

Abstract
The production of mycobactin (MBT) by Mycobacterium tuberculosis is essential for this bacterium to access iron when it is in an infected host. Due to this essential function, there is considerable interest in deciphering the mechanism of MBT assembly, with the goal of targeting select biosynthetic steps for antituberculosis drug development. The proposed scheme for MBT biosynthesis involves assembly of the MBT backbone by a hybrid nonribosomal peptide synthetase (NRPS)/polyketide synthase (PKS) megasynthase followed by the tailoring of this backbone by N6 acylation of the central l-Lys residue and subsequent N6-hydroxylation of the central N6-acyl-l-Lys and the terminal caprolactam. A complete testing of this hypothesis has been hindered by the inability to heterologously produce soluble megasynthase components. Here we show that soluble forms of the NRPS components MbtB, MbtE, and MbtF are obtained when these enzymes are coproduced with MbtH. Using these soluble enzymes we determined the amino acid specificity of each adenylation (A) domain. These results suggest that the proposed tailoring enzymes are actually involved in precursor biosynthesis since the A domains of MbtE and MbtF are specific for N6-acyl-N6-hydroxy-l-Lys and N6-hydroxy-l-Lys, respectively. Furthermore, the preference of the A domain of MbtB for l-Thr over l-Ser suggests that the megasynthase produces MBT derivatives with β-methyl oxazoline rings. Since the most prominent form of MBT produced by M. tuberculosis lacks this β-methyl group, a mechanism for demethylation remains to be discovered. These results suggest revisions to the MBT biosynthesis pathway while also identifying new targets for antituberculosis drug development.
INTRODUCTION
The bacterium Mycobacterium tuberculosis, the primary causative agent of tuberculosis (TB), infects one-third of the world's population and was responsible for 1.4 million deaths in 2010 (46). A significant challenge in treating M. tuberculosis infections is the evolution of strains that are resistant to first- and second-line drugs that are key components of TB treatment (37). To meet this challenge, new anti-TB drugs and new M. tuberculosis drug targets must be identified.
One new target for antitubercular drug development is the biosynthesis of mycobactin (MBT) siderophores in M. tuberculosis. These iron-chelating molecules are attractive drug targets because M. tuberculosis must survive in the phagosomes of macrophages, where it competes with the host for iron (4). Strains of M. tuberculosis that are unable to biosynthesize MBTs or import MBTs with chelated iron are impaired in their ability to grow in the lung or macrophages (35), demonstrating the importance of these molecules in causing disease. Furthermore, the addition of small molecules that disrupt MBT biosynthesis inhibited M. tuberculosis growth in iron-limiting media (10). These data provide strong support for targeting MBT biosynthesis for drug development.
The MBT siderophores are a family of nonribosomal peptide/polyketide hybrid natural products produced by many mycobacterial species and other actinomycetes (13, 30, 39). M. tuberculosis produces both membrane-associated and soluble forms of MBT that have structural differences at two locations (Fig. 1). The most significant structural difference is the variability of acyl group attached to the N6 of the central l-Lys residue. The membrane-associated MBTs have a long fatty acyl chain, which is capped by a methyl group, while the soluble forms of MBT contain a shorter fatty acyl chain, which is capped with either a carboxylate or methyl ester. The fatty acids may also contain desaturation between the α and β carbons. The second region of structural variation is at the β-carbon of the oxazoline ring. This carbon is unmethylated in the predominant MBT produced by M. tuberculosis, MBT T (38), and methylated in a fraction of the soluble MBTs (13). Recent work identified further structural variants of MBT that lack the hydroxyl groups on the N6 of both l-Lys residues (23, 27); thus they are deficient for two moieties that are needed for iron chelation. These molecules were not identified for their iron-chelating proficiency; rather, they were identified as molecules present in the cell wall of M. tuberculosis that stimulate T cell activation (27) or were detected using mass spectrometry (23). These findings were used to suggest that the N6-hydroxylations are the final steps in MBT biosynthesis. Interestingly, the nonhydroxylated structural variants that stimulated T cell activation contain oxazoline rings that are α-methylated, in contrast to MBT T. MBTs containing α-methylated oxazoline rings have also been identified from Nocardia sp. A32030 (43); thus, this form of MBT is also biosynthesized by other actinomycetes, suggesting that M. tuberculosis may also form α-methylated MBT derivatives.
Fig 1.

Chemical structures of the MBTs produced by M. tuberculosis. The membrane-associated MBTs are on the left; the soluble MBTs are on the right. The α and β designations denote the carbons of the oxazoline ring that may contain a methyl group.
The vital role of MBTs in M. tuberculosis physiology and virulence has drawn considerable interest in identifying the genes involved in MBT biosynthesis. Initial genome analysis identified a gene cluster (mbt-1) in M. tuberculosis that codes for salicylate biosynthesis (MbtI), N6-hydroxylation of l-Lys (MbtG), the hybrid nonribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) megasynthase that assembles MBT (MbtA to -F), and two proteins of unknown function (MbtJ and MbtH) (29). Subsequent genetic studies confirmed that this gene cluster was associated with MBT production in M. tuberculosis (4). Analyses of the homologous clusters from Mycobacterium smegmatis (3) and Nocardia farcinica (15) confirmed that they were involved in the biosynthesis of MBT and the analog nocabactin NA, respectively, providing additional support for the idea that these gene clusters are involved the production of MBT and MBT-like siderophores.
As stated above, the megasynthase is a hybrid NRPS/PKS. These enzymes are characterized by a repeating domain architecture that recognizes and covalently tethers each precursor to the megasynthase, followed by directional condensation of the precursors to generate the nonribosomal peptide/polyketide (5). For NRPSs, the core repeating domains include an adenylation (A) domain that recognizes a particular amino or aryl acid and activates it as an acyl-AMP intermediate. These domains are viewed as the “gatekeepers” of NRPS enzymology because they control which aryl or amino acid substrates are incorporated for each position in the peptide (41, 44). The activated carbonyl of the acyl-AMP intermediate is attached to the sulfhydryl of the 4′-phosphopantetheinyl prosthetic group of the neighboring carrier protein (CP) domain, forming an acyl-S-CP intermediate. Condensation (C) domains then catalyze directional amide bond formation between two neighboring acyl-S-CPs. In some cases modified C domains, called cyclization (Cy) domains, are found in place of the C domains and form oxazolidine or thiazoline rings by catalyzing both the condensation and cyclization of Ser, Thr, or Cys residues concomitant with peptide assembly (18, 24). These domains come together to form a module (C[Cy]-A-CP), and the number of modules typically corresponds to the number of acyl or amino acids in the assembled peptide (8). For PKSs, there is an analogous set of repeating domains: (i) acyltransferase (AT) domains that recognize specific thioesterified carboxylic acids and activate them as acyl-O-AT intermediates, (ii) CP domains to which the precursors become tethered via thioesterification, and (iii) ketosynthase (KS) domains that catalyze the directional condensation of CP-linked precursors (19). These core domains (KS-AT-CP) form a basic PKS module analogous to a module in NRPSs. The similarities between NRPS and PKS enzymology are what enables the fusion of these systems to form NRPS/PKS hybrids.
While the genetic analysis of mbt-1 has been fruitful, the same cannot be said for the biochemical analysis of the majority of the megasynthase components (MbtA to -F). The in vitro analysis has been limited to MbtA, an A domain that specifically activates salicylate, and the N-terminal CP domain of MbtB, to which the salicyl moiety is covalently tethered (29). The Cy, A, and second CP domains of MbtB were characterized as part of an artificial hybrid NRPS, confirming that these domains are able to catalyze the formation of an oxazoline or β-methyl oxazoline ring in vitro (6). Further analysis of the megasynthase components has been hampered by the inability to obtain soluble forms of MbtB to -F (29), which has prevented biochemical testing of their assigned functions and amino acid substrate specificity.
The mbt-1 cluster codes for many of the enzymes needed for MBT biosynthesis, but it lacks the genes coding for the enzymes involved in the formation of the fatty acyl chain and its subsequent addition to the N6 of the central l-Lys. A recent study identified an unlinked gene cluster (mbt-2) that codes for the necessary enzymes needed for this aspect of MBT biosynthesis (21). Through a series of elegant in vitro studies, Krithika and colleagues pieced together a potential pathway for the synthesis of the fatty acyl group, its installment on the N6 of the central l-Lys, and hydroxylation of the N6 of the central l-Lys and terminal caprolactam. Thus, it was proposed that the enzymes encoded by the mbt-2 gene cluster, in conjunction the mbt-1-encoded MbtG, function as tailoring enzymes that modify the MBT backbone that is assembled by the NRPS/PKS megasynthase. The authors could not fully test their hypotheses biochemically because of the insolubility of most MBT megasynthase components.
Recently we (7) and others (17, 49) determined that members of the MbtH-like protein superfamily are small proteins that influence the solubility and function of associated NRPS components. One of the most important proposals from these studies is that NRPSs that were previously recalcitrant to in vitro analysis due to insolubility or inactivity issues may become soluble and active when coproduced with their cognate MbtH-like protein. This protein superfamily is named after its founding member, MbtH, a protein of unknown function in MBT biosynthesis (29). It is possible that the prior insolubility problems with the NRPS components MbtB, MbtE, and MbtF were due to heterologous production in the absence of MbtH. Therefore, coproduction of MbtH with these enzymes may provide soluble and active forms of these enzymes for in vitro analysis. This would allow the amino acid specificity of the A domains of MbtB, MbtE, and MbtF to be assessed. Because A domains are the “gatekeepers” of NRPSs and select for the correct substrate to be incorporated into the growing peptide, defining A domain substrate specificity establishes where that particular NRPS fits into the biosynthetic scheme and whether modifications to the amino acids seen in the final product occur before or after peptide synthesis by the NRPS. Here we show that the solubility and activity of MbtB, MbtE, and MbtF are influenced by the presence of MbtH. Furthermore, we define the A domain substrate specificities of each protein, which leads to a revision of the proposed MBT biosynthetic pathway and the possibility that additional steps in MBT biosynthesis have yet to be identified.
MATERIALS AND METHODS
Plasmid construction.
The genes coding for MbtB, MbtE, MbtF, and Rv1347c in M. tuberculosis mc26020 were cloned into the overexpression vector pET28b (EMD Chemicals) using polymerase incomplete primer extension (PIPE)-based cloning (20). Each construct resulted in the production of a protein with an N-terminal hexahistidine tag. PIPE cloning was also used to introduce mbtH into the expression vectors pTEV5 and pACYC-Duet1. The pTEV5 construct produced MbtH with an N-terminal hexahistidine tag that was removable using tobacco etch virus (TEV) protease. The pACYC-Duet1 expression vector overproduced untagged MbtH. The primers used for PCR amplification are listed Table 1.
Table 1.
Primers used for PIPE cloning
| Primer | Sequence (5′→3′) |
|---|---|
| MbtHpTEVPIPEFor | GTATTTTCAGGGCGCTAGCCATATGAGCACCAATCCTTTCGATG |
| MbtHpTEVPIPERev | GCTCGAGAATTCCATGGCTCAGTCCTCGACCATGGCGTCACG |
| pTEVPIPEFor | GCCATGGAATTCTCGAGC |
| pTEVPIPRev | ATGGCTAGCGCCCTGAAAATAC |
| MbtHpACYCNcoIPIPEFor | TTAATAAGGAGATATACCATGAGCACCAATCCTTTCGATG |
| MbtHpACYCHindIIIPIPERev | TTATGCGGCCGCAAGCTTTCAGTCCTCGACCATGGCGTC |
| pACYCNcoIPIPE | TTAATAAGGAGATATACCATG |
| pACYCHindIIIPIPE | TTATGCGGCCGCAAGCTT |
| MbtB-NdeI | CCGCGCGGCAGCCATATGGTGCATGCTACGGCGTGCTCG |
| MbtB-HindIII | GAGTGCGGCCGCAAGCTTTTATCGGACATCGGCACTCAC |
| MbtE-NdeI | CCGCGCGGCAGCCATATGTGGTTCGTGCAGATGGCCGAC |
| MbtE-HindIII | GAGTGCGGCCGCAAGCTTTCACGGCCACTGGTCCCATGA |
| MbtF-NdeI | CCGCGCGGCAGCCATATGGGACCAGTGGCCGTGACGCGA |
| MbtF-HindIII: | GAGTGCGGCCGCAAGCTTTCATGCTGTTATCTCCGCCAG |
Overproduction and purification of MbtB, MbtE, MbtF, Rv1347c, and MbtH.
All proteins in this study were overproduced in Escherichia coli BL21(DE3)ybdZ::aac(3)-IV (7). Strains overproducing MbtB, MbtE, and MbtF also contained either the pACYC-Duet1-MCS empty vector or pACYC-Duet1-MbtH, which overproduced untagged MbtH. In all cases, the overproducing strains were grown in 3 liters of LB containing the appropriate antibiotics (kanamaycin [50 μg/ml], chloramphenicol [15 μg/ml]). The cells were grown at 25°C until an optical density at 600 nm (OD600) of 0.5 was reached, the temperature was shifted to 15°C, and 1 h later isopropyl-β-d-1-thiogalactopyranoside (100 μM) was added to the cultures to induce gene expression. Expression was at 15°C for 15 h. The cells were harvested by centrifugation and resuspended in buffer A (20 mM HEPES [pH 8.0], 300 mM NaCl, 10% [vol/vol] glycerol). MbtB, MbtE, MbtF, and Rv1347c purification followed previously described protocols for NRPS, and MbtH-like protein purification used nickel chelate and ion-exchange chromatography (7, 9). The protein concentrations were determined using the BCA protein assay kit (Pierce), with bovine serum albumin (BSA) as a standard.
Untagged MbtH was purified using nickel chelate chromatography before and after hexahistidine tag cleavage. Briefly, soluble protein from cell extract in buffer A supplemented with imidazole (5 mM) was batch bound to 1 ml of Ni-nitrilotriacetic acid (Ni-NTA) resin (Qiagen) at 4°C for 2 h. The resin was washed with buffer A containing 7.5 mM imidazole, and the resin was poured into a column. The proteins bound to the Ni-NTA resin were eluted using a step gradient of buffer A containing 20, 40, 60, 100, or 250 mM imidazole. Fractions containing MbtH were pooled and dialyzed overnight in buffer B (50 mM Tris-HCl [pH 8.0], 50 mM NaCl, 10% [vol/vol] glycerol) containing 0.3 mg of hexahistidine-tagged TEV protease (34). The dialyzed and TEV-digested protein was passed through 1 ml of Ni-NTA resin to remove hexahistidine-tagged MbtH and TEV protease. The untagged MbtH that did not bind to the resin was collected and concentrated. The protein concentration was determined using the BCA protein assay kit (Pierce), with BSA as a standard.
Chemical synthesis of N6-hydroxyl-l-Lys.
The synthetic scheme for N6-hydroxy-l-Lys is shown in Fig. 2. During the synthesis procedure, 1H nuclear magnetic resonance (NMR) and 13C NMR were recorded on Bruker spectrometers operating at 500 and 125 MHz, respectively. Deuterated chloroform (CDCl3) or perdeuterated methanol (CD3OD) served as the solvent and internal reference. Chemical shifts are reported as ppm, and coupling constants are reported in hertz. All reagents and solvents were obtained from commercial suppliers and used without further purification. All flash chromatography was performed with normal phase silica gel (Sorbent Technologies; 32- to 63-μm particle size, 60-Å pore size), following the general protocol of Still et al. (42).
Fig 2.
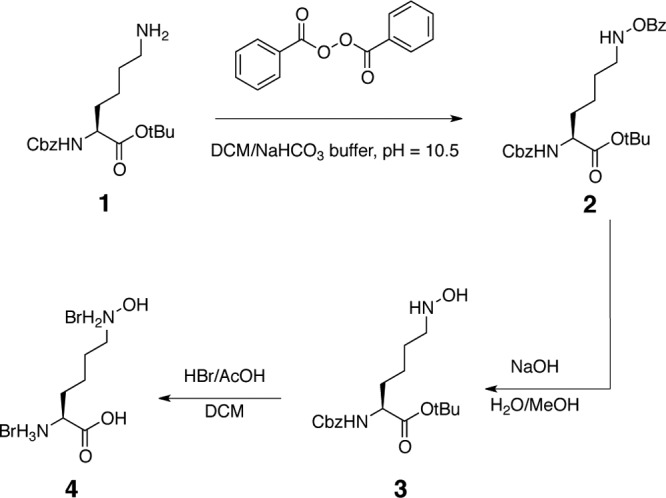
Schematic of the chemical synthesis of N6-hydroxy-l-Lys. Cbz, carbobenzyloxy; tBu, tert-butyl; DCM, dichloromethane; Bz, benzoyl; MeOH, methanol; AcOH, acetic acid.
Synthesis of 2.
Benzoyl peroxide (75%, 706 mg, 2.18 mmol) was added to a stirring biphasic mixture of 1 (16) (490 mg, 1.46 mmol) in buffer (7 ml, 7:3 0.75 M NaHCO3–1.5 M NaOH, pH 10.5) and dichloromethane (DCM; 7 ml). After 3 h, thin-layer chromatography indicated consumption of the starting material. The reaction mixture was diluted with DCM (15 ml) and H2O (15 ml). The layers were separated, and the aqueous portion was extracted with DCM (twice, 15 ml each). The organics were pooled, washed with 10% Na2S2O3 (twice, 15 ml each) and brine (once, 30 ml), and then dried (Na2SO4), filtered, and concentrated. The crude material was chromatographed on silica (1:0→9:1 DCM-ethylacetate [EtOAc]) to afford 2 as a thick syrup (512 mg, 77%).
1H-NMR (500 MHz, CDCl3) data were δ: 8.06 (2 H, dd, J = 6.8 Hz, 0.5 Hz), 7.97 (1 H, s), 7.60 (1 H, m), 7.48 (2 H, m), 7.41 to 7.31 (5 H, m), 5.73 (1 H, d, J = 8 Hz), 5.15 (2 H, s), 4.34 (1 H, m), 3.15 (2 H, t, J = 6 Hz), 1.91 (1 H, m), 1.71 (3 H, m), 1.39 (11 H, m).
13C-NMR (125 MHz, CDCl3) data were δ: 172.9, 167.2, 156.4, 136.9, 133.7, 129.7, 129.5, 128.9, 128.8, 128.7, 128.4, 82.3, 67.1, 55.3, 53.6, 32.2, 27.7, 26.6, 22.1.
Synthesis of 3.
An aqueous solution of NaOH (1.5 M, 550 μl, 0.83 mmol) was added to a solution of 2 (250 mg, 0.55 mmol) in methanol (5 ml) under an N2 atmosphere. After 40 min thin-layer chromatography indicated the loss of the starting material. The reaction mixture was diluted with H2O (15 ml), which produced a white precipitate. The mixture was extracted with diethyl ether (Et2O) (3 times, 10 ml each). The organic fractions were pooled and washed with brine (twice, 20 ml each) and then dried (MgSO4), filtered, and concentrated. Filtration and concentration yielded a colorless clear oil. The crude material was purified on silica with 33:1→30:1 DCM-methanol containing 0.1% NH3 to afford 3 (157 mg, 81%) as a cloudy oil.
1H-NMR (500 MHz, CDCl3) data were δ: 7.35–7.29 (5 H, m), 5.49 (1 H, d, J = 8 Hz), 5.10 (2 H, s), 4.26 (1 H, m), 4.12 (1 H, br s), 3.34 (1 H, br s), 2.89 (2 H, m), 1.80 (1 H, m), 1.70 to 1.50 (3 H, m), 1.45 (9 H, s), 1.42 to 1.30 (2 H, m).
13C-NMR (125 MHz, CDCl3) data were δ: 172.1, 156.4, 136.8, 158.9, 128.54, 128.52, 82.5, 67.3, 54.6, 53.8, 33.2, 28.4, 26.9, 23.0.
Synthesis of N6-hydroxy-l-Lys (4).
3 (38 mg, 0.11 mmol) was dissolved in DCM (1 ml) in a vial under N2. HBr (33% solution in acetic acid [AcOH], 1 ml) was added via syringe down the side of the vial. The resulting brown solution was stirred at room temperature (RT) for 1 h. Volatiles were removed via an N2 stream, and the remaining liquid was removed under reduced pressure. The residue was processed in an azeotropic solution twice with PhMe and then triturated with Et2O and DCM to afford the desired product. Spectroscopic data were as previously described (12).
Enzymatic synthesis of N6-acyl-l-Lys and N6-acyl-N6-hydroxy-l-Lys.
The previously reported reaction conditions for Rv1347c catalysis were used to catalyze N6 acylation of l-Lys and N6-hydroxy-l-Lys (11, 21). Briefly, 3.3 μg of Rv1347c was added to a 350-μl reaction mixture of 100 mM Tris-HCl, pH 8.0, 1 mM decanoyl-coenzyme A (decanoyl-CoA), and 500 μM l-Lys or N6-hydroxy-l-Lys. Reaction mixtures containing N6-hydroxy-l-Lys were incubated at 25°C for 3 h, while reaction mixtures containing l-Lys were incubated at 25°C for 20 h.
Enzymatic synthesis of N6-acyl-N6-hydroxy-l-Lys and N6-acyl-l-Lys was monitored and quantified using a combination of 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB)- and high-pressure liquid chromatography (HPLC)-based assays. Briefly, the progression of the N6-hydroxy-l-Lys-containing reaction was monitored using an established DTNB-based assay that monitors the release of free CoA from decanoyl-CoA after Rv1347c-dependent acylation of N6-hydroxy-l-Lys (11, 21). The amount of N6-acyl-N6-hydroxy-l-Lys formed was determined using the molar extinction coefficient of 5-thio-2-nitrobenzoate (14,150 M−1 cm−1), which is formed when free CoA reacts with DTNB (31). The amount of N6-acyl-N6-hydroxy-l-Lys was determined to be 31.5 nmol (90 μM final concentration) after 3 h of incubation at 25°C. This assay was not reliable for quantifying the amount of N6-acyl-l-Lys produced because the rate of CoA release was near that for the no-amino-acid control.
An HPLC-based assay was used to quantify the amount of N6-acyl-l-Lys produced. Briefly, 50 μl of an Rv1347c-catalyzed reaction mixture was removed and added to an equal volume of o-phthalaldehyde (OPA). The OPA-derivatized reaction components were separated by reverse-phase HPLC using a C18 column (Grace C18 small pore, 250 by 4.6 mm). Separation was carried out using a water-acetonitrile (MeCN) gradient containing 0.1% (vol/vol) trifluoroacetic acid and 0% MeCN for 0 to 5 min, 0 to 100% MeCN for 5 to 20 min, and 100% MeCN for 20 to 30 min. The gradient was run at a flow rate of 1 ml/min. The elution of OPA-derivatized molecules was monitored for absorbance at 340 nm, and the area under the peak associated with derivatized N6-acyl(decanoyl)-l-Lys was quantified using Beckman-Coulter 32K software. The identity of the OPA-derivatized N6-acyl(decanoyl)-l-Lys was confirmed by electrospray ionization mass spectrometry (ESI-MS; theoretical m/z = 415.3 [M-H]−, observed m/z = 415.2 [M-H]−). To quantify the amount of N6-acyl(decanoyl)-l-Lys, known amounts of N6-acyl(decanoyl)-N6-hydroxy-l-Lys were derivatized with OPA and analyzed by reverse-phase HPLC and the area under the eluting peaks was quantified. These data were used estimate the amount of N6-acyl-l-Lys that was enzymatically synthesized. The identity of the OPA-derivatized N6-acyl-N6-hydroxy-l-Lys was confirmed by ESI-MS (theoretical m/z = 431.3 [M-H]−, observed m/z = 431.2 [M-H]−). All MS was performed at the University of Wisconsin Biotechnology Center.
Amino acid-dependent ATP-PPi exchange assays.
Amino acid-dependent ATP-PPi exchange assays were performed as previously described (7, 9). Briefly, each 100-μl reaction mixture contained 75 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 5 mM dithiothreitol, 3.5 mM ATP (pH 7.0), 1 mM [32P]PPi (0.9 Ci/mol), and various concentrations of amino acid and enzyme. For amino acid specificity studies all amino acids were present at 1 mM, with the exception of N6-decanoyl-l-Lys and N6-decanoyl-N6-hydroxy-l-Lys, which were present at 15 μM. Enzyme concentrations used are noted in the appropriate figures. All assays were performed in the linear range for enzyme concentration, with less than 10% substrate-to-product conversion. The pseudo-first-order kinetic parameters for amino acid activation were determined by holding the ATP concentration constant at 3.5 mM while varying the amino acid concentration. For MbtB reactions the l-Thr concentrations were 0.1, 0.3, 0.6, 0.8, 1.25, 3.0, and 5.0 mM. For MbtF reactions, the amino acid concentrations were as follows: l-Lys, 25, 50, 75, 100, 150, 200, and 300 mM; N6-hydroxy-l-Lys, 10, 25, 50, 75, 100, 200, and 300 μM. All kinetic parameters were calculated using nonlinear regression analysis (KaleidaGraph) with data from three independent assays performed in parallel.
RESULTS
MbtH influences the solubility of heterologously produced MbtB, MbtE, and MbtF.
In vitro analysis of the MBT NRPS/PKS megasynthase has been hampered by the inability to heterologously produce soluble forms of these enzymes (29). We (7) and others (17, 49) recently determined that proteins belonging to the MbtH-like protein superfamily influence the solubility and activity of some NRPSs. The MbtH-like protein superfamily is named after the founding member, MbtH, a protein of unknown function encoded by a gene in the mbt-1 biosynthetic gene cluster. This raised the possibility that heterologous coproduction of MbtH with the NRPS components of the megasynthase (MbtB, MbtE, and MbtF) may resolve difficulties with solubility and activity of these enzymes.
MbtB, MbtE, and MbtF containing N-terminal hexahistidine tags were overproduced in E. coli BL21(DE3)ybdZ::aac(3)-IV (7) in the presence and absence of untagged MbtH to assess whether MbtH coproduction influenced NRPS solubility. In contrast to a prior report (29), MbtB was soluble when overproduced without MbtH (Fig. 3A); however, the presence of MbtH did influence MbtB solubility because approximately twice as much soluble MbtB was recovered from cells coproducing MbtH. MbtB overproduced in the presence or absence of MbtH was purified to near homogeneity using nickel chelate and ion-exchange chromatography (Fig. 3). MbtB did not copurify with coproduced MbtH, in contrast to other NRPSs that require MbtH-like proteins for solubility (7, 17, 49). Overproduction of MbtE or MbtF in the absence of MbtH did not result in soluble enzymes (data not shown), consistent with prior reports (29). In contrast, when MbtE and MbtF were coproduced with MbtH, both were soluble and copurified with MbtH through nickel chelate and ion-exchange chromatography (Fig. 3B). From these data we conclude that MbtH interacts with and influences the conformation of MbtB, MbtE, and MbtF, resulting in enhanced protein solubility. A mechanism for how MbtH-like proteins influence the solubility and activity of NRPSs is under investigation.
Fig 3.
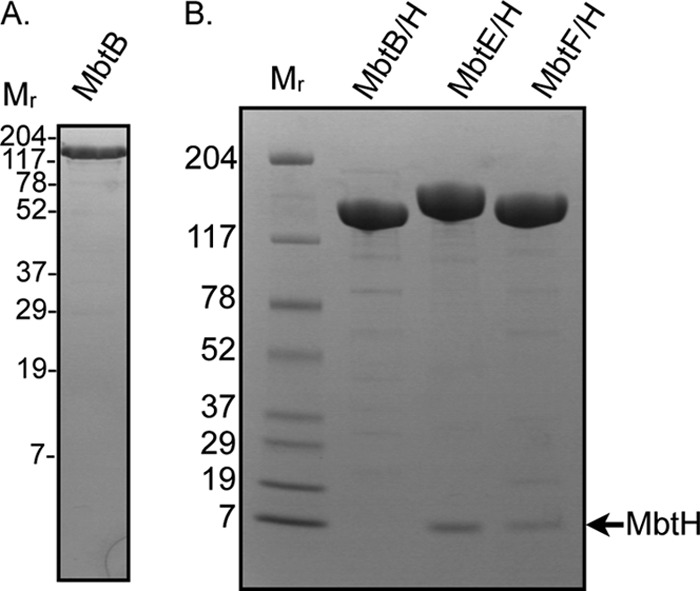
SDS-PAGE (4 to 20% polyacrylamide) and Coomassie staining analysis of purified NRPSs. (A) Purified MbtB (7 μg) overproduced in the absence of MbtH. (B) Purified MbtB, MbtE, and MbtF overproduced in the presence of MbtH. Eleven micrograms of protein was loaded in each lane. Mr, molecular weights (in thousands) of protein standards.
The A domain of MbtB preferentially activates l-threonine.
Structural analysis of membrane-associated MBT, soluble MBT, and MBT fragments that stimulate T cell activation suggests that the oxazoline ring is formed from l-Ser, l-Thr, or α-methyl-l-Ser. These findings, when combined with bioinformatics analyses of the NRPS/PKS megasynthase enzymology, led to the proposal that the A domain of MbtB is responsible for incorporating these amino acids into MBT (4, 29). It is reasonable to hypothesize that l-Ser is the preferred substrate because the majority of the MBT purified from M. tuberculosis contains an oxazoline ring lacking methylation at the α or β carbon.
Standard amino acid-dependent ATP-PPi exchange assays were used to define the amino acid specificity of the A domain of MbtB. The amino acids tested included Gly, l-Ala, l-Ser, l-Thr, α-methyl-l-Ser, l-Lys, and N6-l-Lys. Surprisingly, the A domain of MbtB showed significant preference for l-Thr regardless of whether MbtB was overproduced in the presence or absence of MbtH (Fig. 4A and B). In fact, MbtB that was overproduced in the presence of MbtH was even more selective for l-Thr, with l-Ser activation being comparable to that observed with l-Ala (Fig. 4B). A comparison of the kinetic parameters for l-Thr activation by MbtB overproduced with or without MbtH found they were similar, with a statistically significant change observed only in the Vmax (Table 2), which was consistent with the increased activity noted in Fig. 4. Also, since MbtB did not copurify with MbtH, we tested whether the addition of MbtH to the reaction mixtures containing the MbtB coproduced with MbtH altered the kinetic parameters, but only subtle effects on both the Km and Vmax were observed (Table 2) and there was no change in amino acid specificity.
Fig 4.
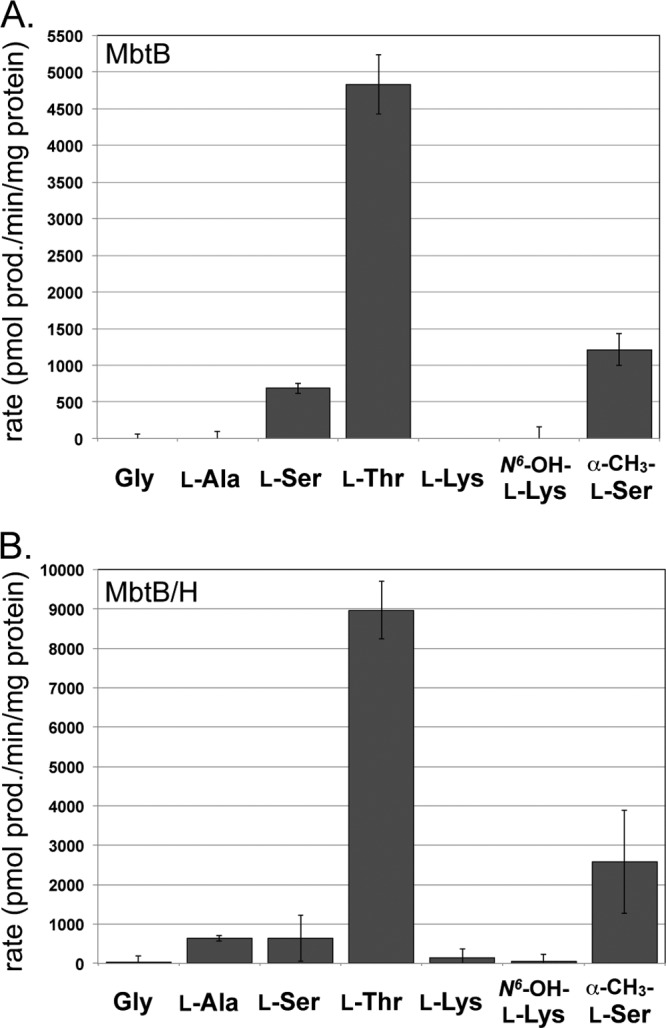
Amino acid specificity of the A domain of MbtB. (A) Amino acid specificity of MbtB overproduced and purified in the absence of MbtH (MbtB). (B) Amino acid specificity of MbtB overproduced and purified in the presence of MbtH (MbtB/H). The results shown are from three independent amino acid-dependent ATP/PPi exchange reactions, with the error bars representing the standard deviations. The reaction mixtures contained either 3.8 μg (MbtB) or 4.0 μg (MbtB/H) of protein and 1 mM the noted amino acid, and the reactions were run for 20 min. A reaction mixture lacking amino acid substrate was used to define the background activation, and this activity was subtracted from that for each amino acid-containing reaction. These conditions were in the linear range of the assays for enzyme concentration, and there was less than 10% substrate-to-product conversion based on the rates of l-Ser, l-Thr, and α-methyl-l-Ser activation. Abbreviations: N6-OH-l-Lys, N6-hydroxy-l-Lys; α-CH3-l-Ser, α-methyl-l-Ser.
Table 2.
Kinetic parameters for amino acid activation by MbtB and MbtFa
| Enzyme | Amino acid | Kmb (μM) | Vmaxc (pmol product/min/mg protein) | Vmax/Km (pmol product/min/mg protein/μM) |
|---|---|---|---|---|
| MbtBd | l-Thr | 344 ± 31 | 11,152 ± 263 | 32 |
| MbtB/He | l-Thr | 444 ± 53 | 18,025 ± 642 | 41 |
| MbtB/H + MbtHf | l-Thr | 388 ± 36 | 15,192 ± 528 | 39 |
| MbtF/Hg | l-Lys | 40,247 ± 8,781 | 18,580 ± 1,126 | 0.46 |
| MbtF/H | N6-hydroxy-l-Lys | 46.7 ± 4.3 | 210,720 ± 6,191 | 4,512 |
See Fig. S1 and S2 in the supplemental material for the Michaelis-Menten plots.
The values are the averages of three independent assays ± the standard errors from the nonlinear regression curve fit.
The values are the averages of three independent assays ± the standard errors from the nonlinear regression curve fit.
MbtB overproduced in the absence of MbtH.
MbtB overproduced in the presence of MbtH.
MbtB overproduced in the presence of MbtH with 100 μM MbtH added to the reaction.
MbtF overproduced in the presence of MbtH.
The A domain of MbtE is specific for N6-acyl-N6-hydroxy-l-lysine.
The l-Lys residue adjacent to the oxazoline ring of MBT is hydroxylated and acylated at the N6 position in the final MBT structures (Fig. 1). Prior biochemical studies suggest that N6-acylation of the l-Lys residue occurs following its incorporation into the MBT backbone and that N6-hydroxylation occurs subsequently (21). Based on these prior results, MbtE was predicted to activate l-Lys, and we used soluble MbtE resulting from coproduction with MbtH to test this hypothesis. Surprisingly, MbtE did not activate l-Lys. These data raised the alternate possibility that l-Lys must be modified by N6-hydroxylation or N6-acylation prior to activation by MbtE. Commercially available N6-acetyl-l-Lys (to mimic N6-acyl-l-Lys) and chemically synthesized N6-hydroxyl-l-Lys were used in ATP-PPi exchange assays to assess whether these modifications were required for MbtE recognition; however, neither acted as a substrate for MbtE. l-Ser, l-Thr, α-methyl-l-Ser, and α-amino-ε-caprolactam (the cyclized form of l-Lys) were also tested as substrates, but none were activated by MbtE.
Two possibilities that would explain the failure to detect MbtE activity with the substrates listed above are that N6-acetyl-l-Lys does not have a long enough fatty acyl group for recognition by MbtE and that the true substrate for MbtE is N6-acyl-N6-hydroxy-l-Lys. To test these hypotheses, the previously characterized M. tuberculosis acyltransferase Rv1347c was used to enzymatically generate N6-decanoyl-l-Lys and N6-decanoyl-N6-hydroxy-l-Lys (Fig. 5A). The decanoyl group was chosen as a mimic of the various acyl groups found on MBT, and prior work on Rv1347c showed that this enzyme efficiently recognized decanoyl-CoA as a surrogate substrate (11). The enzymatically generated substrates (at 15 μM final concentration) were used in ATP-PPi exchange assays, and it was determined that MbtE efficiently activated N6-decanoyl-N6-hydroxy-l-Lys, but only weakly activated N6-decanoyl-l-Lys (Fig. 5B).
Fig 5.

Amino acid specificity of the A domain of MbtE. (A) Schematic showing the substrates and products of the enzymatic synthesis of N6-decanoyl-l-Lys and N6-decanoyl-N6-hydroxy-l-Lys. AT, acyltransferase. (B) Amino acid specificity of the A domain of MbtE. Results shown are from three independent amino acid-dependent ATP/PPi exchange reactions using MbtE overproduced and purified with MbtH. The error bars represent standard deviations. The reaction mixtures contained 9 μg of MbtE/H and 15 μM each acylated amino acid, and the reactions were run for 20 min. These reaction conditions were in the linear range for enzyme concentration, and there was less than 10% substrate-to-product conversion based on N6-decanoyl-N6-hydroxy-l-Lys as the substrate. A reaction mixture lacking Rv1347c was used to define the background activity, and this activity was subtracted from that for each amino acid-containing reaction.
These data strongly support a model whereby the l-Lys residue is fully modified prior to incorporation by MbtE. Furthermore, Rv1347c was determined to acylate approximately half of the N6-hydroxy-l-Lys in a reaction mixture within 1 h, but the same amount of Rv1347c was able to acylate less than 1/10 of the l-Lys in overnight reactions. This suggests that the N6-hydroxylation of l-Lys occurs prior to N6-acylation. While prior analyses of Rv1347c investigated its amino acid specificity (11, 21), neither study tested N6-hydroxyl-l-Lys as a substrate. Providing further support for this model are studies showing that the M. tuberculosis and M. smegmatis hydroxylase, MbtG, hydroxlyates free l-Lys (21, 33). Furthermore, MbtG homologs involved in N5-hydroxylation of l-ornithine catalyze hydroxylation on the free amino acid prior to incorporation by the associated NRPS (25). Finally, a similar progression of N5 acylation followed by N5 hydroxylation of l-ornithine occurs during erythrochelin biosynthesis (32). Based on our data and these precedents, it is reasonable to conclude that l-Lys modification in MBT biosynthesis proceeds as follows: N6-hydroxylation by MbtG, followed by N6-acylation of N6-hydroxy-l-Lys by Rv1347c and subsequent incorporation of N6-acyl-N6-hydroxy-l-Lys by MbtE.
The A domain of MbtF is specific for N6-hydroxy-l-Lys.
The finding that l-Lys must be fully modified prior to recognition by MbtE raised the possibility that MbtF would activate N6-hydroxy-l-Lys rather than l-Lys as previously proposed. The purified MbtF/MbtH complex was assayed for amino acid activation, and a clear preference for N6-hydroxy-l-Lys over l-Lys was observed (Fig. 6). Defining the kinetic parameters of amino acid activation revealed that the A domain showed a >10,000-fold preference (Vmax/Km) for N6-hydroxy-l-Lys over l-Lys (Table 2). l-Ser, l-Thr, α-methyl-l-Ser, and α-amino-ε-caprolactam were not substrates for MbtF; thus, as seen with MbtE, the modified l-Lys is the substrate for the A domain of MbtF.
Fig 6.
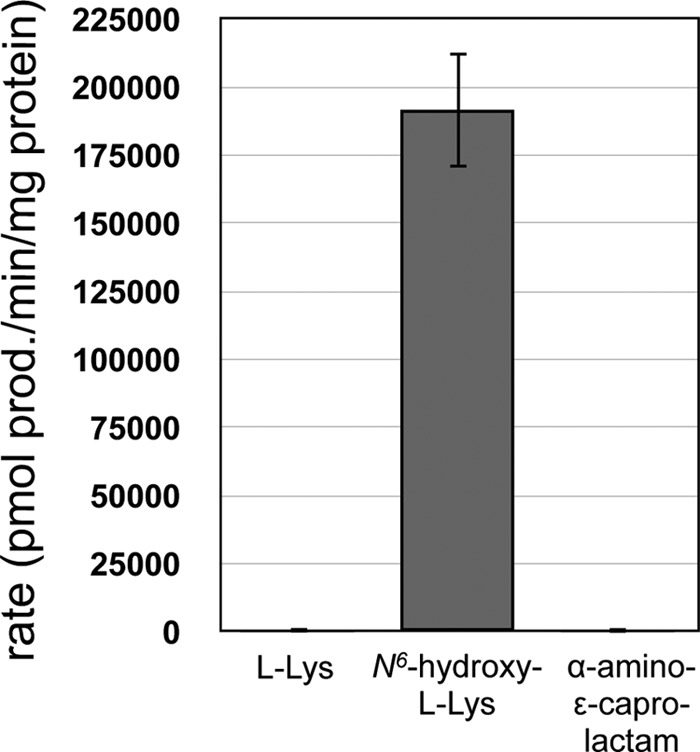
Amino acid specificity of the A domain of MbtF. The results shown are from three independent amino acid-dependent ATP/PPi exchange reactions using MbtF purified with MbtH coproduction. The error bars represent standard deviations. The reaction mixtures contained 1.6 μg of MbtF/H and 1 mM each amino acid, and the reactions were run for 20 min. These reaction conditions were in the linear range for enzyme concentration, and there was less than 10% substrate-to-product conversion based on N6-hydroxy-l-Lys as the substrate.
DISCUSSION
Access to soluble NRPS components of the MBT megasynthase enabled us to test hypotheses concerning the true substrates for the NRPS adenylation domains and the timing of amino acid modification during MBT assembly. Our data suggest the biosynthesis of the l-Thr-containing MBT derivatives occurs as shown in Fig. 7. First, all the necessary precursors for MBT must be biosynthesized. MbtI catalyzes the transformation of isochorismate to salicylate, providing the aryl acid that caps one end of MBT (14, 29, 50). MbtG will catalyze the N6-hydroxylation of l-Lys, generating N6-hydroxy-l-Lys (21, 29). This is the substrate for the A domain of MbtF and also the substrate for the acyltransferase Rv1347c, which catalyzes the acylation of N6-hydroxyl-Lys to form N6-acyl-N6-hydroxy-l-Lys, the substrate for the A domain of MbtE. The l-Thr substrate for the A domain of MbtB is readily available in the cell and does not have to be biosynthesized specifically for MBT assembly. Based on the final structure of MBT, it is reasonable to hypothesize that acetoacetyl-CoA is the substrate for the AT domain of the PKS portion of the megasynthase.
Fig 7.

Proposed MBT biosynthetic pathway. For brevity, only the MBT derivatives with α,β desaturation of the fatty acyl chain are shown. Fully saturated fatty acyl chains are likely to be formed by Rv1347c recognizing substrates tethered to Rv1344 prior to FadE14 catalysis of the α,β desaturation. Enzymes previously noted as MBT tailoring enzymes are proposed to be involved in the precursor biosynthesis that provides the amino acid substrates for the NRPS components of the megasynthase. Assembly of MBT involves the function of the NRPS/PKS megasynthase, with MbtB, MbtE, and MbtF interacting with MbtH. Release of MBT from the megasynthase produces derivatives containing β-methyl oxazoline rings. The mechanism of ring demethylation is not yet known. ACP, acyl carrier protein; A, adenylation domain; PCP, peptidyl carrier protein domain; C, condensation domain; Cy, cyclization domain; TeII, type II thioesterase; KS, ketosynthase; AT, acyltransferase; KR, ketoreductase; E, epimerase.
Once these substrates are assembled, each will be activated and thioesterified to their respective CP domains. The Cy domain of MbtB will catalyze the condensation of salicylate and l-Thr, followed by cyclization to generate the salicyl-methythiazolinyl-MbtB intermediate (6, 29). The N-terminal C domain of MbtE catalyzes the condensation of N6-acyl-N6-hydroxy-l-Lys-MbtE, with the upstream components tethered to MbtB. It is not clear at this point what (if any) role the additional C-CP pair at the C terminus of MbtE has in MBT biosynthesis, and further mutational analysis of MbtE is needed to determine whether these domains play any role in MBT biosynthesis. The β-keto group of the acetoacetyl moiety on MbtD will be reduced by the KR domain, generating the hydroxyl group that attacks the carbonyl of the thioesterified intermediate on MbtE, forming the ether linkage of MBT. The C domain of MbtF condenses the upstream nonribosomal peptide/polyketide hybrid molecule with the N6-hydroxy-l-lysyl-MbtF. Cyclization of the terminal N6-hydroxy-l-lysyl moiety, potentially by the terminal domain of MbtE, releases MBT from the megasynthase.
The described biosynthetic pathway differs from that previously proposed in the timing of the modifications to the l-Lys residues. Prior models proposed that these modifications occur after the synthesis of the backbone of MBT; thus, they are tailoring functions. Our data suggest that these enzymes are not involved in MBT tailoring; rather, they are precursor biosynthetic enzymes that generate the substrates for the NRPS components of the megasynthase. As the manuscript was being prepared, Madigan et al. published data that they interpreted as providing evidence that the hydroxylations of the l-Lys residues are the final steps in siderophore biosynthesis (23). A key piece of these data was the finding that M. tuberculosis still produced dideoxycarboxymycobactin (soluble MBT derivatives lacking the l-Lys hydroxylations) when mbtG was deleted. We interpret these data differently when we compare the relative amounts of all MBT derivatives produced in the wild-type strain versus the ΔmbtG strain. According to their published data, the ΔmbtG strain produces ≈2% of the amount of MBT derivatives that were detected in the wild-type strain, a result that is inconsistent with the model that hydroxylation is the final step in MBT biosynthesis. If hydroxylation were the final step, nearly equivalent amounts of total MBT derivatives produced in both strains would be expected, with all of the MBT in the ΔmbtG strain being the dideoxy derivative, while the wild-type strain would produce a distribution of the monodeoxy, dideoxy, and fully hydroxylated MBT derivatives. Taking into account our results whereby the hydroxylated substrates are the preferred substrates, we reinterpret the substantial reduction in MBT production as being consistent with our model, whereby the loss of MbtG results in the absence of the natural substrates for MbtE and MbtF. In the absence of these substrates, the structurally similar substrates l-Lys and N6-acyl-l-Lys, respectively, are used with a lower efficiency, resulting in lower product yields. This is analogous to our findings during the analysis of viomycin biosynthesis, whereby the absence of a precursor biosynthesis enzyme resulted in the incorporation of a structurally similar, but incorrect, substrate. This change resulted in the production of a viomycin derivative at a level ≈1% of viomycin in the wild-type strain (1).
Our work provides a pathway for the biosynthesis of MBT derivatives with a β-methylated oxazoline ring but does not explain how the nonmethylated or α-methylated derivatives are generated; thus, the mechanism for removal of the β methyl group or its migration to the α carbon remains an open question. We do not favor the hypothesis that the A domain specificity of MbtB is somehow altered in vivo to favor l-Ser over l-Thr or α-methyl-l-Ser. Instead, we favor the hypothesis that there is a yet to be identified MBT biosynthetic enzyme(s). There is no enzyme coded within the known MBT biosynthesis gene clusters (mbt-1 and mbt-2) that is a candidate for catalyzing such reactions. It is reasonable to hypothesize that a member of the radical S-adenosylmethionine (SAM) enzyme family is a candidate for catalyzing such demethylation or methyl group migration reactions (2, 47, 48). Interestingly, a gene (locus tag Rv2388c) encoding a class III radical SAM enzyme is upstream of mbtI in all M. tuberculosis strains as well as many other Mycobacterium species. Furthermore, the homolog of this gene in M. smegmatis (locus tag MSMEG_4525) may be in an operon with mbtI, raising the possibility that the encoded protein is involved in MBT biosynthesis. We note, however, that this open reading frame (ORF) is annotated as hemN, which codes for a radical SAM enzyme that may be involved in heme biosynthesis (22, 36); therefore, its association with MBT biosynthesis genes may be unrelated to its possible direct role in MBT assembly. Further analysis of this gene product and investigations into other potential enzyme candidates, along with labeling studies to analyze l-Thr or l-Ser incorporation into MBT, will provide additional insight into this significant question in MBT biosynthesis.
Finally, MBT biosynthesis has already proven to be a desirable target for drug development (10, 26, 45). The use of salicyl-AMP analogs initially developed by Quadri and Tan (10) and extended by others (28, 40) as inhibitors of M. tuberculosis growth provides a template for targeting the A domains of the MBT megasynthase for drug development. The focus on salicyl-AMP analogs to inhibit the function of MbtA was viable because these analogs were unlikely to inhibit host metabolic processes. In contrast, using l-Ser-AMP or l-Lys-AMP analogs would likely result in problematic inhibition of host tRNA synthetases. Our discovery that the A domains of MbtE and MbtF activate N6-acyl-N6-hydroxy-l-Lys and N6-hydroxy-l-Lys, not l-Lys, reveals that these A domains are targets for inactivation by amino acid-AMP analogs of these nonproteinogenic amino acids. Our discovery that MbtH influences the solubility and function of MbtB, MbtE, and MbtF suggests that disrupting the interactions between MbtH and these NRPSs may provide another avenue for development of the next generation of antituberculosis drugs. We (7), and others (17, 49) have shown that eliminating MbtH-like proteins from siderophore biosynthetic pathways abolishes the ability of the associated organism to grow under iron-limiting conditions due to the disruption of siderophore biosynthesis. Importantly, MbtH is the only member of the MbtH-like protein superfamily to be encoded by M. tuberculosis; thus, cross-complementation by MbtH-like proteins from other NRPS systems is not an issue, as has been observed in M. smegmatis (3). Finally, the described work validates the utility of MbtH-like proteins for reinvestigating medically or industrially important NRPSs that have previously proven to be recalcitrant to in vitro analysis.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the NIH grant AI065850 (to M.G.T.).
We thank Adel Talaat and Sarah Marcus for the M. tuberculosis mc26020 chromosomal DNA. We thank Hyunjun Park for technical assistance during the analysis of MbtB.
Footnotes
Published ahead of print 23 March 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Barkei JJ, Kevany BM, Felnagle EA, Thomas MG. 2009. Investigations into viomycin biosynthesis by using heterologous production in Streptomyces lividans. Chembiochem 10:366–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Booker SJ. 2009. Anaerobic functionalization of unactivated C-H bonds. Curr. Opin. Chem. Biol. 13:58–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chavadi SS, et al. 2011. Mutational and phylogenetic analyses of the mycobacterial mbt gene cluster. J. Bacteriol. 193:5905–5913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Voss JJ, et al. 2000. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. U. S. A. 97:1252–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Du L, Sanchez C, Shen B. 2001. Hybrid peptide-polyketide natural products: biosynthesis and prospects toward engineering novel molecules. Metab. Eng. 3:78–95 [DOI] [PubMed] [Google Scholar]
- 6. Duerfahrt T, Eppelmann K, Muller R, Marahiel MA. 2004. Rational design of a bimodular model system for the investigation of heterocyclization in nonribosomal peptide biosynthesis. Chem. Biol. 11:261–271 [DOI] [PubMed] [Google Scholar]
- 7. Felnagle EA, et al. 2011. MbtH-like proteins as integral components of bacterial nonribosomal peptide synthetases. Biochemistry 49:8815–8817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Felnagle EA, et al. 2008. Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol. Pharm. 5:191–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Felnagle EA, Podevels AM, Barkei JJ, Thomas MG. 2011. Mechanistically distinct nonribosomal peptide synthetases assemble the structurally related antibiotics viomycin and capreomycin. Chembiochem 12:1859–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. 2005. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol. 1:29–32 [DOI] [PubMed] [Google Scholar]
- 11. Frankel BA, Blanchard JS. 2008. Mechanistic analysis of Mycobacterium tuberculosis Rv1347c, a lysine Nε-acyltransferase involved in mycobactin biosynthesis. Arch. Biochem. Biophys. 477:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Genet JP, Thorimbert S, Mallart S, Kardos N. 1993. A short route to (±)-N-hydroxylysine and (±)-laminine by palladium-catalyzed reactions. Synthesis 1993:312–324 [Google Scholar]
- 13. Gobin J, et al. 1995. Iron acquisition by Mycobacterium tuberculosis: isolation and characterization of a family of iron-binding exochelins. Proc. Natl. Acad. Sci. U. S. A. 92:5189–5193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harrison AJ, et al. 2006. The structure of MbtI from Mycobacterium tuberculosis, the first enzyme in the biosynthesis of the siderophore mycobactin, reveals it to be a salicylate synthase. J. Bacteriol. 188:6081–6091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hoshino Y, et al. 2011. Identification of nocobactin NA biosynthetic gene clusters in Nocardia farcinica. J. Bacteriol. 193:441–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hu J, Miller MJ. 1997. Total synthesis of a mycobactin S, a siderophore and growth promoter of Mycobacterium smegmatis, and determination of its growth inhibitory activity against Mycobacterium tuberculosis. J. Am. Chem. Soc. 119:3462–3468 [Google Scholar]
- 17. Imker HJ, Krahn D, Clerc J, Kaiser M, Walsh CT. 2010. N-acylation during glidobactin biosynthesis by the tridomain nonribosomal peptide synthetase module GlbF. Chem. Biol. 17:1077–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Keating TA, Miller DA, Walsh CT. 2000. Expression, purification, and characterization of HMWP2, a 229 kDa, six domain protein subunit of Yersiniabactin synthetase. Biochemistry 39:4729–4739 [DOI] [PubMed] [Google Scholar]
- 19. Keating TA, Walsh CT. 1999. Initiation, elongation, and termination strategies in polyketide and polypeptide antibiotic biosynthesis. Curr. Opin. Chem. Biol. 3:598–606 [DOI] [PubMed] [Google Scholar]
- 20. Klock HE, Koesema EJ, Knuth MW, Lesley SA. 2008. Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins 71:982–994 [DOI] [PubMed] [Google Scholar]
- 21. Krithika R, et al. 2006. A genetic locus required for iron acquisition in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 103:2069–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Layer G, Verfurth K, Mahlitz E, Jahn D. 2002. Oxygen-independent coproporphyrinogen-III oxidase HemN from Escherichia coli. J. Biol. Chem. 277:34136–34142 [DOI] [PubMed] [Google Scholar]
- 23. Madigan CA, et al. 2012. Lipidomic discovery of deoxysiderophores reveals a revised mycobactin biosynthesis pathway in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 109:1257–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marshall CG, Hillson NJ, Walsh CT. 2002. Catalytic mapping of the vibriobactin biosynthetic enzyme VibF. Biochemistry 41:244–250 [DOI] [PubMed] [Google Scholar]
- 25. Meneely KM, Lamb AL. 2007. Biochemical characterization of a flavin adenine dinucleotide-dependent monooxygenase, ornithine hydroxylase from Pseudomonas aeruginosa, suggests a novel reaction mechanism. Biochemistry 46:11930–11937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Monfeli RR, Beeson C. 2007. Targeting iron acquisition by Mycobacterium tuberculosis. Infect. Disord. Drug Targets 7:213–220 [DOI] [PubMed] [Google Scholar]
- 27. Moody DB, et al. 2004. T cell activation by lipopeptide antigens. Science 303:527–531 [DOI] [PubMed] [Google Scholar]
- 28. Neres J, et al. 2008. Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: structure-activity relationships of the nucleobase domain of 5′-O-[N-(salicyl)sulfamoyl]adenosine. J. Med. Chem. 51:5349–5370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT. 1998. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem. Biol. 5:631–645 [DOI] [PubMed] [Google Scholar]
- 30. Ratledge C, Snow GA. 1974. Isolation and structure of nocobactin NA, a lipid-soluble iron-binding compound from Nocardia asteroides. Biochem. J. 139:407–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Riener CK, Kada G, Gruber HJ. 2002. Quick measurement of protein sulfhydryls with Ellman's reagent and with 4,4′-dithiodipyridine. Anal. Bioanal. Chem. 373:266–276 [DOI] [PubMed] [Google Scholar]
- 32. Robbel L, Helmetag V, Knappe TA, Marahiel MA. 2011. Consecutive enzymatic modification of ornithine generates the hydroxamate moieties of the siderophore erythrochelin. Biochemistry 50:6073–6080 [DOI] [PubMed] [Google Scholar]
- 33. Robinson R, Sobrado P. 2011. Substrate binding modulates the activity of Mycobacterium smegmatis G, a flavin-dependent monooxygenase involved in the biosynthesis of hydroxamate-containing siderophores. Biochemistry 50:8489–8496 [DOI] [PubMed] [Google Scholar]
- 34. Rocco CJ, Dennison KL, Klenchin VA, Rayment I, Escalante-Semerena JC. 2008. Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli. Plasmid 59:231–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rodriguez GM, Smith I. 2006. Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J. Bacteriol. 188:424–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seehra JS, Jordan PM, Akhtar M. 1983. Anaerobic and aerobic coproporphyrinogen III oxidases of Rhodopseudomonas spheroides. Mechanism and stereochemistry of vinyl group formation. Biochem. J. 209:709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shah NS, et al. 2007. Worldwide emergence of extensively drug-resistant tuberculosis. Emerg. Infect. Dis. 13:380–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Snow GA. 1965. Isolation and structure of mycobactin T, a growth factor from Mycobacterium tuberculosis. Biochem. J. 97:166–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Snow GA. 1970. Mycobactins: iron-chelating growth factors from mycobacteria. Bacteriol. Rev. 34:99–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Somu RV, et al. 2006. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem. 49:31–34 [DOI] [PubMed] [Google Scholar]
- 41. Stachelhaus T, Mootz HD, Marahiel MA. 1999. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 6:493–505 [DOI] [PubMed] [Google Scholar]
- 42. Still WC, Kahn M, Mitra M. 1978. Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 43:2923–2925 [Google Scholar]
- 43. Tsukamoto M, et al. 1997. BE-32030 A, B, C, D and E, new antitumor substances produced by Nocardia sp. A32030. J. Antibiot. (Tokyo) 50:815–821 [DOI] [PubMed] [Google Scholar]
- 44. Turgay K, Krause M, Marahiel MA. 1992. Four homologous domains in the primary structure of GrsB are related to domains in a superfamily of adenylate-forming enzymes. Mol. Microbiol. 6:529–546 [DOI] [PubMed] [Google Scholar]
- 45. Vasan M, et al. 2010. Inhibitors of the salicylate synthase (MbtI) from Mycobacterium tuberculosis discovered by high-throughput screening. Chemmedchem 5:2079–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. World Health Organization 2011. Global tuberculosis control: WHO report 2011. World Health Organization, Geneva, Switzerland: http://www.who.int/tb/publications/global_report/en/ [Google Scholar]
- 47. Wu SC, Zhang Y. 2010. Active DNA demethylation: many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 11:607–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Q, van der Donk WA, Liu W. 18 November 2011, posting date Radical-mediated enzymatic methylation: a tale of two SAMS. Acc. Chem. Res. doi:10.1021/ar200202c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang W, Heemstra Jr, Jr, Walsh CT, Imker HJ. 2010. Activation of the pacidamycin PacL adenylation domain by MbtH-like proteins. Biochemistry 49:9946–9947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zwahlen J, Kolappan S, Zhou R, Kisker C, Tonge PJ. 2007. Structure and mechanism of MbtI, the salicylate synthase from Mycobacterium tuberculosis. Biochemistry 46:954–964 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.