

Abstract
Mycobacterium tuberculosis survives and replicates in macrophages, where it is exposed to reactive oxygen and nitrogen species that damage DNA. In this study, we investigated the roles of UvrA and UvrD1, thought to be parts of the nucleotide excision repair pathway of M. tuberculosis. Strains in which uvrD1 was inactivated either alone or in conjunction with uvrA were constructed. Inactivation of uvrD1 resulted in a small colony phenotype, although growth in liquid culture was not significantly affected. The sensitivity of the mutant strains to UV irradiation and to mitomycin C highlighted the importance of the targeted genes for nucleotide excision repair. The mutant strains all exhibited heightened susceptibility to representatives of reactive oxygen intermediates (ROI) and reactive nitrogen intermediates (RNI). The uvrD1 and the uvrA uvrD1 mutants showed decreased intracellular multiplication following infection of macrophages. Most importantly, the uvrA uvrD1 mutant was markedly attenuated following infection of mice by either the aerosol or the intravenous route.
INTRODUCTION
Tuberculosis remains a major world health problem, causing 1.75 million deaths annually (54). Furthermore, it has been estimated that one-third of the world's population is latently infected with Mycobacterium tuberculosis (16). Approximately 10% of those latently infected will develop active disease, but this risk is significantly increased by factors weakening the immune response, such as HIV infection (52). If latent infection in individuals at high risk of reactivation could be treated, transmission and levels of disease could be reduced dramatically. For the development of drugs active against latent bacteria, it is necessary to identify targets with functions required by the bacteria under these conditions (2).
One such function may be the ability to repair damaged DNA. In mice the continued synthesis of NO maintains the persistent state (24), in which bacterial numbers remain constant; administration of an inducible nitric oxide synthase (iNOS) inhibitor during infection results in a rapid increase in bacterial numbers (20). Thus, the bacteria must be constantly exposed to NO, which readily crosses cell membranes. Although the role of NO in human disease is less clear, it has been reported that iNOS is present and active in human tuberculosis lesions (7, 37). NO and related reactive nitrogen intermediates (RNI) damage a range of macromolecules within the cell, but it is damage to DNA that is most likely to be lethal (5, 35). These observations suggest that DNA damage must be repaired to allow the bacteria to replicate when presented with favorable conditions.
Like other bacteria, M. tuberculosis possesses a number of mechanisms to repair DNA: recombination, nonhomologous end joining, base excision repair, and nucleotide excision repair (NER) (14). Studies in other organisms have indicated that nucleotide excision repair is active on the kind of DNA damage produced by RNI (34, 45) and reactive oxygen intermediates (ROI) (10, 29). Furthermore, it has been shown that NER is important for resistance to NO in vitro in Mycobacterium smegmatis (22, 27) and M. tuberculosis (11, 12), providing the first evidence that NER performs a role in mycobacterial pathogenesis.
The process of NER begins with recognition of the damaged nucleotide by UvrA and UvrB, following which UvrA is released and UvrC is recruited by UvrB. Dual incisions of the DNA backbone either side of the damage are introduced by UvrC. Finally, release of the resulting single-stranded oligonucleotide is facilitated by the helicase UvrD, permitting resynthesis to occur (51). As well as orthologues of the three excinuclease components UvrA, UvrB, and UvrC, M. tuberculosis possesses two homologues of the helicase UvrD: UvrD1 and UvrD2 (8). UvrD1 is most similar to Escherichia coli UvrD at the amino acid sequence level (BLAST score of 1 × e−124 for UvrD1 compared with 6 × e−53 for UvrD2). UvrD2 also differs from most UvrD proteins in possessing a HRDC domain more commonly associated with RecQ family helicases (47). Furthermore, purified M. tuberculosis UvrD1 protein has been shown to be highly active on a substrate resembling an NER intermediate in vitro (9). Therefore, we hypothesized that UvrD1 is most likely functional in NER.
We targeted uvrD1 for deletion in M. tuberculosis and assessed its phenotype along with that of a uvrA mutant described elsewhere (40). While UvrA functions solely within the NER pathway, in E. coli UvrD also plays a role in the mismatch repair pathway (23), which is absent in M. tuberculosis (49), and in recombination repair at blocked replication forks (19, 30). Our study reveals that although elimination of uvrA has only modest effects on pathogenicity, elimination of uvrD1 significantly affects the chronic stage of infection, and the combined loss of both functions severely impacts the ability of M. tuberculosis to replicate and persist in a mouse model of infection.
MATERIALS AND METHODS
Bacterial strains, media, and culture conditions.
Standard procedures were adopted for cloning using Escherichia coli (42). The M. tuberculosis wild-type strain was 1424, a rpsL (StrR) derivative of M. tuberculosis H37Rv (13). M. tuberculosis cultures were grown in albumin, dextrose, catalase (ADC)-enriched Middlebrook 7H9 medium or modified Dubos (Difco) supplemented with albumin and 0.2% glycerol at 37°C in a rolling incubator at 2 rpm. Generation times were calculated from optical density measurements at 600 nm (OD600) of cultures in the logarithmic growth phase. When appropriate, antibiotics were added at the following concentrations: hygromycin, 50 μg/ml; kanamycin, 25 μg/ml; streptomycin, 100 μg/ml; and gentamicin, 15 μg/ml. All procedures with M. tuberculosis were carried out under containment level 3 conditions. The strains and plasmids used in this study are listed in Table 1.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic | Source or reference |
|---|---|---|
| M. tuberculosis strain 1424 | M. tuberculosis wild-type H37Rv derivative, streptomycin resistant | 13 |
| ΔuvrA | uvrA deletion mutant | 40 |
| ΔuvrD1 | uvrD1 deletion mutant | This study |
| ΔuvrA uvrD | uvrA uvrD1 double deletion mutant | This study |
| pUvrD1::gm-rpsL | Targeting plasmid for removal of base pairs 299-1633 of uvrD1 ORF | This study |
| Z205 | uvrD1 complementing vector, uvrD1 in pMV361 used for in vitro experiments | This study |
| pAW33 | uvrD1 complementing vector, uvrD1 in pKP201 used for in vivo experiments | This study |
| uvrD1comp | ΔuvrD1 plus either Z205 or pAW33 | This study |
Isolation and complementation of mutant strains of M. tuberculosis.
M. tuberculosis mutants were generated by allelic replacement (43). For disruption of uvrD1 (Rv0949), a 5.1-kb ApaI/EcoRV fragment containing the uvrD1 gene was isolated from bacterial artificial clone Rv103 (4) and cloned into plasmid ptrpA-1-rpsL (44) previously digested with ApaI/EcoRV to give puvrD1-rpsL. A 1.4-kb fragment (SalI-AatII) was deleted and substituted by a 3-kb gentamicin resistance cassette originally derived from plasmid pML10 (28), resulting in plasmid puvrD1::gm-rpsL. The deletion allele lacks base pairs 299 to 1683 of the 2,316-base pair uvrD1 open reading frame (ORF).
To isolate mutant strains, the uvrD1 targeting construct was transformed into M. tuberculosis strain 1424 or into the uvrA mutant selecting for chromosomal integration of the targeting plasmid using gentamicin. Following the identification of a single crossover by Southern blot analysis, counter selection on streptomycin in the presence of gentamicin was used to isolate a double crossover. Southern analysis was performed to identify uvrD1 mutant strains using a 2,411-bp 5′ probe (HpaI/SacI) with SacI-digested DNA.
For in vitro complementation experiments, the uvrD1 gene plus 362-bp upstream and 641-bp downstream sequences was cloned from puvrD1-rpsL into the HpaI site of the integrative vector pMV361 (50), such that uvrD1 would be transcribed in the opposite direction to the vector hsp60 promoter. For in vivo complementation experiments, uvrD1 plus 299-bp upstream and 102-bp downstream DNA was cloned into the EcoRV of the integrative vector pKP201 (21) that lacks the integrase to ensure high-level stability of the plasmid in M. tuberculosis, yielding pAW33. This plasmid was introduced into the uvrD1 and the uvrA uvrD1 strains by cotransformation with pBluescriptint, a suicide plasmid bearing a copy of the integrase gene (48).
Susceptibility to DNA damaging agents, ROI and RNI.
To assess the susceptibilities of the mutant strains to both UV irradiation and in vitro stress, cultures were compared with that of the wild-type parental strain as previously described (40), but the cultures also included menadione (250 μM) in addition to sodium nitrite (2 mM), mitomycin C (0.1 μg/ml), and t-butyl hydroperoxide (0.1 mM). The OD of all cultures was determined at the start of the experiment, and approximately equal levels of inoculum were used for all strains. Serial dilutions were prepared and plated in duplicate for CFU determination. Survival was calculated by the ratio of CFU of the treated cultures compared to the CFU of the untreated controls. Three independent biological replicates were performed.
Bone marrow macrophage isolation and infection.
Bone marrow derived macrophages (BMDMs) were generated from 6- to 8-week-old BALB/c mice in RPMI 1640 (Gibco) containing 10% fetal calf serum, 20 μM l-glutamine, 1 mM sodium pyruvate, 10 μM HEPES, and 50 nM β-mercaptoethanol. The cells were grown and differentiated in complete RPMI 1640 supplemented with 20% L929 cell supernatant for 6 days at 37°C in 5% CO2. The differentiated cells were seeded at a density of 2 × 105 cells/well in 1 ml complete RPMI 1640 supplemented with 5% L929 cell supernatant; where cells were stimulated with gamma interferon (IFN-γ), media were supplemented with IFN-γ at a concentration of 10 ng/ml. After seeding, all cells were incubated overnight prior to infection.
M. tuberculosis strains were grown to an OD600 of 0.5 to 0.8 and inocula prepared by washing and resuspending the cultures in phosphate-buffered saline (PBS). Strains were then spun slowly at 50 × g to remove any clumps and produce a single-cell suspension. This was used to infect BMDMs at a multiplicity of infection of 1:10. After 4 h, the cells were washed to remove all extracellular bacilli, the medium was replaced, and incubation continued. Macrophages were lysed with water-0.05% Tween 80 to release intracellular bacteria at specific time points postinfection. Released bacilli were serially diluted in PBS-Tween and plated on 7H11-oleic acid-albumin-dextrose-catalase (OADC) for enumeration of surviving bacteria.
Infection of mice.
Experiments involving mice were conducted in strict accordance with the United Kingdom Animal (Scientific Procedures) Act 1986 under licenses 80/2236, 80/2379, and 80/1927, and all efforts were made to minimize suffering. Female, specific-pathogen-free BALB/c mice (6- to 8-weeks-old) were obtained from the breeding facility at the National Institute for Medical Research (Mill Hill, United Kingdom), and the studies were performed in containment level 3 animal facilities.
For intravenous infection, logarithmically growing cultures were diluted in saline and approximately 106 CFU injected into the tail vein. The progress of the infection was assessed by determining organ CFU at various time points using four mice per strain per time point. Organs were homogenized in a FastPrep instrument (MP Biomedicals) in 5 ml of water-0.05% Tween 80 using 1/4-in. ceramic beads at a setting of 4.0 m/s for 25 s in a chilled holder.
For aerosol infection, logarithmically growing cultures were diluted to approximately 105 CFU/ml and mice were infected using an inhalation exposure system (Glas-Col) to deliver approximately 100 bacilli per mouse. Organs were homogenized in 1.2 ml of saline using the FastPrep 120 at a setting of 6.0 for 20 s. Serial dilutions of organ homogenates from five mice per strain per time point were plated on 7H11-OADC to quantify CFU.
RNA isolation and purification.
To isolate mycobacterial RNA from in vitro cultures, 50 ml of culture at an OD of 0.5 to 0.8 was pelleted at 3,000 rpm for 15 min. RNA was extracted from the resulting pellet using a FastRNA Pro Blue kit (Qbiogene). To isolate mycobacterial RNA from infected mice lungs, tissue was homogenized in TRIzol using a beadbeater with (1/4-in.) beads at a setting of 6.0 for 20 s. This was then passed through a 70-μm cell strainer before being centrifuged at 13,000 rpm for 5 min. TRIzol containing host material was then removed and the resulting pellet resuspended in 1 ml TRIzol and homogenized using 150 to 200 μm glass beads at a setting of 6.0 for 40 s.
Additional RNA purification, removal of contaminating DNA, conversion to cDNA and quantitative real-time PCR (qRT-PCR) were performed as described previously (18).
Statistical analyses.
Data were log transformed prior to statistical testing so that they would more closely follow a normal distribution. Statistical significance of the difference between experimental groups was then assessed by one-way analysis of variance (ANOVA) applying Tukey's multiple comparison test using Prism 4 (GraphPad), except in the case of the UV survival experiment where two-way ANOVA was performed followed by Bonferroni posttests.
RESULTS
Isolation of mutant strains and complementation.
The strains containing mutations in uvrD1 alone or in conjunction with uvrA were constructed (43) as described in Materials and Methods. For each mutant strain, the deletion was confirmed by Southern analysis (Fig. 1). The uvrD1 gene was expressed from its own promoter in an integrating plasmid for complementation, as detailed in Materials and Methods.
Fig 1.

Analysis of the uvrD1 and uvrA uvrD1 mutant genotypes by Southern blot. (A) Schematic illustration of the uvrD1 locus and the Southern blot strategy. Shown are the genomic organization of the wild type (WT) and the mutated genomic uvrD1 region in the double crossover mutant (DCO). Fragments detected by the probe specific for the 5′ flanking region are shown in bold. (B) Southern blot hybridized with a uvrD1-specific probe. Genomic DNA from the M. tuberculosis wild type (lane 1), the uvrD1 mutant (lane 2), and the complemented uvrD1 mutant (lane 3) was digested with SacI and probed with a 2,411-bp HpaI/SacI uvrD1 gene fragment. The presence of a single 5.3-kb fragment instead of a 3.7-kb fragment demonstrates successful deletion of uvrD1 coding sequences. Complementation of uvrD1 is indicated by an additional hybridization signal at 2.4 kbp. Inactivation of the uvrA locus in the uvrA uvrD1 mutant was also reconfirmed by Southern blot analysis (data not shown).
The M. tuberculosis uvrD1 and uvrA uvrD1 mutants have altered colony morphology.
The uvrD1 and uvrA uvrD1 mutants differed from the wild type by changes in colony morphology, most noticeably in colony size (Fig. 2A). These colonies were markedly smaller than those of the wild-type or the complemented uvrD1 strain. Despite the differences in colony size, growth in liquid culture, as assessed by optical density measurements, displayed no statistically significant difference between the strains (Fig. 2B).
Fig 2.

In vitro growth characteristics of the uvrD1 and uvrA uvrD1 mutant strains. (A) The colony size and morphology of the uvrD1 and uvrA uvrD1 strains are compared with the wild type, the uvrD1 complement, and a uvrA mutant, demonstrating the reduced colony size caused by inactivation of uvrD1. In each case the white bar in the image represents 3 mm, and the images were taken after 3 weeks of growth at 37°C. (B) Growth of the strains in liquid culture was compared with that of the wild-type parental strain by measuring optical density.
The M. tuberculosis NER mutants have enhanced susceptibility to DNA damaging agents.
As M. tuberculosis contains two UvrD homologues, of which UvrD1 is most similar to UvrD of E. coli, we undertook experiments to confirm that UvrD1 is involved in NER. NER is particularly important for the repair of intrastrand cross-links, such as cyclobutane pyrimidine dimers commonly formed as a result of UV irradiation, and interstrand cross-links caused by bifunctional chemical reagents, such as mitomycin C (39). Therefore, the susceptibilities of the mutant strains to UV light and mitomycin C were assessed and compared with those of the parental strain.
The uvrA mutant was highly sensitive to UV light at doses which had only minor effects on the wild-type strain, and inactivation of uvrD1 in addition to uvrA resulted in enhanced susceptibility to UV irradiation (P < 0.001) (Fig. 3A). The uvrD1 mutant also exhibited greater sensitivity to UV than the wild type, although to a lesser degree than the uvrA mutant. The difference in sensitivity from the wild type was statistically significant at the higher doses tested (P < 0.001) (Fig. 3A). Survival of the uvrD1 mutant was restored by complementation (P > 0.05 compared with the wild type).
Fig 3.
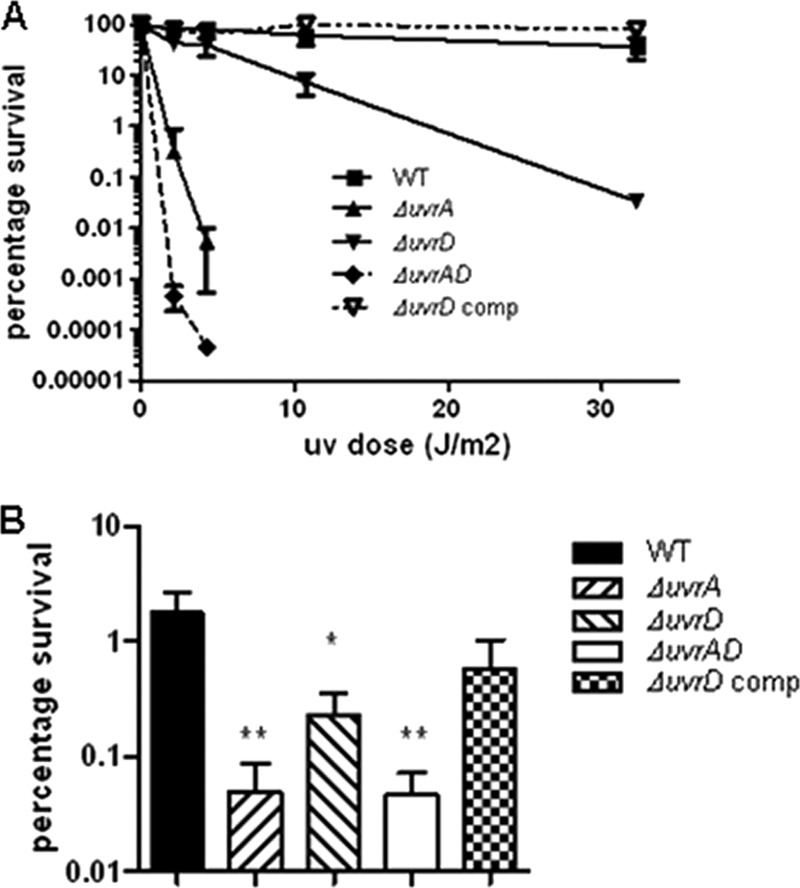
Sensitivity of the uvrD1 and uvrA uvrD1 mutant strains to DNA damage. Survival of the uvrD1 and uvrA uvrD1 strains following DNA damage caused by UV irradiation (A) or mitomycin C (B) was compared with that of the wild type, the uvrD1 complement, and a uvrA mutant. Survival following exposure to the indicated agent was assessed as described in Materials and Methods. In each case the surviving fraction was calculated by comparison with an untreated control. The data shown are the means of duplicate (UV) or triplicate (mitomycin C) assays from three biological replicates; the error bars represent standard deviations. One hundred percent survival in each case corresponds to approximately 107 CFU. The data in panel A for the uvrA and uvrA uvrD1 strains at all UV doses were significantly different from the wild type (P < 0.001; ANOVA). Survival of each mutant strain in panel B was significantly different from the wild type (*, P < 0.05; **, P < 0.01; ANOVA).
Similarly, the uvrA and uvrA uvrD1 mutant strains were significantly (P < 0.001) more sensitive to mitomycin C than the wild-type strain (Fig. 3B). Again, the uvrD1 mutant exhibited an intermediate level of susceptibility to this form of DNA damage compared with the uvrA mutant and the wild type (P < 0.05 compared with the wild type), and survival similar to the wild type was restored by complementation with uvrD1.
These results parallel the reported phenotypes for uvrA and uvrD mutants in E. coli (25, 53) and suggest that uvrD1 is the homologue involved in NER in M. tuberculosis.
The M. tuberculosis NER mutants exhibit elevated sensitivity to ROI and RNI.
During infection, DNA damage can be caused by ROI and/or RNI. Therefore, we investigated whether the mutant strains were more susceptible to representative agents generating such stresses.
The uvrA and uvrA uvrD1 mutant strains were highly sensitive to nitrosative stress (Fig. 4A), with survival being reduced by 500-fold and 2,000-fold, respectively, relative to that of the wild type (P < 0.001). Similar to the observations reported above for the standard DNA damaging agents, the susceptibility of the uvrD1 mutant to nitrosative stress was less severe than for the other mutants, although it was still 40-fold more sensitive than the wild type (P < 0.01); this enhanced sensitivity was restored to a near-wild-type level by complementation (P > 0.05 compared with the wild type).
Fig 4.
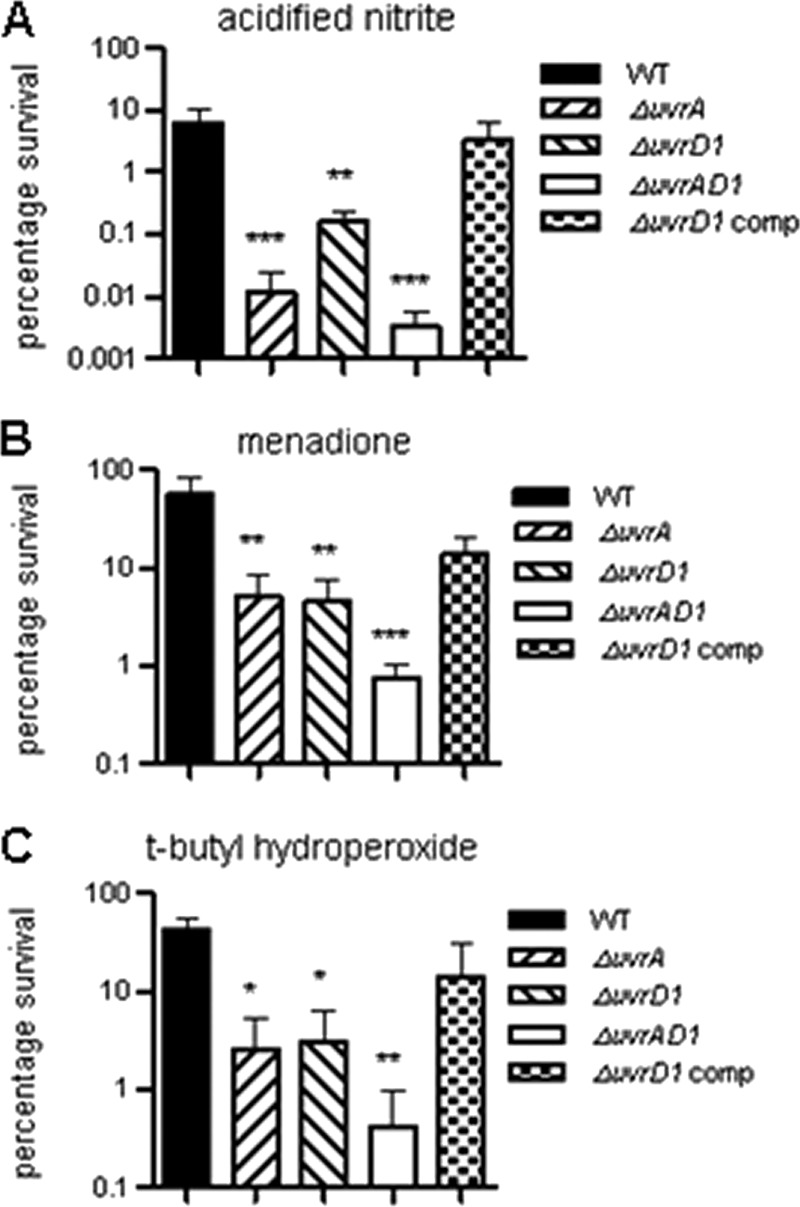
Susceptibility of the uvrD1 and uvrA uvrD1 mutant strains to nitrosative and oxidative stresses. Survival of the uvrD1 and uvrA uvrD1 strains following exposure to acidified sodium nitrite (A), t-butyl hydroperoxide (B), or menadione (C) was compared with that of the wild type, the uvrD1 complement, and a uvrA mutant. Survival following exposure to the indicated agent was assessed as described in Materials and Methods. In each case the surviving fraction was calculated by comparison with an untreated control. The data shown are the means of triplicate assays from three biological replicates; the error bars represent standard deviations. One hundred percent survival in each case corresponds to approximately 107 CFU. The data for each mutant was significantly different from that for the wild type for each stress condition (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ANOVA).
All three of the mutant strains were also more sensitive than the wild type to both t-butyl hydroperoxide and menadione but to a lesser extent than was the case for nitrosative stress (Fig. 4B and C). In the case of oxidative stress, the susceptibilities of the uvrA and uvrD1 mutants were similar to each other, with survival being reduced 11- to 12-fold in the case of menadione (P < 0.01) and 14- to 16-fold with t-butyl hydroperoxide (P < 0.05) relative to the wild type. In contrast, the uvrA uvrD1 strain was 73-fold (P < 0.001) and 98-fold (P < 0.01) more sensitive than the wild type to menadione and t-butyl hydroperoxide. Again, the phenotype of the uvrD1 mutant was restored to a near-wild-type level by complementation (P > 0.05 compared with the wild type).
Thus, NER in M. tuberculosis is important for the repair of damage caused by RNI and to a lesser extent by ROI and so might be expected to contribute to survival during infection.
The uvrD1 and the uvrA uvrD1 mutants showed decreased intracellular multiplication following infection of macrophages.
To investigate the function of NER during infection, we initially used a cell-based model. Both naïve and IFN-γ activated macrophages were infected with each strain, and the progress of infection was monitored.
The wild-type strain reproducibly replicated to levels corresponding to an increase of at least 100-fold the initial bacterial number during the course of the experiment. Growth and survival of the uvrA mutant was very similar to that of the wild type, but the uvrD1 and uvrA uvrD1 strains were unable to reach such high bacterial loads (Fig. 5A and B). At the end of the experiment, the bacterial numbers recovered from naïve infections with the uvrD1 and the uvrA uvrD1 mutants were consistently lower than those for the wild type (12- to 18-fold and 16- to 50-fold lower, respectively) (P < 0.001). When macrophages were IFN-γ stimulated, bacterial numbers were lower for all strains with the uvrD1 and the uvrA uvrD1 mutants remaining less able to replicate than the wild type. Bacterial numbers for the uvrD1 and the uvrA uvrD1 mutants were 3- to 6-fold (P < 0.05) and 14- to 29-fold (P < 0.001) lower, respectively, than for the wild type. Complementation of the uvrD1 mutant restored the ability to survive in macrophages to wild-type levels (Fig. 5C).
Fig 5.
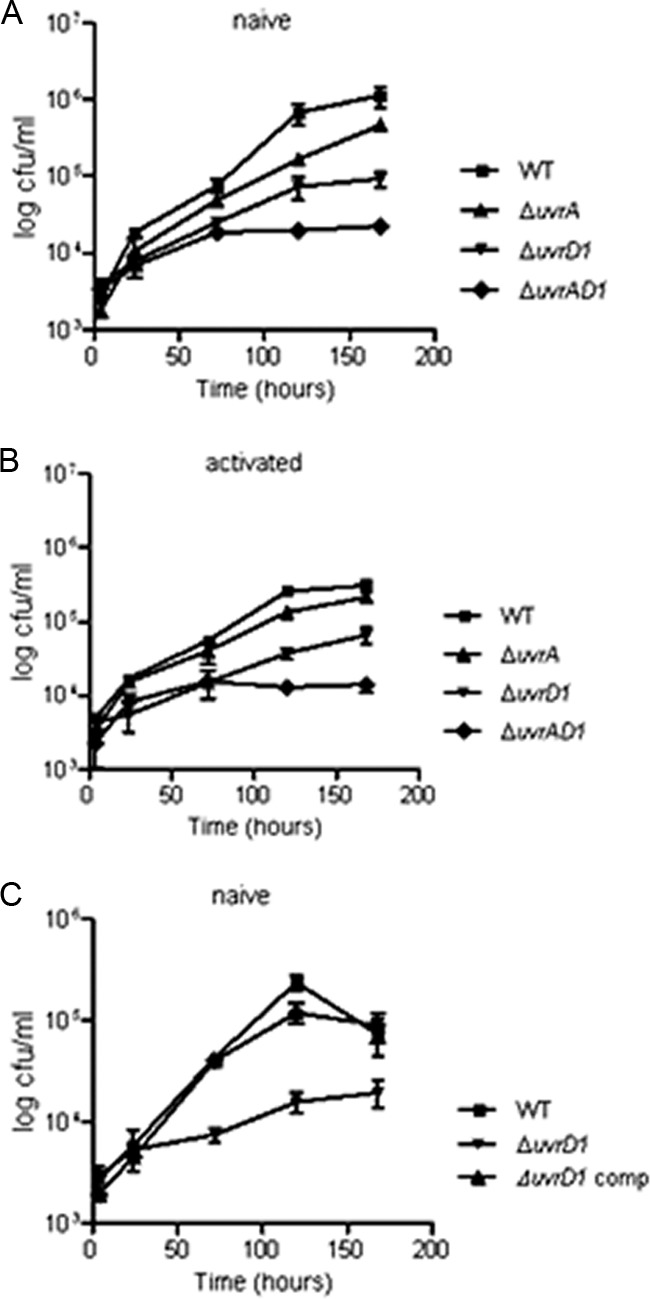
Attenuation of the uvrD1 and uvrA uvrD1 mutant strains in a macrophage model of infection. Growth and survival of the uvrA, uvrD1, and uvrA uvrD1 strains were compared with those of the wild type following infection of naïve macrophages (A and C) and IFN-γ-activated mouse bone-marrow-derived macrophages (B). Macrophages were isolated and infected as detailed in Materials and Methods. Bacterial numbers were determined at specific time points up to a maximum of 7 days postinfection. Each experiment was performed at least three times with similar results; the results of a representative experiment are shown, for which the data are the means of CFU determinations performed on triplicate infections, and the error bars represent standard deviations. The data from the uvrD1 and uvrA uvrD1 mutants were significantly different from those for the wild type from day 3 onwards (P < 0.05; ANOVA).
The uvrD1 and the uvrA uvrD1 mutants are attenuated following infection of mice.
To determine the role of NER in pathogenesis in whole animals, two mouse models of infection were used: a low-dose aerosol challenge model and an intravenous challenge model to assess complementation.
BALB/c mice were infected with each strain by the aerosol route (Fig. 6A and B). The bacterial load of the uvrA mutant was only slightly less than that of the wild type throughout the course of infection. However, the uvrD1 mutant exhibited a strongly attenuated phenotype, with consistently lower CFU after day 50 in both the lungs and the spleen (P < 0.001 compared with the wild type), indicating attenuation at the later stages of infection. Strikingly, the uvrA uvrD1 mutant was highly attenuated throughout the course of infection (P < 0.001 compared with the wild type), showing very limited replication in the lungs and with CFU in the spleen being close to the limit of detection at all time points analyzed.
Fig 6.

Attenuation of the uvrD1 and uvrA uvrD1 mutant strains in mouse models of infection. Growth and survival of the uvrD1 and uvrA uvrD1 strains were compared with the wild type and a uvrA mutant following infection of BALB/c mice as detailed in Materials and Methods. (A and B) Infection by the aerosol route; CFU were enumerated from the lungs and spleens of five mice for each time point. The data for the uvrA uvrD1 mutant were significantly different from the corresponding values for the wild type (P < 0.001), the uvrA mutant (P < 0.001), and the urvD1 mutant (P < 0.01) at all time points in the lungs and at each time point after day 50 in the spleens. The data for the uvrD1 mutant were significantly different from those for the wild type in both organs at each time point after day 50 (P < 0.001). (C and D) Infection by the intravenous route; additional strains in which the urvD1 and uvrA uvrD1 mutants were complemented with uvrD1 were included in the analysis. Bacterial loads were determined from the lungs and spleens of four mice for each time point. The data for the uvrA uvrD1 mutant were significantly different from the corresponding values for the wild type (P < 0.001), the uvrA mutant (P < 0.001), the urvD1 mutant (P < 0.01), and both the complemented strains (P < 0.001) at all time points in both organs. The data for the uvrD1 mutant were significantly different from those for the wild type and its complemented strain in both organs from day 42 onwards (P < 0.01). In each case the mutant strains showed similar phenotypes in a second independent experiment; the results shown are the means, and the error bars represent standard deviations.
To confirm that inactivation of uvrD1 in the uvrA uvrD1 double mutant was responsible for the attenuation observed, uvrD1 was complemented and an intravenous infection of mice was performed (Fig. 6C and D). The patterns of growth for the strains were similar to those seen in the aerosol infection experiment. The uvrD1 mutant failed to reach as high a bacterial burden as the wild type (P < 0.01), and CFU declined at later time points in the spleens, but this phenotype was restored upon complementation (P > 0.05 compared with the wild type). The uvrA uvrD1 mutant again was severely defective in its ability to establish an infection (P < 0.001 compared with the wild type). In contrast, complementation of uvrD1 restored the ability of the bacteria to replicate in the mouse to a level similar to that observed for the uvrA mutant (P > 0.05). This demonstrates that the severe attenuation of the uvrA uvrD1 strain is due to inactivation of uvrD1 in the absence of a functional NER system.
Expression of uvrD1 is increased in late infection.
To address the importance of UvrD1 in persistence, uvrD1 expression was assessed by qRT-PCR from 3 and 22 weeks infection of mice with wild-type M. tuberculosis. These time points correspond to acute and chronic stages of infection, respectively (Fig. 7). It was observed that uvrD1 was upregulated 5.6-fold in early infection compared to in vitro expression levels. Expression of uvrD1 was then further upregulated in late infection, increasing to 128-fold the in vitro levels. These observations support the importance of UvrD1 during infection, particularly in the chronic stage.
Fig 7.
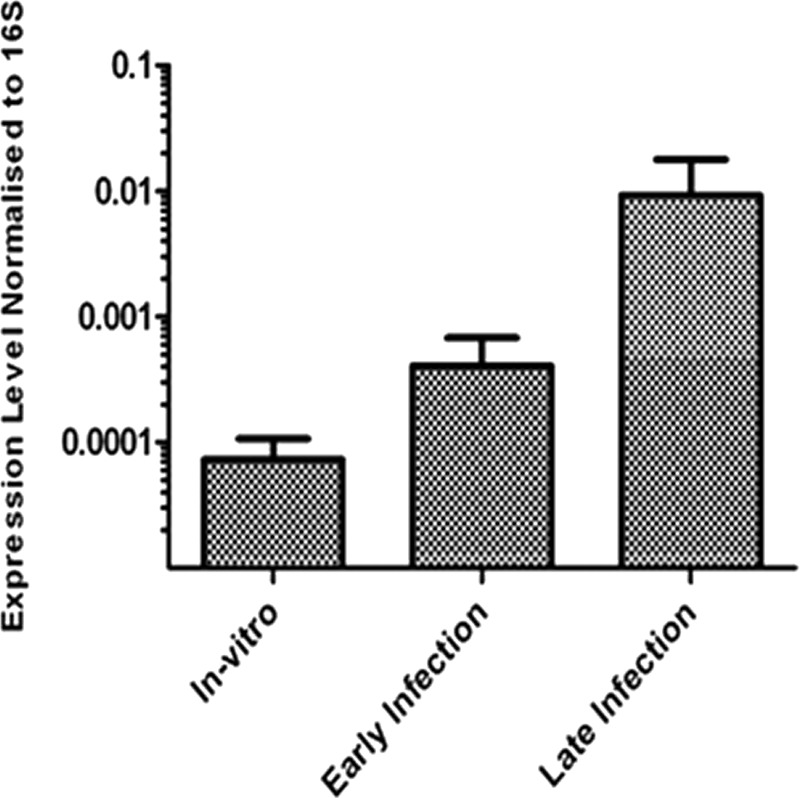
Expression of uvrD1 during mouse infection by qRT-PCR. Expression level of uvrD1 was measured from in vitro exponential cultures, early mouse infection (3 weeks), and late mouse infection (22 weeks). Expression level was normalized to that of 16S. The results shown are the means of 3 biological replicates, and the error bars represent standard deviations.
DISCUSSION
This study set out to address the role of NER in the pathogenicity of M. tuberculosis by targeting uvrD1 for inactivation in both a wild-type strain and an isogenic uvrA mutant strain. UvrA is part of the damage recognition complex while UvrD is the helicase involved in the final stages of oligonucleotide displacement prior to repair synthesis (51).
Published data demonstrate that E. coli UvrD is required for mismatch repair (23) (which is absent in mycobacteria [49]), NER (26), replication restart, and recombination (19, 30). Up until now there has been no direct evidence for the involvement of UvrD1 in nucleotide excision repair in M. tuberculosis. However, biochemical studies of M. tuberculosis UvrD1 have shown it to be a helicase with 3′-5′ polarity and with an unwinding preference for nicked DNA duplexes that resemble that of NER intermediates in addition to substrates resembling stalled replication forks (9), and a recent study has implicated a role for M. smegmatis UvrD1 in recombination (22).
UvrD1, having the closest sequence similarity to E. coli UvrD, was selected for inactivation. Inactivation of uvrD1 led to a pronounced reduction in colony size. However, growth in liquid culture was not affected, with no significant difference in doubling times between strains. Our demonstration of enhanced susceptibility of the uvrD1 mutant to UV and mitomycin C, which generate DNA damage classically repaired by NER, along with the restoration of this phenotype to that of the wild type upon complementation, confirms that UvrD1 is involved in the NER pathway. The intermediate sensitivity of the uvrD1 strain compared with the uvrA mutant and the wild-type strains to both these stresses is similar to the phenotype of uvrD mutants in E. coli (25, 53).
To investigate the role of NER in the repair of DNA damage generated by more physiologically relevant agents, susceptibilities to ROI and RNI were assessed. The agents used were t-butyl hydroperoxide, a membrane-permeant oxidant that is more stable than hydrogen peroxide (33), and menadione, a generator of superoxide stress (6, 38), for ROI and acidified sodium nitrite for RNI. While all the mutant strains exhibited increased sensitivity to these stresses, in each case the uvrA uvrD1 double mutant was most affected, suggesting that either UvrA or, more likely, UvrD1 plays a role in another process in addition to NER.
The enhanced susceptibility of the mutants was most pronounced for the nitrosative stress. This may be a consequence of deaminated bases resulting from NO exposure reacting with other cellular components to form adducts that require NER for their repair (34), while base damage caused by oxidative stress is also subject to repair by base excision repair (29). An M. tuberculosis uvrB mutant has been reported to exhibit a dramatic increase in susceptibility to nitrosative stress generated by acidified sodium nitrite (11, 12). The same authors found no significant increase in sensitivity to ROI (hydrogen peroxide, cumene hydroperoxide, or plumbagin) in vitro for their uvrB strain, as assessed using a disk diffusion assay. In contrast, uvrB mutants of M. smegmatis have been reported to exhibit enhanced susceptibility to ROI as well as RNI (22, 27), similar to the findings reported here for M. tuberculosis. However, the difference could be due to the different assays used in the studies.
There was no significant difference in the growth and survival of the uvrA mutant and that of the wild type within macrophages, indicating that NER is not required under these conditions. This could either be because significant DNA damage does not occur or, more likely, because other DNA repair mechanisms can compensate for the lack of NER. The M. tuberculosis uvrB mutant studied previously likewise exhibited only a slightly reduced ability to survive in BMDMs (12), although the infection conditions used in that study differed from those used here. In contrast, both the uvrD1 and the uvrA uvrD1 mutants exhibited reduced replication compared with the wild type following infection of macrophages.
The uvrA uvrD1 mutant phenotype was significantly reversed by complementation of uvrD1 alone. This would indicate that either both UvrA and UvrD must be inactivated to block the NER pathway, and/or there is an additional function for UvrD1 outside NER that is important for replication under intracellular conditions. This alternate function for UvrD1 during infection is supported by the less-pronounced phenotype that is observed for the uvrD1 mutant to in vitro RNI stress compared to the uvrA mutant, in contrast to the relative ability of these strains to survive during infection.
Reactive nitrogen species produced by inducible nitric oxide synthase (iNOS) from IFN-γ stimulation (17) play a strong role in controlling infection. Treatment of M. tuberculosis-infected mice with iNOS inhibitors given during acute or chronic infection caused mice to succumb to the disease (20, 36). Thus, there is a requirement for a functional NER and other DNA damage repair mechanisms during all stages of infection. Both types of mouse infection experiments conducted in this study demonstrated a striking attenuation throughout the course of infection when both uvrA and uvrD1 were inactivated. The uvrD1 single mutant showed attenuation at the later time points of infection, indicating a requirement for this protein in persistence, while the uvrA mutant was only modestly affected. In the study by Darwin and Nathan (12), the uvrB mutant exhibited a modest reduction in bacterial load during a mouse infection, analogous to that seen here for the uvrA mutant. The uvrB mutant was also shown to be attenuated for growth in primate lungs (15), indicating the importance for NER during infection.
When M. tuberculosis from infected mouse lungs was analyzed by qRT-PCR for uvrD1 expression, it was observed that there were increased transcript levels during infection compared to that found during in vitro growth. In particular, there was a dramatic increase of ca. 100-fold during late infection. As the requirement for uvrD1 is increased in chronic infection, this would explain why the uvrD1 mutant lacks the ability to persist at later time points.
Thus, it appears that UvrD1 is involved in processes separate from NER that are important for persistence in the absence of NER, while deletion of uvrD1 alone is insufficient to fully inactivate NER.
The requirement for UvrD1 in pathogenesis in the absence of NER may reflect a role in replication restart or recombination. UvrD in E. coli or its homologue PcrA in Bacillus subtilis is important for replication restart (3, 19). It is thought that this action is at least partly mediated by the ability of these enzymes to restrict the actions of RecA at blocked replication forks (1, 31). Indeed, biochemical studies have shown that M. tuberculosis UvrD1 suppresses DNA strand exchange reactions catalyzed by RecA in vitro (46). UvrD homologues can also act as translocases, and in this way they are able to displace proteins bound to DNA (1). It is possible that translocase activity of M. tuberculosis UvrD1 could contribute to clearing RecA from the DNA at fork structures (9, 46). This behavior would be similar to that of the E. coli RecG helicase, which has been shown to be involved in the recovery of stalled replication forks (41). Although UvrD and RecG proteins are not homologues by sequence, they could have overlapping functions.
The studies described here suggest that targeting UvrD1 might be a potential strategy to combat persistence and that an inhibitor of UvrD1 would be particularly useful in combination with a compound such as one recently described (32) that targets an essential component of NER. Importantly, these results validate DNA repair processes in M. tuberculosis as potential new drug targets.
ACKNOWLEDGMENTS
This work was supported by the European Community (CSI_LTB LSHP-CT-2007-037235), the United Kingdom Medical Research Council (program number U1175 32056 to E.O.D.), and the University of Zurich (to E.C.B.).
We thank Bosco Chan for performing some preliminary phenotyping experiments and Belinda Dagg, James Keeble, and Biological Services at both NIMR and NIBSC for help with the mouse infection experiments.
Footnotes
Published ahead of print 30 March 2012
REFERENCES
- 1. Anand SP, Zheng H, Bianco PR, Leuba SH, Khan SA. 2007. DNA helicase activity of PcrA is not required for the displacement of RecA protein from DNA or inhibition of RecA-mediated strand exchange. J. Bacteriol. 189:4502–4509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barry CE, III, et al. 2009. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 7:845–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bidnenko V, Lestini R, Michel B. 2006. The Escherichia coli UvrD helicase is essential for Tus removal during recombination-dependent replication restart from Ter sites. Mol. Microbiol. 62:382–396 [DOI] [PubMed] [Google Scholar]
- 4. Brosch R, et al. 1998. Use of a Mycobacterium tuberculosis H37Rv bacterial artificial chromosome library for genome mapping, sequencing, and comparative genomics. Infect. Immun. 66:2221–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burney S, Caulfield JL, Niles JC, Wishnok JS, Tannenbaum SR. 1999. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. 424:37–49 [DOI] [PubMed] [Google Scholar]
- 6. Cadenas E, Sies H. 1985. Oxidative stress: excited oxygen species and enzyme activity. Adv. Enzyme Regul. 23:217–237 [DOI] [PubMed] [Google Scholar]
- 7. Chan ED, Chan J, Schluger NW. 2001. What is the role of nitric oxide in murine and human host defense against tuberculosis? Current knowledge. Am. J. Respir. Cell Mol. Biol. 25:606–612 [DOI] [PubMed] [Google Scholar]
- 8. Cole ST, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544 [DOI] [PubMed] [Google Scholar]
- 9. Curti E, Smerdon SJ, Davis EO. 2007. Characterization of the helicase activity and substrate specificity of Mycobacterium tuberculosis UvrD. J. Bacteriol. 189:1542–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Czeczot H, Tudek B, Lambert B, Laval J, Boiteux S. 1991. Escherichia coli Fpg protein and UvrABC endonuclease repair DNA damage induced by methylene blue plus visible light in vivo and in vitro. J. Bacteriol. 173:3419–3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. 2003. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302:1963–1966 [DOI] [PubMed] [Google Scholar]
- 12. Darwin KH, Nathan CF. 2005. Role for nucleotide excision repair in virulence of Mycobacterium tuberculosis. Infect. Immun. 73:4581–4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davis EO, et al. 2002. DNA damage induction of recA in Mycobacterium tuberculosis independently of RecA and LexA. Molecular Microbiology 46:791–800 [DOI] [PubMed] [Google Scholar]
- 14. Davis EO, Forse LN. 2009. DNA repair: key to survival?, p 79–117 In Parish T, Brown A. (ed), Mycobacterium genomics and molecular biology. Caister Academic Press, Wymondham, United Kingdom [Google Scholar]
- 15. Dutta NK, et al. 2010. Genetic requirements for the survival of tubercle bacilli in primates. J. Infect. Dis. 201:1743–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. 1999. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA 282:677–686 [DOI] [PubMed] [Google Scholar]
- 17. Ehrt S, Schnappinger D. 2009. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol. 11:1170–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fivian-Hughes AS, Davis EO. 2010. Analyzing the regulatory role of the HigA antitoxin within Mycobacterium tuberculosis. J. Bacteriol. 192:4348–4356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Florés MJ, Sanchez N, Michel B. 2005. A fork-clearing role for UvrD. Mol. Microbiol. 57:1664–1675 [DOI] [PubMed] [Google Scholar]
- 20. Flynn JL, Scanga CA, Tanaka KE, Chan J. 1998. Effects of aminoguanidine on latent murine tuberculosis. J. Immunol. 160:1796–1803 [PubMed] [Google Scholar]
- 21. Frota CC, Papavinasasundaram KG, Davis EO, Colston MJ. 2004. The AraC family transcriptional regulator Rv1931c plays a role in the virulence of Mycobacterium tuberculosis. Infect. Immun. 72:5483–5486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Güthlein C, et al. 2009. Characterization of the mycobacterial NER system reveals novel functions of the uvrD1 helicase. J. Bacteriol. 191:555–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iyer RR, Pluciennik A, Burdett V, Modrich PL. 2006. DNA mismatch repair: functions and mechanisms. Chem. Rev. 106:302–323 [DOI] [PubMed] [Google Scholar]
- 24. Jung YJ, LaCourse R, Ryan L, North RJ. 2002. Virulent but not avirulent Mycobacterium tuberculosis can evade the growth inhibitory action of a T helper 1-dependent, nitric oxide synthase 2-independent defense in mice. J. Exp. Med. 196:991–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuemmerle NB, Masker WE. 1980. Effect of the uvrD mutation on excision repair. J. Bacteriol. 142:535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kumura K, Sekiguchi M, Steinum AL, Seeberg E. 1985. Stimulation of the UvrABC enzyme-catalyzed repair reactions by the UvrD protein (DNA helicase II). Nucleic Acids Res. 13:1483–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kurthkoti K, Kumar P, Jain R, Varshney U. 2008. Important role of the nucleotide excision repair pathway in Mycobacterium smegmatis in conferring protection against commonly encountered DNA-damaging agents. Microbiology 154:2776–2785 [DOI] [PubMed] [Google Scholar]
- 28. Labes M, Puhler A, Simon R. 1990. A new family of RSF1010-derived expression and lac-fusion broad-host-range vectors for gram-negative bacteria. Gene 89:37–46 [DOI] [PubMed] [Google Scholar]
- 29. Laval J. 1996. Role of DNA repair enzymes in the cellular resistance to oxidative stress. Pathol. Biol. 44:14–24 [PubMed] [Google Scholar]
- 30. Lestini R, Michel B. 2007. UvrD controls the access of recombination proteins to blocked replication forks. EMBO J. 26:3804–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mahdi AA, Buckman C, Harris L, Lloyd RG. 2006. Rep. and PriA helicase activities prevent RecA from provoking unnecessary recombination during replication fork repair. Genes Dev. 20:2135–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mazloum NA, et al. 2011. Identification of a chemical that inhibits the mycobacterial UvrABC complex in nucleotide excision repair. Biochemistry 50:1329–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mitić-Culafić D, et al. 2009. Protective effect of linalool, myrcene and eucalyptol against t-butyl hydroperoxide induced genotoxicity in bacteria and cultured human cells. Food Chem. Toxicol. 47:260–266 [DOI] [PubMed] [Google Scholar]
- 34. Nakano T, et al. 2005. Repair activity of base and nucleotide excision repair enzymes for guanine lesions induced by nitrosative stress. Nucleic Acids Res. 33:2181–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nathan C, Shiloh MU. 2000. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. U. S. A. 97:8841–8848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nathan C. 2002. Inducible nitric oxide synthase in the tuberculous human lung. Am. J. Respir. Crit. Care Med. 166:130–131 [DOI] [PubMed] [Google Scholar]
- 37. Nicholson S, et al. 1996. Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J. Exp. Med. 183:2293–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Palyada K, et al. 2009. Characterization of the oxidative stress stimulon and PerR regulon of Campylobacter jejuni. BMC Genomics 10:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reardon JT, Sancar A. 2005. Nucleotide excision repair. Prog. Nucleic Acid Res. Mol. Biol. 79:183–235 [DOI] [PubMed] [Google Scholar]
- 40. Rossi F, et al. 2011. The biological and structural characterization of Mycobacterium tuberculosis UvrA provides novel insights into its mechanism of action. Nucleic Acids Res. 39:7316–7328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Runyon GT, Bear DG, Lohman TM. 1990. Escherichia coli helicase II (UvrD) protein initiates DNA unwinding at nicks and blunt ends. Proc. Natl. Acad. Sci. U. S. A. 87:6383–6387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sambrook J, Fritsch E, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 43. Sander P, Meier A, Bottger EC. 1995. rpsL+: a dominant selectable marker for gene replacement in mycobacteria. Mol. Microbiol. 16:991–1000 [DOI] [PubMed] [Google Scholar]
- 44. Sander P, et al. 2001. Mycobacterium bovis BCG recA deletion mutant shows increased susceptibility to DNA-damaging agents but wild-type survival in a mouse infection model. Infect. Immun. 69:3562–3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sidorkina O, Saparbaev M, Laval J. 1997. Effects of nitrous acid treatment on the survival and mutagenesis of Escherichia coli cells lacking base excision repair (hypoxanthine-DNA glycosylase-ALK A protein) and/or nucleotide excision repair. Mutagenesis 12:23–28 [DOI] [PubMed] [Google Scholar]
- 46. Singh P, et al. 2010. Mycobacterium tuberculosis UvrD1 and UvrA proteins suppress DNA strand exchange promoted by cognate and noncognate RecA proteins. Biochemistry 49:4872–4883 [DOI] [PubMed] [Google Scholar]
- 47. Sinha KM, Stephanou NC, Unciuleac MC, Glickman MS, Shuman S. 2008. Domain requirements for DNA unwinding by mycobacterial UvrD2, an essential DNA helicase. Biochemistry 47:9355–9364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Springer B, Sander P, Sedlacek L, Ellrott K, Bottger EC. 2001. Instability and site-specific excision of integration-proficient mycobacteriophage L5 plasmids: development of stably maintained integrative vectors. Int. J. Med. Microbiol. 290:669–675 [DOI] [PubMed] [Google Scholar]
- 49. Springer B, et al. 2004. Lack of mismatch correction facilitates genome evolution in mycobacteria. Mol. Microbiol. 53:1601–1609 [DOI] [PubMed] [Google Scholar]
- 50. Stover CK, et al. 1991. New use of BCG for recombinant vaccines. Nature 351:456–460 [DOI] [PubMed] [Google Scholar]
- 51. Truglio JJ, Croteau DL, Van Houten B, Kisker C. 2006. Prokaryotic nucleotide excision repair: the UvrABC system. Chem. Rev. 106:233–252 [DOI] [PubMed] [Google Scholar]
- 52. Tufariello JM, Chan J, Flynn JL. 2003. Latent tuberculosis: mechanisms of host and bacillus that contribute to persistent infection. Lancet Infect. Dis. 3:578–590 [DOI] [PubMed] [Google Scholar]
- 53. Washburn BK, Kushner SR. 1991. Construction and analysis of deletions in the structural gene (uvrD) for DNA helicase II of Escherichia coli. J. Bacteriol. 173:2569–2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. World Health Organization 2009. Global tuberculosis control: epidemiology, strategy, financing. WHO report 2009. World Health Organization, Geneva, Switzerland [Google Scholar]