

Abstract
It is known that Ca2+ influx plays an important role in the modulation of inositol trisphosphate-generated Ca2+ oscillations, but controversy over the mechanisms underlying these effects exists. In addition, the effects of blocking membrane transport or reducing Ca2+ entry vary from one cell type to another; in some cell types oscillations persist in the absence of Ca2+ entry (although their frequency is affected), whereas in other cell types oscillations depend on Ca2+ entry. We present theoretical and experimental evidence that membrane transport can control oscillations by controlling the total amount of Ca2+ in the cell (the Ca2+ load). Our model predicts that the cell can be balanced at a point where small changes in the Ca2+ load can move the cell into or out of oscillatory regions, resulting in the appearance or disappearance of oscillations. Our theoretical predictions are verified by experimental results from HEK293 cells. We predict that the role of Ca2+ influx during an oscillation is to replenish the Ca2+ load of the cell. Despite this prediction, even during the peak of an oscillation the cell or the endoplasmic reticulum may not be measurably depleted of Ca2+.
In response to an increased concentration of inositol trisphosphate (IP3), oscillations in the concentration of free intracellular calcium (Ca2+) occur in many cell types and are important for the control of many cellular functions (1–4). In nonexcitable cells, such as epithelial cells, these oscillations occur as the result of Ca2+ flux into and out of the endoplasmic reticulum (ER). However, although the oscillations result from the cycling of Ca2+ between the ER and the cytoplasm, the transport of Ca2+ across the cell membrane can have a dramatic effect on these oscillations.
Although Ca2+ influx is known to be important, disagreement exists, first, over the mechanisms by which Ca2+ influx is modulated during a Ca2+ oscillation, and, second, over the role played by Ca2+ influx. The capacitative entry hypothesis proposes that depletion of ER Ca2+ causes enhanced entry of Ca2+ across the plasma membrane (5–9). Often, although not necessarily, in this scenario, Ca2+ entry is necessary for the refilling of the ER, and thus oscillation frequency is controlled by the refilling time (10, 11). Although capacitative entry is certainly an important factor during the response to a maximal stimulus, other investigators have pointed to a lack of direct evidence that it plays an important role during smaller stimuli that generate oscillatory behavior (12). They maintain that Ca2+ influx, under these conditions, is controlled by a noncapacitative pathway involving arachidonic acid and that the purpose of the influx is to increase the likelihood that low levels of IP3 will induce Ca2+ release from internal stores (13). This controversy is complicated by the fact that, in some cell types, Ca2+ oscillations persist in the absence of Ca2+ influx (14–16), whereas in other cell types, oscillations depend absolutely on influx (17, 18). Even in a single cell type, the effect of Ca2+ influx on agonist-induced oscillations varies according to the agonist (19), and the effect of removal of extracellular Ca2+ depends on when it is removed (17).
Using a combination of theoretical and experimental work, we attempt to resolve this controversy by studying how Ca2+ transport across the cell membrane can affect ER-based oscillations. We conclude that small changes in Ca2+ load can have a large impact on Ca2+ oscillations. By Ca2+ load we mean the total amount of Ca2+ in the cell, including the ER, the cytoplasm, and the Ca2+ buffers. (For the purposes of the model here, we neglect mitochondrial Ca2+, but its inclusion would have no qualitative effect on our results.) The amount of Ca2+ in the ER is directly related to the Ca2+ load, and thus our model predicts that Ca2+ oscillations will also be highly sensitive to ER Ca2+ concentration.
Our results are independent of the mechanism by which Ca2+ influx is controlled and thus cannot be used to distinguish between capacitative entry or noncapacitative, arachidonic acid-dependent entry, but they provide a way in which seemingly contradictory experimental results can be understood. In addition, they show that direct control of Ca2+ influx by the cytosolic or ER Ca2+ concentration is not necessary for Ca2+ influx to play a crucial role in the control of oscillations. Finally, our results do not depend on the precise details of the model, but they occur in at least four different models of Ca2+ oscillations.
The Model
Our model for Ca2+ oscillations is based on a dynamic model of the IP3 receptor (IPR) (20) and assumes that oscillations in [IP3] are not necessary for the generation of Ca2+ oscillations. A schematic diagram of the cell is given in Fig. 1. From there we see that the conservation equations for Ca2+ are
 |
[1] |
 |
[2] |
where c and ce denote [Ca2+] in the cytoplasm and ER, respectively, γ is the cytoplasmic-to-ER volume ratio, and δ is a dimensionless parameter that controls the relative magnitudes of membrane and ER fluxes without changing the steady-state concentrations.
Fig. 1.

Schematic diagram of the model fluxes.
Calcium release through the IPR is described by the model of ref. 20, slightly modified for the context of a whole-cell model. Thus,
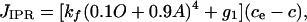 |
[3] |
where g1 models a constant background leak from the ER at zero [IP3]. O and A denote the probability the IPR is in the open and activated state, respectively, and are governed by differential equations with parameters determined by fitting to experimental data from the type II hepatocyte IPR (21).
Calcium pumping into the ER (Jserca) and extrusion across the plasma membrane (Jpm) are modeled by
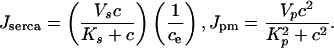 |
[4] |
These expressions are the ones used by ref. 22 and are discussed in detail there. Ca2+ entry is modeled as a linear, increasing function of [IP3] (p), and thus
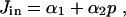 |
[5] |
where α1 and α2 are constants. This form for Jin is entirely arbitrary; we know that Jin must be an increasing function of p but do not know the exact relationship. Thus, we just choose the simplest possible form. Using a more complex function for Jin makes no qualitative difference to the model results.
Buffering is taken into account by assuming each flux to be an effective flux, scaled by the effects of fast linear buffers. Inclusion of slower nonlinear buffers will not alter the qualitative results of our analysis.
The parameter values are given in the legend to Fig. 2.
Fig. 2.

Blue lines are the bifurcation diagram of the closed-cell model with ct as the bifurcation parameter. Parameter values of the IPR model are taken from ref. 20. Other parameter values (adapted from ref. 22) are γ = 5.4, kf = 0.96 s–1, g1 = 0.002 s–1, Vs = 120 μM2·s–1, Ks = 0.18 μM, Vp = 28 μM·s–1, Kp = 0.42 μM, α1 = 0.03 μM·s–1, and α2 = 0.2 s–1. The dotted red line is a solution of the open-cell model superimposed on the bifurcation diagram of the closed-cell model and calculated by using p = 10 and δ = 0.01. ss, steady states; max osc, maximum of the oscillation.
Analysis of the Model
Introduction of the new variable, ct = c + (1/γ)ce gives the system
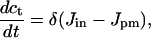 |
[6] |
 |
[7] |
The new variable ct is the total number of moles of Ca2+ in the cell divided by the cytoplasmic volume. We shall call ct the Ca2+ load of the cell. When δ is small, ct can change only slowly, and in the limit, as δ → 0, the cell becomes closed, i.e., when δ = 0 no Ca2+ exchange occurs with the external medium.
We study the model by treating ct as a bifurcation parameter and analyzing the model's behavior for a range of fixed values of ct, a procedure that was used by refs. 23–25 (for a general introduction, see ref. 26) in a classic study of bursting oscillations, and by refs. 27 and 28 in the context of Ca2+ oscillations in neurosecretory cells. If ct is constant, we replace ce by γ(ct – c) and consider just Eq. 1. We call this the closed-cell model. The bifurcation diagram is given in Fig. 2 (blue curve). Stable oscillations exist only for an intermediate range of values of ct between the homoclinic (HC) and saddle node of periodics (SNP) bifurcations. In other words, when Ca2+ transport across the plasma membrane is blocked, oscillations can occur only if neither too much nor too little Ca2+ is inside the cell.
When ct is allowed to vary, we obtain the open-cell model. A typical solution (drawn in the c, ct phase plane and superimposed on the closed-cell bifurcation diagram) is sketched in Fig. 2 (red dotted curve). Suppose that, when p = 0 and δ = 0.01, ct ≈ 12 at rest (see IPR_open.ode, which is published as supporting information on the PNAS web site). Raising p to 10 will cause immediate Ca2+ oscillations, with a gradually decreasing baseline as ct decreases (Fig. 2, red dotted line). These oscillations are pseudosteady; the solution is trying to follow the oscillatory solutions of the closed-cell model, but cannot exactly, because δ is nonzero. Once ct gets low enough, the oscillations change their character. During the interspike intervals, the solution approximately follows the lower stable steady state. (As δ → 0 the solution follows the lower stable steady state more closely. The agreement in Fig. 2 is not exact, but it is the general trend we are considering here.) Because the lower steady state is below the line where dct/dt = 0, ct is increasing there. Once the solution reaches the limit point (the bend) it can no longer follow this branch, and “falls off,” moving toward the long period limit cycle close to the HC orbit. However, once on the other side of the dct/dt = 0 line, ct starts to decrease, the solution returns to the lower stable steady state, and the cycle repeats.
Thus, in response to an increase in [IP3], the model cell exhibits transient pseudosteady oscillations that tend to a stable steady-state limit cycle, in which the oscillation is a degenerate bursting oscillation having only a single peak during each bursting phase. This slow modulation of the oscillations is caused by a gradual decrease in ct, and so the speed with which the model reaches the steady-state oscillation is governed by the value of δ, which controls the speed at which ct reaches a steady-state limit cycle.
If an open cell exhibits Ca2+ oscillations, how will these oscillations be affected if membrane transport is stopped? This is most easily studied with the bifurcation diagram shown in Fig. 3. The loci of HC and SNP bifurcations as p and ct vary in the closed-cell model are shown by the red curves. Thus, for each value of p, the closed-cell model will exhibit oscillations only when ct lies between these two curves. Superimposed on this we plot bifurcation diagrams of the open-cell model (broken curves), plotting the maximum and minimum of ct over an oscillation as a function of p. For each value of p where steady oscillations exist in the open-cell model, ct moves between the appropriate broken curves. (For instance, when δ = 0.1, the broken curves are labeled max and min, and denote, respectively, the maximum and minimum values of ct over an oscillation.) It follows that oscillations can occur in both the closed-cell and open-cell models only when the broken curves lie between the solid curves.
Fig. 3.

The broken lines are the maximum (max) and minimum (min) of oscillations in the open-cell model for different values of δ. The curves labeled HC and SNP are two-parameter continuations of the points labeled HC and SNP in Fig. 2. Thus, for each value of δ, oscillations exist in the closed-cell model only for values of ct between the HC and SNP curves.
Consider the behavior at p = 1 and δ = 0.01. If the open-cell model is allowed to reach its steady-state oscillation, ct will be moving between the blue short-dashed curves (over the point labeled A). If δ is now reduced to zero, i.e., membrane transport is shut off, ct will now be held constant at point A and will not lie between the HC and SNP curves. Hence, the closed-cell model cannot oscillate and the oscillations stop. To restore the oscillations, ct could be increased until it crosses the HC curve (point B). Alternatively, oscillations can be restored by increasing p (point C).
When p = 40 and δ = 0.01, we see that stable oscillations coexist in both the open-cell and closed-cell model, but for only a relatively small region (region D). If p and ct are such that the cell sits in region D, blockage of membrane transport will not eliminate Ca2+ oscillations. When δ = 0.001, i.e., when membrane transport in the open-cell model is very slow, the open-cell bifurcation curves (the broken lines) begin to superimpose on the HC curve of the closed-cell model, thus showing how, in the limit as δ → 0, the open-cell oscillations get progressively restricted to planes of constant ct. In addition, the p interval for which stable-steady oscillations coexist in the open-cell and the closed-cell models gets wider (region E).
From Fig. 2 we see that, immediately after an increase in [IP3], the open-cell model exhibits oscillations with a gradually decreasing baseline and with a decreasing average ct. This finding suggests that the effects of blocking membrane transport will depend on the timing of the block. If the block occurs soon after, or before, the increase in [IP3], oscillations can continue because ct has not yet fallen far enough to make oscillations impossible. However, if the block occurs later, after ct has decreased past HC, then the block will be expected to eliminate oscillations.
Influx of Ca2+ from outside the cell is known to affect the frequency of Ca2+ oscillations and waves. We investigate this in the open-cell model by varying the background rate of Ca2+ influx, α1. In Fig. 4 we plot lines in the α1 – p plane that separate regions where Ca2+ oscillations occur from regions where they do not. For a fixed p, an increase in Ca2+ influx will lead to oscillations or to a decrease in the oscillation period. Similarly, for a fixed α1, an increase in p will have the same effect. This occurs even when membrane transport is slow (δ = 0.001). Oscillations can be eliminated by a decrease in influx but can then be restored by increasing p.
Fig. 4.

Two-parameter bifurcation diagram of the open-cell model. The lines (for three different values of δ) divide the α1 – p plane into regions for which oscillations do or do not exist.
Physiological Interpretation of the Analysis
A model cell can fire a Ca2+ spike only if its Ca2+ load (ct) is above threshold. This is because enough cytosolic Ca2+ must be present to activate the IP3 receptor, and enough ER Ca2+ must be available to provide sufficient driving force for efflux from the ER. Once ct drops below threshold, these conditions are violated and the cell cannot fire a Ca2+ spike.
At rest, the model cell has a high resting Ca2+ load (ct). On agonist stimulation, the rise in cytosolic Ca2+ causes a net loss of Ca2+ from the cell (as it is removed by the plasma membrane pumps), even though the influx of Ca2+ has been increased. Thus, ct gradually declines, resulting in oscillations with a gradually decreasing baseline. If membrane transport is blocked during this phase of the response, ct is still high enough to maintain oscillations, which would then occur purely by recycling Ca2+ to and from the ER.
Eventually, ct declines to a new steady-state average level. Now, at the peak of each oscillation, the high cytosolic concentration causes a net loss of Ca2+ from the cell. The Ca2+ load thus drops below threshold, and the cell cannot fire another spike until this Ca2+ loss is restored by Ca2+ influx. Because each oscillation involves the loss and reuptake of Ca2+ from the cell (possibly only a small amount), these oscillations depend on Ca2+ influx and will be eliminated by blockage of membrane transport.
An increase in Ca2+ influx will increase oscillation frequency, as it increases the rate at which the Ca2+ lost during each spike is regained. In addition, an increase in Ca2+ influx will increase the average steady-state Ca2+ load, which will also increase oscillation frequency, even when the oscillations are not dependent on Ca2+ influx. A decrease in Ca2+ influx can eliminate oscillations, which can then be restored by an increase in [IP3].
When membrane transport is slow enough, Ca2+ oscillations can persist in the absence of membrane transport, although their frequency is affected by Ca2+ influx.
A closed cell needs a higher Ca2+ load to support an oscillation than does an open cell. In the closed cell, because the Ca2+ load is constant, oscillations are possible only if the Ca2+ load is always over threshold. In the open cell this constraint is relaxed. For a large part of the oscillatory cycle, the Ca2+ load can be below threshold. It is only when Ca2+ influx from the outside drives the Ca2+ load over threshold that a spike occurs. Some of the Ca2+ released from the ER is then pumped out of the cell, the Ca2+ load drops below threshold, and the cycle repeats. Thus, when averaged over one cycle, the Ca2+ load is lower for the open cell than for the closed cell. For open-cell oscillations, Ca2+ influx serves to increase the Ca2+ load to a level where another oscillation can occur, and an increase in Ca2+ influx increases the oscillation frequency. However, although it is cell refilling that is governing the oscillation period, the amount of Ca2+ actually lost from the cell during each oscillation can be small and not easily detectable.
From our analysis we predict the following:
Prediction 1. Blocking membrane transport can eliminate Ca2+ oscillations. An increase in the Ca2+ load of the cell will restore oscillations that have been eliminated by the block.
Prediction 2. The effects of blocking membrane transport will depend on the timing of the block. If the block occurs soon after, or before, the increase of [IP3], it will not eliminate oscillations, whereas if it occurs after the cell has reached a steady-state oscillation, it will eliminate the oscillations.
Experimental Verification
We tested our predictions experimentally in the following way. HEK293 cells were stably transfected with the m3 human muscarinic receptor and loaded with fluo-4. They were imaged on a TILL Photonics system with a monochromator (TILL Photonics, Planegg, Germany) and a progressive line-scan digital camera (Imago, Scientific Instruments, Madison, WI). The cells were stimulated with 0.5 μM carbamylcholine (CCh) to induce Ca2+ oscillations, then 1 mM La3+ was added to block both Ca2+ entry and the plasma membrane Ca2+ pump (29–33). Fig. 5 shows the effects of the addition of La3+ at two different times: after steady oscillations had appeared (Upper) and immediately before the addition of CCh (Lower). As predicted, oscillations were eliminated in the first case but not in the second (20 cells in four separate experiments). In the first case, the effect of La3+ was reversible. This result is similar to previous results showing that removal of extracellular Ca2+ before application of CCh to pancreatic acinar cells did not prevent oscillations, whereas removal of extracellular Ca2+ from a cell exhibiting Ca2+ oscillations terminated oscillations (17).
Fig. 5.

Addition of La3+ to HEK293 cells after addition of CCh eliminates oscillations (Upper), whereas addition of La3+ before CCh does not prevent oscillations (Lower).
In a second set of experiments, the cells were also loaded with caged Ca2+. Then, after the addition of La3+, intracellular Ca2+ was increased by uncaging the Ca2+ with the “strobe” mode on the TILL flash unit. This mode releases Ca2+ by brief low-energy flashes of UV light at 12 Hz. This strobing typically lasted for ≈1–1.5 min (Fig. 6). Then the La3+ was washed out, still in the presence of 0.5 μM CCh. A typical result (from seven cells in three separate experiments) is shown in Fig. 6. On addition of CCh, the cell exhibited Ca2+ oscillations, which terminated on blockage of membrane transport, even though the Ca2+ load of the cell remained unchanged by the addition of La3+. Thus, it is not the Ca2+ load alone that is controlling the oscillations, it is the combination of the Ca2+ load and the transport across the plasma membrane. An increase in ct by the strobe then leads to the resumption of Ca2+ oscillations, which persist even when the strobe is turned off. Thus, once ct is increased sufficiently, Ca2+ oscillations persist even in the absence of membrane transport, as predicted by the model. When membrane transport is restored by the removal of La3+, the oscillations continue.
Fig. 6.

Addition of CCh initiates oscillations that terminate on addition of La3+. Increasing ct by strobed photorelease of cytosolic Ca2+ restores oscillations even during continued blockage of membrane transport (blue lines). This is equivalent to moving from point A to point B in Fig. 3. The red lines show the effect of the strobe in the presence of La3+ alone (done in a separate experiment, but plotted here on the same graph for ease of comparison). Note how, when the strobe is removed, [Ca2+] does not decline until the La3+ is removed.
Application of the strobe in the absence of CCh, but the presence of La3+, resulted in a small increase in resting [Ca2+] (Fig. 6, red line). On removal of the strobe, [Ca2+] remained unchanged, because the La3+ prevented it from leaving the cell; on removal of the La3+, [Ca2+] returned to baseline. Application of the strobe in the absence of caged Ca2+ has no effect on oscillations (34).
Discussion
Our results suggest that membrane transport of Ca2+ controls oscillations by controlling the Ca2+ load of the cell. It is already well known that Ca2+ influx affects oscillation frequency in many cell types (10, 13, 35–37). In Xenopus oocytes, which have a very small surface-to-volume ratio and thus a small δ, Ca2+ oscillations persist in the absence of external Ca2+, as found in the model. Nevertheless, an increase in Ca2+ influx increases oscillation frequency and wave speed (16, 35). In pancreatic acinar cells application of acetylcholine gives rise to Ca2+ oscillations, which disappear on reduction of external Ca2+. However, these oscillations reappear when the concentration of acetylcholine is increased (38), as expected from the results in Fig. 4. The effects of Ca2+ load on oscillations and waves have also been well characterized in cardiac cells (39–41).
Our model is also consistent with the results of ref. 42, which showed that blockage of Ca2+ influx during the interspike period eliminated further spikes, whereas, if the block occurred during a spike, the spike occurred as usual. According to our model, this is because only a small amount of Ca2+ influx during the interspike period is needed to push the Ca2+ load of the cell over threshold, whereas the majority of the Ca2+ in a spike is released from the ER.
We have shown that it is not the Ca2+ load per se that is the controlling factor, but it is rather the interplay between Ca2+ load and membrane transport. In HEK293 cells, addition of CCh stimulates Ca2+ oscillations with a gradually decreasing baseline (Fig. 6). If membrane transport is then blocked, the oscillations terminate, even though the cell's Ca2+ load remains unchanged. A subsequent increase in the Ca2+ load restores oscillations (Fig. 6), even though membrane transport of Ca2+ still does not occur. Furthermore, the effects of blocking membrane transport depend on the timing of the block. If the block occurs while the Ca2+ load is high, i.e., before application of agonist, an increase in [IP3] will cause oscillations, whereas if the block occurs later, after the Ca2+ load has declined, agonist-induced oscillations will be eliminated (Fig. 5). Thus, our model can also explain previous puzzling results of ref. 17, which showed that removal of extracellular Ca2+ before the application of CCh did not eliminate oscillations, whereas, if Ca2+ oscillations had already developed, they were eliminated by the removal of extracellular Ca2+.
That small changes in Ca2+ load can have large effects on oscillations suggests a possible resolution of the controversy over the role of Ca2+ entry. Since experimental data have been unable to show conclusively that the ER is significantly depleted during an oscillation (43), it has been argued that ER depletion, and hence capacitative Ca2+ entry, does not play a significant role (12). In contrast, our model shows that cell depletion (i.e., a decrease in cytosolic and ER Ca2+) can play a crucial role, even when depletion is so small as to be unmeasurable. Our analysis also provides a consistent framework for understanding why removal of extracellular Ca2+ has such different effects, depending on the cell type, and it can explain why the timing of removal is so important (17, 42).
The same qualitative behavior occurs in at least three other models of Ca2+ oscillations: the Li–Rinzel model (44), and modified versions of the Atri model (45) and the LeBeau model (38) (computations not shown), all of which have different models for the IP3 receptor and Ca2+ pumps. This finding suggests that our predictions do not depend on the precise model details, but they are instead a generic feature of models that have the structure shown in Fig. 1. We hypothesize that every model to follow this basic structure will exhibit the same behavior, no matter what the exact details are of the various pumps and release processes.
Supplementary Material
Acknowledgments
We thank Robert T. Dirksen and Vivien Kirk for helpful discussions. J.S. and K.T.-A. were supported by the Marsden Fund of the Royal Society of New Zealand. D.I.Y. was supported by National Institutes of Health Grants DE14756 and DK54568. T.J.S. was supported by National Institutes of Health Grant GM40457.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: IP3, inositol (1,4,5)-trisphosphate; IPR, IP3 receptor; ER, endoplasmic reticulum; HC, homoclinic bifurcation; SNP, saddle node of periodics bifurcation; CCh, carbamylcholine.
References
- 1.Sanderson, M. J., Charles, A. C., Boitano, S. & Dirksen, E. R. (1994) Mol. Cell. Endocrinol. 98, 173–187. [DOI] [PubMed] [Google Scholar]
- 2.Clapham, D. (1995) Cell 80, 259–268. [DOI] [PubMed] [Google Scholar]
- 3.Thomas, A. P., Bird, G. S. J., Hajnóczky, G., Robb-Gaspers, L. D. & Putney, J. W. J. (1996) FASEB J. 10, 1505–1517. [PubMed] [Google Scholar]
- 4.Berridge, M. J. (1997) J. Physiol. 499, 291–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Putney, J. W., Jr. (1990) Cell Calcium 11, 611–624. [DOI] [PubMed] [Google Scholar]
- 6.Putney, J. W., Jr., Broad, L. M., Braun, F. J., Lievremont, J. P. & Bird, G. S. (2001) J. Cell Sci. 114, 2223–2229. [DOI] [PubMed] [Google Scholar]
- 7.Hoth, M. & Penner, R. (1992) Nature 355, 353–356. [DOI] [PubMed] [Google Scholar]
- 8.Hoth, M. & Penner, R. (1993) J. Physiol. 465, 359–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berridge M. J. (1995) Biochem. J. 312, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawanishi T., Blank, L. M., Harootunian, A. T., Smith, M. T. & Tsien, R. Y. (1989) J. Biol. Chem. 264, 12859–12866. [PubMed] [Google Scholar]
- 11.Berridge, M. J. (1993) Nature 361, 315–325. [DOI] [PubMed] [Google Scholar]
- 12.Shuttleworth, T. J. (1999) Cell Calcium 25, 237–246. [DOI] [PubMed] [Google Scholar]
- 13.Shuttleworth, T. J. & Thompson, J. L. (1996) Biochem. J. 313, 815–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rooney, T. A., Sass, E. J. & Thomas, A. P. (1989) J. Biol. Chem. 264, 17131–17141. [PubMed] [Google Scholar]
- 15.Lechleiter, J. D. & Clapham, D. E. (1992) Cell 69, 283–294. [DOI] [PubMed] [Google Scholar]
- 16.Girard, S. & Clapham, D. (1993) Science 260, 229–232. [DOI] [PubMed] [Google Scholar]
- 17.Yule, D. I. & Gallacher, D. V. (1988) FEBS Lett. 239, 358–362. [DOI] [PubMed] [Google Scholar]
- 18.Martin, S. C. & Shuttleworth, T. J. (1994) FEBS Lett. 352, 32–36. [DOI] [PubMed] [Google Scholar]
- 19.Yule, D. I., Lawrie, A. M. & Gallacher, D. V. (1991) Cell Calcium 12, 145–151. [DOI] [PubMed] [Google Scholar]
- 20.Sneyd, J. & Dufour, J. F. (2002) Proc. Natl. Acad. Sci. USA 99, 2398–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dufour, J.-F., Arias, I. M. & Turner, T. J. (1997) J. Biol. Chem. 272, 2675–2681. [DOI] [PubMed] [Google Scholar]
- 22.Sneyd, J., Tsaneva-Atanasova, K., Bruce, J. I., Straub, S. V., Giovannucci, D. R. & Yule, D. I. (2003) Biophys. J. 85, 1392–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rinzel, J. (1985) in Ordinary and Partial Differential Equations, eds. Sleeman, B. D. & Jarvis, R. J. (Springer, New York).
- 24.Rinzel, J. & Lee, Y. S. (1986) in Nonlinear Oscillations in Biology and Chemistry. Lecture Notes in Biomathematics, ed. Othmer, H. G. (Springer, New York), Vol. 66.
- 25.Rinzel, J. (1987) in Mathematical Topics in Population Biology, Morphogenesis, and Neurosciences. Lecture Notes in Biomathematics, eds. Teramoto, E. & Yamaguti, M. (Springer, Berlin), Vol. 71.
- 26.Keener, J. P. & Sneyd, J. (1998) Mathematical Physiology (Springer, New York).
- 27.Li,Y.-X., Stojilković, S. S., Keizer, J. & Rinzel, J. (1997) Biophys. J. 72, 1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sherman A. S., Li, Y.-X. & Keizer, J. E. (2002) in Computational Cell Biology, eds. Fall, C. P., Marland, E. S., Wagner, J. M. & Tyson, J. J. (Springer, New York).
- 29.Pandol, S. J., Schoeffield, M. S., Fimmel, C. J. & Muallem, S. (1987) J. Biol. Chem. 262, 16963–16968. [PubMed] [Google Scholar]
- 30.Kwan, C. Y., Takemura, H., Obie, J. F., Thastrup, O. & Putney, J. W., Jr. (1990) Am. J. Physiol. 258, C1006–C1015. [DOI] [PubMed] [Google Scholar]
- 31.Shimizu, H., Borin, M. L. & Blaustein, M. P. (1997) Cell Calcium 21, 31–41. [DOI] [PubMed] [Google Scholar]
- 32.Herscher, C. J. & Rega, A. F. (1997) Ann. N. Y. Acad. Sci. 834, 407–409. [DOI] [PubMed] [Google Scholar]
- 33.Mignen, O. & Shuttleworth, T. J. (2000) J. Biol. Chem. 275, 9114–9119. [DOI] [PubMed] [Google Scholar]
- 34.Wagner L. E., II, Li, W. H. & Yule, D. I. (2003) J. Biol. Chem., 10.1074/jbc.M306270200.
- 35.Yao, Y. & Parker, I. (1994) J. Physiol. 476, 17–28. [PMC free article] [PubMed] [Google Scholar]
- 36.Bootman, M. D., Young, K. W., Young, J. M., Moreton, R. B. & Berridge, M. J. (1996) Biochem. J. 314, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X., Liao, D. & Ambudkar, I. S. (2001) J. Membr. Biol. 181, 185–193. [DOI] [PubMed] [Google Scholar]
- 38.LeBeau, A. P., Yule, D. I., Groblewski, G. E. & Sneyd, J. (1999) J. Gen. Physiol. 113, 851–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng, H., Lederer, M. R., Lederer, W. J. & Cannell, M. B. (1996) Am. J. Physiol. 270, C148–C159. [DOI] [PubMed] [Google Scholar]
- 40.Satoh, H., Blatter, L. A. & Bers, D. M. (1997) Am. J. Physiol. 272, H657–H668. [DOI] [PubMed] [Google Scholar]
- 41.Bers, D. M., Li, L., Satoh, H. & McCall, E. (1998) Ann. N.Y. Acad. Sci. 853, 157–177. [DOI] [PubMed] [Google Scholar]
- 42.Thorn, P. (1995) J. Physiol. 482, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park, M. K., Petersen, O. H. & Tepikin, A. V. (2000) EMBO J. 19, 5729–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li, Y.-X. & Rinzel, J. (1994) J. Theor. Biol. 166, 461–473. [DOI] [PubMed] [Google Scholar]
- 45.Atri, A., Amundson, J., Clapham, D. & Sneyd, J. (1993) Biophys. J. 65, 1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.