS3 translocation, luciferase assays, ELISAs, and bimolecular fluorescence complementation (BiFC) data were analyzed statistically using one-way analysis of variance (ANOVA). C. rodentium colonization and mouse survival data were analyzed using the Kruskal-Wallis and log rank tests, respectively. RT-PCR data were analyzed with unpaired t tests. P values of <0.05 were considered significant.
RESULTS AND DISCUSSION
EHEC and EPEC NleH effectors have similar functions.
Bacterial infection ultimately results in the induction of an inflammatory response after the host recognizes pathogen-associated molecular patterns (PAMPs). Toll-like receptor (TLR) signaling causes activation of the IκB kinase complex (IKK), which phosphorylates IκBα, subsequently promoting its ubiquitination and degradation, liberating NF-κB for nuclear translocation. Bacterial pathogens have evolved T3SS effectors that inhibit NF-κB activation to reduce proinflammatory cytokine expression.
RPS3 was identified as a non-Rel NF-κB subunit that confers regulatory specificity on NF-κB (26). Knockdown of RPS3 alters the expression of a subset of NF-κB-dependent genes (26). For example, p65 binding to κB sites in the IκBα and interleukin-8 (IL-8) gene promoters is reduced in RPS3 knockdown cells (26), whereas RPS3-independent genes (e.g., CD25 and CD69) are unaffected. Moreover, RPS3 was recently identified as a novel substrate for IKKβ (27). Although bearing no consensus IKK recognition motif, RPS3 was phosphorylated at a casein kinase (CK2) recognition motif through the alternative substrate specificity of IKKβ (27). Phosphorylation of RPS3 S209 promotes its nuclear translocation, coordinating the nuclear import of this subunit with the p50-p65 NF-κB heterodimer (27).
Some debate has emerged as to the mechanism by which the NleH effectors present in A/E pathogens inhibit NF-κB activation. We previously showed that EHEC NleH1 and NleH2 bind to the N terminus of RPS3 (6). NleH1, but not NleH2, inhibits the nuclear translocation of RPS3 by reducing the extent of RPS3 S209 phosphorylation by IKKβ (27). However, based upon NleH sequence similarity to that of the Shigella OspG effector, a protein kinase that binds to ubiquitinylated ubiquitin-conjugating enzymes (E2s) to block IκBα degradation (10), and upon experimental data obtained using EPEC (21), it has been suggested that both NleH1 and NleH2 may instead inhibit IκBα phosphorylation/degradation (21). Others have also shown that both EPEC NleH1 and NleH2 can inhibit NF-κB activity when IKKβ is overexpressed (21), calling into question our previous data indicating that NleH1 and NleH2 have opposing functions (6).
To begin to resolve the potential discrepancies in these data, we first examined whether the EHEC and EPEC NleH effectors have similar functions. We quantified RPS3 nuclear translocation in the presence of either EHEC or EPEC NleH effectors after stimulating 293T cells with TNF. We also quantified PARP to normalize nuclear protein concentrations. As expected, EHEC NleH1, but not EHEC NleH2, inhibited RPS3 translocation (Fig. 1A and B). Similarly, EPEC NleH1, but not EPEC NleH2, also inhibited RPS3 translocation (Fig. 1A and B). Thus, we conclude that the EHEC and EPEC NleH effectors are functionally equivalent in their ability to affect RPS3 nuclear translocation.
Fig 1.

EHEC and EPEC NleH proteins have similar functions. (A) RPS3 nuclear translocation. Immunoblotting of cytoplasmic and nuclear fractions of 293T cells transfected with EHEC or EPEC NleH1, NleH2, or an HA-epitope control, with (+) or without (−) TNF (20 ng/ml; 30 min). Blots were probed with RPS3, HA, tubulin, and PARP monoclonal antibodies. (B) Nuclear RPS3. Quantification (n = 4) of the fold increase in nuclear RPS3 after TNF treatment. RPS3 signal intensity was normalized to nuclear PARP. Asterisks indicate significant differences compared with HA transfection. (C) NF-κB activity. Relative NF-κB activity determined using luciferase reporter assays in 293T cells transfected with the indicated NleH plasmids (n = 3). Cells were stimulated with TNF (20 ng/ml; 30 min) or cotransfected with IKKβ, as indicated. Cells were also transfected with a firefly luciferase construct driven by a consensus κB site and with a renilla luciferase plasmid. Asterisks indicate significantly different pairwise comparisons between TNF and IKKβ stimulations. (D) Neither NleH1 nor NleH2 alters IκBα dynamics. Immunoblot analysis of total and phosphorylated IκBα after TNF (20 ng/ml; 30 min) treatment in the presence of indicated NleH constructs. (E) NleH2 does not inhibit RPS3 phosphorylation. Immunoblotting of HeLa cell lysates after a 3-h infection with C. rodentium strains complemented with the indicated EHEC NleH expression plasmids. Cells were treated with TNF (20 ng/ml; 30 min) prior to harvest. (F) PDZ-binding motif of NleH regulates its function in innate immunity. Relative NF-κB activity determined using luciferase reporter assays in 293T cells transfected with the indicated NleH plasmids. Experiments were conducted as described for panel C. Asterisks indicate significantly different pairwise comparisons between wild-type and ΔLSKI constructs.
We next evaluated the functional significance of inhibiting RPS3 nuclear translocation on NF-κB activity. To do this, we quantified NF-κB activation using a luciferase reporter proven responsive to RPS3 (6). As expected, EHEC NleH1, but not EHEC NleH2, inhibited NF-κB activity after TNF stimulation (Fig. 1C). Similarly, EPEC NleH1, but not EPEC NleH2, also inhibited NF-κB activity (Fig. 1C).
We next determined whether the previously reported differences in NleH activities (6, 21) might be attributable to the mechanism used to stimulate the NF-κB pathway. We therefore also evaluated NF-κB luciferase reporter activity in the context of IKKβ overexpression. In this case, all NleH effectors (both NleH1 and NleH2 from both EHEC and EPEC) functioned as inhibitors of NF-κB (Fig. 1C), consistent with other reports (21). Thus, we conclude that while NleH1 and NleH2 have strikingly differing abilities to inhibit NF-κB following TNF stimulation, they both function as inhibitors of the pathway when IKKβ is overexpressed (Fig. 1C) (21). The biochemical basis for this difference in NleH activities depending upon the mechanism of NF-κB activation is currently unknown.
It has also been reported that rather than, or in addition to, targeting RPS3, EPEC NleH1 and NleH2 alter IκBα phosphorylation/degradation to more generally subvert the NF-κB pathway (21). We also evaluated the kinetics of IκBα stability and phosphorylation after stimulating 293T cells with TNF. As expected, TNF induced the rapid phosphorylation and subsequent degradation of IκBα (Fig. 1D). In agreement with our previous studies (6, 27), but in disagreement with results from others (21), we did not observe an ability of any NleH effector to alter the kinetics of IκBα phosphorylation/degradation (Fig. 1D).
We previously showed that IKKβ-mediated phosphorylation of RPS3 S209 was inhibited by the kinase activity of NleH1 (27). To determine whether EHEC NleH2 also alters RPS3 phosphorylation, we complemented C. rodentium ΔnleH with EHEC NleH2 or the kinase mutant NleH2(K169A). As expected, TNF significantly increased RPS3 S209 phosphorylation and was inhibited by C. rodentium expressing EHEC NleH1 (Fig. 1E). Consistent with its mild stimulatory activity toward NF-κB, complementing C. rodentium ΔnleH with EHEC NleH2 elevated the extent of RPS3 S209 phosphorylation. Mutating the active-site lysine (K169) to alanine (K169A) reduced RPS3 phosphorylation levels to those observed in cells infected with ΔnleH (Fig. 1E). Thus, the kinase activity of NleH2 is also essential for its ability to stimulate RPS3 phosphorylation.
It was previously shown that the PDZ-binding motif of NleH1 is responsible for its interaction with NHERF2 (12). To test the hypothesis that NleH1/NleH2 recruitment to other host proteins via their PDZ-binding motif might contribute to differences in their activities against NF-κB, we assessed the activities of wild-type and mutant forms of NleH1 and NleH2 in which we deleted the C-terminal PDZ-binding domain (residues 290 to 293 in NleH1 and 300 to 303 in NleH2 [ΔLSKI]). Transfecting these constructs and performing luciferase assays indicated that the PDZ-binding motif was indeed essential for NleH function in the NF-κB pathway. Deleting LSKI from the C terminus of NleH1 (NleH1 ΔLSKI) substantially blocked its ability to function as an NF-κB inhibitor (Fig. 1F). Remarkably, NleH2 ΔLSKI inhibited NF-κB luciferase activity to a magnitude similar to that of wild-type NleH1. These data suggest that the PDZ-binding domain may contribute to functional differences between NleH1 and NleH2 in regulating NF-κB activation.
NleH1 and NleH2 bind to each other in vitro and in vivo.
Because of the overlapping substrate specificity yet opposing functions of NleH1 and NleH2 (6), we considered the possibility that these two effectors might also interact with one another, providing E. coli with an additional mechanism by which to regulate the extent and duration of NF-κB activation via RPS3 interaction. We expressed and purified both GST- and His-tagged forms of NleH1 and NleH2, as well as GST-RPS3 (Fig. 2A). Using gel overlays, we first verified the specific binding of RPS3 to both NleH1 and NleH2 (Fig. 2B). We then quantified NleH binding interactions using ELISAs. As expected, GST-RPS3 bound to immobilized His-NleH1 in a concentration-dependent manner (Fig. 2C), with an apparent dissociation constant (Kd) of 17 ± 5 μM under the conditions of the ELISA (Table 2). Notably, we also observed that GST-NleH2, but not GST-NleH1, was able to bind His-NleH1.
Fig 2.

NleH1 and NleH2 bind to each other. (A) Purification of GST- and His-tagged proteins. RPS3, NleH1, and NleH2 were expressed as GST and His fusions and purified using affinity chromatography. Protein purity was assessed by Coomassie blue staining after electrophoresis through 12% SDS-PAGE. (B) Gel overlays of NleH and RPS3. GST-NleH1 and NleH2 (5 μg) were resolved by SDS-PAGE, transferred to nitrocellulose, and overlaid with 2.5 μg of purified His-RPS3. (C) Binding to NleH1 in vitro. Immulon-2 plates were coated with 2 μg His-NleH1. Coated plates were overlaid with increasing amounts of GST, GST-RPS3, GST-NleH1, and GST-NleH2. GST fusion protein binding was detected using an anti-GST antibody and 1-Step Ultra TMB-ELISA solution. Absorbance at 450 nm was measured. The results are shown as the means ± standard errors (n = 4). The asterisk indicates significant differences in binding to His-NleH1 compared with the GST control. (D) Binding to NleH2 in vitro. Experiments were conducted as described for panel C, except that plates were coated with 2 μg His-NleH2. The asterisk indicates significant differences in binding to His-NleH1 compared with the GST control. (E) Competitive ELISAs using excess NleH1. RPS3 binding to His-NleH1 was assessed in the presence of a 5-fold molar excess of NleH1 during RPS3 titrations. The asterisk indicates significant reduction in RPS3 binding due to the excess soluble NleH1. (F) Competitive ELISAs using specific versus nonspecific competitors. RPS3 binding to His-NleH1 was assessed in the presence of increasing amounts of either NleH1 or 6-phosphofructose kinase. The asterisk indicates significant differences in RPS3 binding due to competition from NleH1, compared with competition from 6-PFK. (G) BiFC. Relative fluorescence intensity after cotransfecting the indicated NleH1- and NleH2-eYFP plasmid combinations (n = 3). Asterisks indicate significantly increased fluorescence compared with Venus fluorescence protein controls (VN + VC). (H) Cotransfection. Relative NF-κB activity determined using luciferase reporter assays in 293T cells cotransfected with the indicated molar ratios of NleH plasmids (n = 3). Asterisks indicate significantly different pairwise comparisons.
Table 2.
Binding affinities of RPS3 and NleH proteins for either NleH1 or NleH2a
| Protein |
Kd (μM) for prebound: |
|
|---|---|---|
| His-NleH1 | His-NleH2 | |
| GST | ND | ND |
| GST-RPS3 | 17 ± 5 | 15 ± 4 |
| GST-NleH1 | ND | 28 ± 8 |
| GST-NleH2 | 25 ± 4 | 24 ± 6 |
Dissociation constants (Kd) were estimated through curve fitting to the equation θ = ligand/(ligand +Kd), as described in Materials and Methods, and are given as means ± standard deviations (n = 4). ND, not detected.
We also quantified protein-protein interactions using prebound His-NleH2 (Fig. 2D). As expected, GST-RPS3 bound to immobilized His-NleH2 with an affinity similar to that of His-NleH1 (Table 2). Both GST-NleH1 and GST-NleH2 bound to His-NleH2, albeit with slightly lesser affinity than did RPS3. Thus, NleH2 is capable of forming both homotypic and heterotypic complexes with NleH1, whereas NleH1 forms only heterotypic complexes with NleH2.
To confirm the specificity of these interactions, we performed a subsequent experiment in which we used soluble NleH1 to compete for RPS3 binding with the prebound His-NleH1 substrate. Since NleH1 binds RPS3 but does not interact with itself, it can serve as a competitor to distinguish between specific versus nonspecific binding in ELISAs. Adding a 5-fold molar excess of NleH1 simultaneously with RPS3 caused a significant reduction in RPS3 binding to the His-NleH1 substrate (Fig. 2E). Similarly, titrating the amount of NleH1 competitor in the presence of a constant amount of RPS3 (0.8 μM), but not titrating an unrelated protein, 6-phosphofructokinase, decreased RPS3 binding to the prebound His-NleH1 (Fig. 2F).
We also used bimolecular fluorescence complementation (BiFC) assays to determine whether NleH1 and NleH2 interact when they are coexpressed in mammalian cells. In support of ELISA data, NleH1-NleH2 as well as NleH2-NleH2 coexpression, but not NleH1-NleH1 coexpression, reconstituted two fragments of the enhanced yellow fluorescent protein (eYFP), resulting in quantifiable increases in fluorescence (Fig. 2G). Thus, we conclude that NleH1 and NleH2 are capable of binding to each other in vitro and in vivo. To assess the biological relevance of NleH1 and NleH2 interaction, we performed NF-κB luciferase assays after TNF stimulation with cells cotransfected with different molar ratios of NleH1 and NleH2. Whereas cells expressing a 4:1 ratio of NleH1/NleH2 displayed little NF-κB luciferase activity, this trend was reversed by increasing the molar ratio of NleH2 (Fig. 2H). While the underlying functional significance of these interactions is currently unclear, they raise the intriguing possibility that interactions between NleH1 and NleH2, and perhaps other effectors targeting the NF-κB pathway, after their translocation into host cells, may profoundly impact the extent and duration of host innate responses to bacterial infection.
E. coli O157:H7 NleH proteins alter C. rodentium colonization and mouse survival.
We performed infection experiments with C. rodentium strains complemented with either EHEC NleH1 or NleH2 to determine if their demonstrated in vitro differences would also contribute to differences in bacterial virulence. Consistent with other reports, the virulence of C. rodentium ΔnleH was attenuated compared with that of wild-type C. rodentium in mouse infections (7, 9). Wild-type C. rodentium caused severe disease in C3H/HeJ mice, to the extent that all mice succumbed to infection within 5 days (Fig. 3A). In contrast, deleting nleH from C. rodentium delayed and reduced mouse mortality. Complementing ΔnleH with NleH1 restored Citrobacter virulence to wild-type levels, whereas complementing with NleH2 reduced Citrobacter virulence. Consistent with our in vitro data, the kinase activity of NleH was essential for maximal Citrobacter virulence in C3H/HeJ mice. Mice infected with C. rodentium ΔnleH complemented with NleH1(K159A) displayed significantly increased survival over the time course of infection (Fig. 3A).
Fig 3.
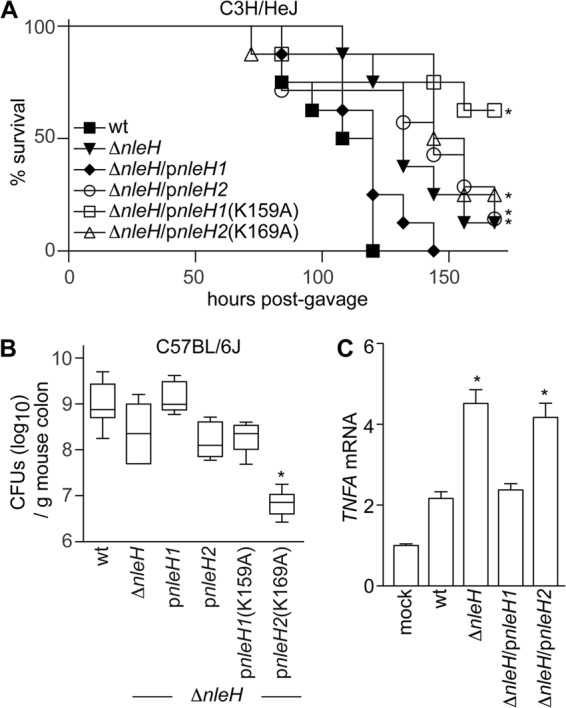
Expressing EHEC NleH effectors alters Citrobacter virulence. (A) C3H/HeJ survival. The survival (percentage of initial population) of C3H/HeJ mice is plotted as a function of time after oral gavage with indicated C. rodentium strains (n = 7 or 8/group). Asterisks indicate significantly different survival rates compared with wild-type infection. (B) Citrobacter colonization. Colonization (CFU/g mouse colon) of indicated C. rodentium strains (7 days postgavage) in C57BL/6J mice (n = 8/group). The asterisk indicates significantly different colonization magnitudes. (C) TNF expression. Relative TNF mRNA expression levels from colons obtained from C57BL/6J mice (7 days postgavage) infected with the indicated C. rodentium strains. Asterisks indicate significantly different TNF expressions compared with wild-type infection.
Infecting C57BL/6J mice, a strain that is relatively resistant to Citrobacter, indicated that Citrobacter colonization was reduced ∼10-fold upon deleting NleH (Fig. 3B). Whereas complementing ΔnleH with NleH1 restored Citrobacter colonization to wild-type levels, complementing with NleH2 had no measurable impact (Fig. 3B). Furthermore, complementing ΔnleH with NleH1(K159A) did not enhance bacterial colonization, further emphasizing the importance of NleH kinase activity to bacterial virulence. Interestingly, complementing ΔnleH with NleH2(K159A) severely dampened Citrobacter colonization.
We also quantified changes in mouse TNF gene transcription (TNFA) using RT-PCR from RNA isolated from C57BL/6J mouse colons 7 days after infection. Deleting C. rodentium NleH significantly increased host TNF transcription, compared with TNF expression in mice infected with wild-type C. rodentium (Fig. 3C). Again, complementing ΔnleH with EHEC NleH1, but not with NleH2, restored mouse TNF transcription to levels similar to those in mice infected with wild-type C. rodentium, indicating that NleH1 plays a key role in regulating the NF-κB/TNF response in vivo. Thus, differences between NleH1 and NleH2 are clearly evident from our in vivo studies using C. rodentium, and similar to what is shown by in vitro data, the kinase activities of the NleH effectors are essential in modulating host phenotypes in vivo.
ACKNOWLEDGMENTS
This work was supported by the U.S. National Institutes of Health (AI087686 and AI076227 to P.R.H. and CA137171 to F.W.).
We thank Minzhao Huang for technical assistance.
Footnotes
Published ahead of print 26 March 2012
REFERENCES
- 1. Baruch K, et al. 2011. Metalloprotease type III effectors that specifically cleave JNK and NF-kappaB. EMBO J. 30:221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coombes BK, et al. 2008. Molecular analysis as an aid to assess the public health risk of non-O157 Shiga toxin-producing Escherichia coli strains. Appl. Environ. Microbiol. 74:2153–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cornelis GR. 2010. The type III secretion injectisome, a complex nanomachine for intracellular ‘toxin’ delivery. Biol. Chem. 391:745–751 [DOI] [PubMed] [Google Scholar]
- 4. Deng W, Li Y, Vallance BA, Finlay BB. 2001. Locus of enterocyte effacement from Citrobacter rodentium: sequence analysis and evidence for horizontal transfer among attaching and effacing pathogens. Infect. Immun. 69:6323–6335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deng W, et al. 2004. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc. Natl. Acad. Sci. U. S. A. 101:3597–3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gao X, et al. 2009. Bacterial effector binding to ribosomal protein s3 subverts NF-kappaB function. PLoS Pathog. 5:e1000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garcia-Angulo VA, Deng W, Thomas NA, Finlay BB, Puente JL. 2008. Regulation of expression and secretion of NleH, a new non-locus of enterocyte effacement-encoded effector in Citrobacter rodentium. J. Bacteriol. 190:2388–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hemrajani C, et al. 2010. NleH effectors interact with Bax inhibitor-1 to block apoptosis during enteropathogenic Escherichia coli infection. Proc. Natl. Acad. Sci. U. S. A. 107:3129–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hemrajani C, et al. 2008. Role of NleH, a type III secreted effector from attaching and effacing pathogens, in colonization of the bovine, ovine, and murine gut. Infect. Immun. 76:4804–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim DW, et al. 2005. The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. U. S. A. 102:14046–14051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levine MM, et al. 1978. Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet i:1119–1122 [DOI] [PubMed] [Google Scholar]
- 12. Martinez E, et al. 2010. Binding to Na(+)/H(+) exchanger regulatory factor 2 (NHERF2) affects trafficking and function of the enteropathogenic Escherichia coli type III secretion system effectors Map, EspI and NleH1. Cell. Microbiol. 12:1718–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. 1995. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. U. S. A. 92:1664–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mead PS, et al. 1999. Food-related illness and death in the United States. Emerg. Infect. Dis. 5:607–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muhlen S, Ruchaud-Sparagano MH, Kenny B. 2011. Proteasome-independent degradation of canonical NFkappaB complex components by the NleC protein of pathogenic Escherichia coli. J. Biol. Chem. 286:5100–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nadler C, et al. 2010. The type III secretion effector NleE inhibits NF-kappaB activation. PLoS Pathog. 6:e1000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Newton HJ, et al. 2010. The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kappaB p65. PLoS Pathog. 6:e1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pearson JS, Riedmaier P, Marches O, Frankel G, Hartland EL. 2011. A type III effector protease NleC from enteropathogenic Escherichia coli targets NF-kappaB for degradation. Mol. Microbiol. 80:219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Petty NK, et al. 2010. The Citrobacter rodentium genome sequence reveals convergent evolution with human pathogenic Escherichia coli. J. Bacteriol. 192:525–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robinson KS, et al. 2010. The enteropathogenic Escherichia coli effector NleH inhibits apoptosis induced by Clostridium difficile toxin B. Microbiology 156:1815–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Royan SV, et al. 2010. Enteropathogenic E. coli non-LEE encoded effectors NleH1 and NleH2 attenuate NF-kappaB activation. Mol. Microbiol. 78:1232–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ruchaud-Sparagano MH, Muhlen S, Dean P, Kenny B. 2011. The enteropathogenic E. coli (EPEC) Tir effector inhibits NF-kappaB activity by targeting TNFalpha receptor-associated factors. PLoS Pathog. 7:e1002414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schauer DB, Falkow S. 1993. Attaching and effacing locus of a Citrobacter freundii biotype that causes transmissible murine colonic hyperplasia. Infect. Immun. 61:2486–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shames SR, et al. 2011. The pathogenic Escherichia coli type III secreted protease NleC degrades the host acetyltransferase p300. Cell. Microbiol. 13:1542–1557 [DOI] [PubMed] [Google Scholar]
- 25. Tzipori S, Sheoran A, Akiyoshi D, Donohue-Rolfe A, Trachtman H. 2004. Antibody therapy in the management of Shiga toxin-induced hemolytic uremic syndrome. Clin. Microbiol. Rev. 17:926–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wan F, et al. 2007. Ribosomal protein S3: a KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell 131:927–939 [DOI] [PubMed] [Google Scholar]
- 27. Wan F, et al. 2011. IKKbeta phosphorylation regulates RPS3 nuclear translocation and NF-kappaB function during infection with Escherichia coli strain O157:H7. Nat. Immunol. 12:335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]