

Highlights
► SiRNAs inhibit adenovirus multiplication. ► Adenoviral DNA replication is a key target for siRNA-mediated inhibition. ► SiRNAs can delay the death of adenovirus-infected cells.
Abbreviations: RNAi, RNAinterference; siRNA, small interfering RNA
Keywords: Adenovirus, Infection, RNA interference, siRNA
Abstract
Human adenoviruses are a common threat to immunocompromised patients, e.g., HIV-positive individuals or solid-organ and, in particular, allogeneic stem cell transplant recipients. Antiviral drugs have a limited effect on adenoviruses, and existing treatment modalities often fail to prevent fatal outcome. Silencing of viral genes by short interfering RNAs (siRNAs) holds a great promise in the treatment of viral infections. The aim of the present study was to identify adenoviral candidate targets for RNA interference-mediated inhibition of adenoviral replication. We investigated the impact of silencing of a set of early, middle, and late viral genes on the replication of adenovirus 5 in vitro. Adenovirus replication was inhibited by siRNAs directed against the adenoviral E1A, DNA polymerase, preterminal protein (pTP), IVa2, hexon, and protease genes. Silencing of early and middle genes was more effective in inhibiting adenovirus multiplication than was silencing of late genes. A siRNA directed against the viral DNA polymerase mRNA decreased viral genome copy numbers and infectious virus progeny by several orders of magnitude. Since silencing of any of the early genes directly or indirectly affected viral DNA synthesis, our data suggest that reducing viral genome copy numbers is a more promising strategy for the treatment of adenoviral infections than is reducing the numbers of proteins necessary for capsid generation. Thus, adenoviral DNA replication was identified as a key target for RNAi-mediated inhibition of adenovirus multiplication. In addition, the E1A transcripts emerged as a second important target, because its knockdown markedly improved the viability of cells at late stages of infection.
1. Introduction
Human adenoviruses are double-stranded DNA (dsDNA) viruses associated with a wide range of human diseases. They are mainly responsible for self-limiting respiratory and intestinal infections, and predominantly affect children and young adults (Lenaerts et al., 2008). However, more severe manifestations, including hemorrhagic cystitis, nephritis, pneumonia, hepatitis, enterocolitis, and disseminated disease, are observed in immunocompromised patients, such as solid-organ and, in particular, allogeneic stem cell transplant recipients (Echavarria, 2008; Ison, 2006; Kojaoghlanian et al., 2003). These manifestations can be life-threatening or even lethal. In the case of disseminated disease, mortality rates as high as 80% have been reported (Blanke et al., 1995; Hale et al., 1999; Howard et al., 1999; Lion et al., 2003; Munoz et al., 1998). Severe manifestations are most commonly associated with serotypes from species B and C (Kojaoghlanian et al., 2003), with a high prevalence of species C in certain geographical areas (Ebner et al., 2006; Lion et al., 2003, 2010).
In the immunocompetent host, a severe manifestation of adenovirus infection is epidemic keratoconjunctivitis (EKC). This is predominantly associated with serotypes 8, 19, and 37 (all belonging to species D), is highly contagious, and can have severe consequences on visual acuity (Gordon et al., 1996). Besides, EKC is generally associated with significant morbidity, which results in considerable economic losses.
The most common agents for treating adenovirus infections are ribavirin and cidofovir. However, apparent clinical efficacy has been demonstrated only for cidofovir. Although cidofovir is widely used, its activity is limited and insufficient to completely prevent fatal outcomes among hematopoietic stem cell transplant recipients (Lenaerts et al., 2008; Lindemans et al., 2010; Ljungman et al., 2003; Symeonidis et al., 2007; Yusuf et al., 2006). Furthermore, concomitant recovery of the immune system may be necessary for complete adenovirus clearance (Chakrabarti et al., 2002; Heemskerk et al., 2005; Lindemans et al., 2010). Cidofovir displays significant nephrotoxicity and limited bioavailability, and this has prompted the development of improved derivatives. However, the effectiveness of these compounds is still under evaluation (Hartline et al., 2005; Paolino et al., 2011). Thus, in view of the growing numbers of immunosuppressed patients, the development of alternative anti-adenovirus treatment options is required to decrease adenovirus-mediated mortality among immunocompromised patients, and also to decrease economic losses caused by milder forms of adenovirus-related disease.
RNA interference (RNAi) is a post-transcriptional mechanism of gene silencing conserved among eukaryotic cells (Carthew and Sontheimer, 2009; Ghildiyal and Zamore, 2009; Huntzinger and Izaurralde, 2011; Hutvagner and Simard, 2008; Kawamata and Tomari, 2010). It is mediated through small double-stranded RNAs (dsRNAs), of ∼21–25 nt in length, which guide the RNA-induced silencing complex (RISC) to the respective target mRNAs (Fire et al., 1998). Depending on the degree of complementarity between the so-called antisense (or guide) strand of the dsRNA and target mRNA, RNAi can bring about the cleavage of the mRNA (in the case of full or nearly full complementarity), accelerated degradation (as a consequence of deadenylation), or translational repression. Following the discovery that the introduction of synthetic small interfering RNAs (siRNAs) into cells can trigger RNAi (Elbashir et al., 2001), this mechanism was rapidly harnessed as a tool to silence disease-associated human, and also viral genes (Davidson and McCray, 2011). Since then, siRNA-mediated silencing of viral genes has been employed to inhibit the replication of a variety of DNA and RNA viruses, in vitro and also in vivo (Arbuthnot, 2010; Haasnoot et al., 2007; Zhou and Rossi, 2011).
Adenoviruses contain a linear dsDNA genome, ∼36 kb long. The first gene to be expressed during the infection cycle is E1A. This gene has a central role, because it reprograms the cell in a way that promotes efficient virus replication (Berk, 2005; Pelka et al., 2008; Zhao et al., 2003). Deletion of E1A renders adenoviruses replication deficient. E1A expression ultimately leads to the activation of other early and late promoters and triggers the onset of viral DNA replication. Viral DNA replication is dependent on three viral proteins: the viral DNA polymerase; the preterminal protein (pTP); and the DNA-binding protein (DBP) (de Jong et al., 2003). Besides creating dsDNAs for packaging into capsids (accomplished with the help of the IVa2 protein) (Zhang and Imperiale, 2003), replication of the adenoviral genome activates the expression of other viral genes, e.g., IVa2 (Flint, 1986; Iftode and Flint, 2004) and genes transcribed from the major late promoter (MLP) (Shaw and Ziff, 1980). Upregulation of major late (ML) gene expression also involves the IVa2 protein (Tribouley et al., 1994), and results in the synthesis of gene products that primarily constitute structural components of the virion or are involved in its assembly. The major component of the capsid is the hexon protein (Russell, 2009). Capsid maturation requires the action of adenovirus protease, also called adenain (Webster et al., 1989). This protein, which is an integral part of the mature virion, is also required for disassembly of the virion upon virus entry, and consequently for release of the viral DNA (Greber et al., 1996).
In vitro silencing of adenoviral genes by siRNAs has been demonstrated for an adenovirus (Ad) 11 strain (2K2/507/KNIH; species B; isolated in Korea) (Chung et al., 2007), and also for a mutant strain of Ad5 (species C) lacking the E1B and E3 genes (Eckstein et al., 2010). In the case of Ad11, siRNAs directed against E1A were reported to result in an overall reduction of plaque-forming capacity. For the Ad5 mutant strain, siRNAs targeting the E1A, IVa2, and hexon mRNAs were evaluated, and the IVa2 mRNA-targeting siRNA was reported to most efficiently decrease virus production. A protective effect on cell viability was observed only when the IVa2 mRNA-targeting siRNA was combined with an E1A mRNA-directed siRNA and administered at high concentration. The Ad5 mutant virus used represented a rather artificial test system, in that it lacked the E1B genes which, when present, prevent premature cell death, thereby prolonging virus replication and promoting viral late mRNA export from the nucleus (Blackford and Grand, 2009; Flint and Gonzalez, 2003; Subramanian et al., 1995; Woo and Berk, 2007). Together with the fact that the E1A gene was expressed from an artificial minimal CMV promoter autoactivated by E1A (Fechner et al., 2003), these differences from the wild-type virus make it somewhat difficult to accurately assess the potential of siRNA-mediated adenovirus gene silencing as a strategy for inhibiting adenovirus multiplication.
Here, we investigated the impact of siRNA-mediated adenovirus gene silencing on the replication of wild-type adenovirus. We expanded the panel of potential adenoviral targets, by evaluating siRNAs directed against the Ad5 E1A, DNA polymerase, pTP, IVa2, hexon, and protease mRNAs. Based on our in vitro results, we propose that the adenoviral mRNAs originating from genes which are essential for viral DNA replication (i.e., the DNA polymerase and pTP (and potentially the DBP) genes are promising targets for RNAi-mediated inhibition of adenovirus multiplication. Moreover, we demonstrate that highly potent E1A mRNA-directed siRNAs, which are also able to inhibit virus replication (albeit to a lesser extent than the DNA polymerase mRNA-directed siRNA), are capable of concomitantly delaying cell death, without the need for combination with other siRNAs. This distinct mode of inhibition may be exploited in vivo for siRNA-mediated attenuation of virus release and, consequently, virus spread.
2. Materials and methods
2.1. Cell culture and virus production
HEK293 (human embryonic kidney; ATCC CRL-1573) and A549 (human epithelial lung carcinoma; ATCC CCL-185) cells were cultivated in Dulbecco’s Modified Eagles Medium (DMEM) with stabilized glutamine (PAA Laboratories, Pasching, Austria) supplemented with 10% fetal bovine serum (FBS; PAA Laboratories) in a humidified 5% CO2 atmosphere at 37 °C. Ad1 (ATCC VR-1), Ad2 (ATCC VR-846), and Ad6 (ATCC-VR6), were amplified in A549 cells; Ad5 (ATCC VR-5) was amplified in HEK293 cells. Virus purifications were performed by standard CsCl density gradient ultracentrifugation. Infectious virus particle titers were determined on A549 cells by 50% tissue culture infective dose (TCID50) assays.
2.2. Vector construction
For the construction of vectors employed in dual-luciferase assays, parts of the Ad5 genome were amplified by PCR using primers specific for E1A (E1A-f1 5′-CGACACCGGGTTTAAACATGAGACATATTATCTGCCAC-3′ and E1A-r1 5′-CAACTCATTGTTTAAACAAAGGCGTTAACCA-3′; annealing temperature [Ta]: 50 °C), DNA polymerase (Pol-f1 5′-ACTCATATGGCCTTGGCTCAAGCTCACCGGGC-3′ and Pol-r1 5′-ACTAGATCTACGGCATCTCGATCCAGCATATC-3′; Ta: 55 °C), pTP (pTP-f3 5′-CTTTTGCACGGTCTCGAGCGTCAACGATTGCGC-3′ and pTP-r3 5′-GTGTCCTTGGATGCGGCCGCTAAAAGCGGTGACGCGGG-3′; Ta: 65 °C), IVa2 (IVa2-f1 5′-CACCGGCTCGTTTAAACCAGAGGGCGAAGAC-3′ and IVa2-r1 5′-AAACATAAAGTTTAAACCAGACTCTGTTTGGAT-3′; Ta: 50 °C), hexon (Hex-f1 5′-CCGCTTCTCGAGATGGCTACCCCTTCGATGATG-3′ and Hex-r1 5′-TGTTGCGCGGCCGCTTATGTTGTGGCGTTGCCGG-3′; Ta: 57 °C), and protease (Prot-f1 5′-CAAGCAACAGTTTAAACAGCTGCCGCCATGG-3′ and Prot-r1 5′-AAATAAGTTTAAACGCCTTTATTGAAAGTGTCTC-3′; Ta: 50 °C). The PCR reactions were performed in a total volume of 50 μL containing 10x PCR buffer (Peqlab), 400 μM dNTPs, 1 μM of each primer, 4 mM MgSO4 and 2.5 U of Pwo-DNA-Polymerase (Peqlab). The cycling parameters consisted of a total of 30 cycles of denaturing at 95 °C for 1 min, followed by annealing at the appropriate temperature for 1 min and extension at 72 °C for 2 min. The PCR products were subjected to agarose gel electrophoresis, stained with ethidium bromide, and visualized on a UV transilluminator. The PCR fragments were inserted into the PmeI site (E1A, IVa2, protease fragments), XhoI and NotI sites (pTP, hexon), or NdeI and BglII sites (DNA polymerase) of psiCHECK-2 (Promega, Mannheim, Germany), all located within the 3′ UTR of the Renilla luciferase gene. The resulting vectors were named psiCHECK-E1A, psiCHECK-pol, psiCHECK-pTP, psiCHECK-IVa2, and psiCHECK-hex. Restriction enzymes and DNA-modifying enzymes were purchased from Fermentas (St. Leon-Rot, Germany) or New England Biolabs (Frankfurt am Main, Germany). PCR reactions were performed with Pwo DNA polymerase obtained from Roche Diagnostics (Vienna, Austria).
2.3. Nucleic acid extraction
Circular plasmid DNA was extracted with QIAprep® Spin Miniprep Kits (QIAGEN, Hilden, Germany), EasyPrep® Pro Plasmid Miniprep Kits (Biozym, Oldendorf, Germany), or HiSpeed® Plasmid Midi Kits (QIAGEN). PCR products were purified with a QIAquick® PCR Purification Kit (QIAGEN). Adenoviral DNA was isolated from cells using a QIAamp DNA Blood Mini Kit (QIAGEN). Total RNA was isolated using an RNeasy® Mini Kit (QIAGEN).
2.4. SiRNAs
With the exception of pTP-si1, pTP-si2, pTP-si3, and pTP-si4, all siRNAs (Table 1) were obtained from Invitrogen (LifeTechnologies Austria, Vienna, Austria). They represented 25-mer, blunt-ended siRNAs carrying the Invitrogen “Stealth” modification. Due to the type of chemical modification, only the antisense strand can participate in RNAi, thus avoiding not only unwanted, sense strand-mediated, off-target effects but also preventing any possible interference of the sense strand with adenoviral transcripts generated from the opposite viral DNA strand not intended to be targeted. Besides, this type of modification (frequently present in similar versions in commercial siRNAs) can increase the intracellular half-life of siRNAs and reduce their cytotoxicity. The pTP-si1 to pTP-si4 siRNAs (obtained from Ambion/LifeTechnologies Austria, Vienna, Austria) were 21-mer, unmodified siRNAs carrying two nucleotide (nt) TT overhangs at their 3′ ends and were also included in our experiments. As negative controls, two distinct universal non-targeting siRNAs (Invitrogen, Ambion), matching the type of design of the respective targeting siRNAs, were employed. SiRNAs were designed using the Invitrogen BLOCK-iT™ RNAi Designer or Dharmacon siDESIGN tools and target site accessibility, as calculated by RNAxs (http://rna.tbi.univie.ac.at/cgi-bin/RNAxs), was taken into account.
Table 1.
siRNA sequences.
| Name | Sequence (sense/antisense) | Target site in Ad5a | Target | Serotype specificityb |
|---|---|---|---|---|
| E1A-si1 | 5′-AACGAGGAGGCGGUUUCGCAGAUUU-3′ | 731–755 | E1A | 1, 2, 5, 6 |
| 5′-AAAUCUGCGAAACCGCCUCCUCGUU-3′ | ||||
| E1A-si2 | 5′-CAGGAAGGGAUUGACUUACUCACUU-3′ | 782–806 | E1A | 1, 5 |
| 5′-AAGUGAGUAAGUCAAUCCCUUCCUG-3′ | ||||
| E1A-si3 | 5′-CCUGUGUCUAGAGAAUGCAAUAGUA-3′ | 1333–1357 | E1A | 2, 5, 6 |
| 5′-UACUAUUGCAUUCUCUAGACACAGG-3′ | ||||
| E1A-si3_scr | 5′-CCUUGUCGAGAAAGUACUAAUGGUA-3′ | – | – | – |
| 5′-UACCAUUAGUACUUUCUCGACAAGG-3′ | ||||
| Pol-si1 | 5′-CAAGACCCGCUUACCUUCUACUGCA-3′ | 7782–7806 | DNA pol | 1, 2, 5, 6 |
| 5′-UGCAGUAGAAGGUAAGCGGGUCUUG-3′ | ||||
| Pol-si2 | 5′-CAACGUCUUCCAGCGUCCAACCAUA-3′ | 6928–6952 | DNA pol | 1, 2, 5, 6 |
| 5′-UAUGGUUGGACGCUGGAAGACGUUG-3′ | ||||
| Pol-si3 | 5′-TGUCUCAGAGUGGUCCGAGUUUCUA-3′ | 5818–5842 | DNA pol | 5 |
| 5′-UAGAAACUCGGACCACUCUGAGACA-3′ | ||||
| Pol-si4 | 5′-CGUCUUCCAGCGUCCAACCAUAUCA-3′ | 6925–6949 | DNA pol | 1, 2, 5, 6 |
| 3′ UGAUAUGGUUGGACGCUGGAAGACG-3′ | ||||
| Pol-si5 | 5′-CAGCGUCCAACCAUAUCAUCCAACU-3′ | 6918–6942 | DNA pol | 2, 5, 6 |
| 5′-AGUUGGAUGAUAUGGUUGGACGCUG-3′ | ||||
| Pol-si6 | 5′-GCGUCCAACCAUAUCAUCCAACUCA-3′ | 6916–6940 | DNA pol | 2, 5, 6 |
| 5′-UGAGUUGGAUGAUAUGGUUGGACGC-3′ | ||||
| Pol-si2_scr | 5′-CAACUUUCCGAGCCUACCACGCAUA-3′ | – | – | – |
| 5′-UAUGCGUGGUAGGCUCGGAAAGUUG-3′ | ||||
| IVa2-si1 | 5′-ACAUGCGAGUCAGGGACAUGCUUAA-3′ | 5013–5037 | IVa2 | 5 |
| 5′-UUAAGCAUGUCCCUGACUCGCAUGU-3′ | ||||
| IVa2-si2 | 5′-AAAUACAGUCCAAGAUGCAUCUCAU-3′ | 4410–4434 | IVa2 | 1, 2, 5, 6 |
| 5′-AUGAGAUGCAUCUUGGACUGUAUUU-3′ | ||||
| IVa2-si3 | 5′-CAUCCCAGCUUAACCGCUUUGUAAA-3′ | 4368–4392 | IVa2 | 1, 2, 5, 6 |
| 5′-UUUACAAAGCGGUUAAGCUGGGAUG-3′ | ||||
| Hex-si1 | 5′-UAGAAACUCGGACCACUCUGAGACA-3′ | 5818–5842 | Hexon | 5 |
| 5′-AUUUAUACCAGAAUAAGGCGCCUGC-3′ | ||||
| Hex-si2 | 5′-GAGAACUAAUGGGCCAACAAUCUAU-3′ | 19776–19800 | Hexon | 5 |
| 5′-AUAGAUUGUUGGCCCAUUAGUUCUC-3′ | ||||
| Hex-si3 | 5′-GCCUCAGAAGUUCUUUGCCAUUAAA-3′ | 20527–20551 | Hexon | 5 |
| 5′-UUUAAUGGCAAAGAACUUCUGAGGC-3′ | ||||
| Hex-si4 | 5′-CCGUCAGGUGGUGGAUGAUACUAAA-3′ | 21247–21271 | Hexon | 5 |
| 5′-UUUAGUAUCAUCCACCACCUGACGG-3′ | ||||
| Prot-si1 | 5′-GAGCAGGAACUGAAAGCCAUUGUCA-3′ | 21745–21769 | Protease | 2, 5, 6 |
| 5′-UGACAAUGGCUUUCAGUUCCUGCUC-3′ | ||||
| Prot-si2 | 5′-AAGAUCUUGGUUGUGGGCCAUAUUU-3′ | 21770–21794 | Protease | 2, 5, 6 |
| 5′-AAAUAUGGCCCACAACCAAGAUCUU-3′ | ||||
| Prot-si3 | 5′-UCAAGCAGGUUUACCAGUUUGAGUA-3′ | 21972–21995 | Protease | 1, 2, 5, 6 |
| 5′-UACUCAAACUGGUAAACCUGCUUGA-3′ | ||||
| pTP-si1 | 5′-CCGCCUACUUUAAUUACAUTT-3′ | 9805–9823 | pTP | 1, 2, 5, 6 |
| 5′-AUGUAAUUAAAGUAGGCGGTT-3′ | ||||
| pTP-si2 | 5′-GAGGAGAUUGAAGAAGAAGTT-3′ | 9359–9377 | pTP | 1, 2, 5, 6 |
| 5′-CUUCUUCUUCAAUCUCCUCTT-3′ | ||||
| pTP-si3 | 5′-GGUAGAAAGGCUCAUGCAATT-3′ | 9945–9963 | pTP | 1, 2, 5, 6 |
| 5′-UUGCAUGAGCCUUUCUACCTT-3′ | ||||
| pTP-si4 | 5′-CGAAAUUGAUUCUGUCGAATT-3′ | 8796–8814 | pTP | 1, 2, 5, 6 |
| 5′-UUCGACAGAAUCAAUUUCGTT-3′ | ||||
| pTP-si5 | 5′-GACUACGUAUUUGACUCGAGGGCUU-3′ | 10289–10313 | pTP | 5 |
| 5′-AAGCCCUCGAGUCAAAUACGUAGUC-3′ | ||||
| pTP-si6 | 5′-CGGUAGAAAGGCUCAUGCAAGACUA-3′ | 9940–9964 | pTP | 1, 2, 5, 6 |
| 5′-UAGUCUUGCAUGAGCCUUUCUACCG-3′ | ||||
| pTP-si7 | 5′-ACUACCUCUUUCAGCGCCUGCGAAA-3′ | 9103–9127 | pTP | 1, 2, 5, 6 |
| 5′-UUUCGCAGGCGCUGAAAGAGGUAGU-3′ | ||||
| pTP-si8 | 5′-GAAAUUGAUUCUGUCGAACUCUCUU-3′ | 8789–8813 | pTP | 1, 2, 5, 6 |
| 5′-AAGAGAGUUCGACAGAAUCAAUUUC-3′ | ||||
GenBank ID: AC_000008.
Serotypes displaying 100% target site complementarity.
2.5. Dual-luciferase assay-based screening for functional siRNAs
1.4e+05 HEK293 and 3e+04 A549 cells were seeded into the wells of 96-well plates, and reverse transfected with 50 ng of individual dual-luciferase reporter vectors and 30 nM targeting or non-targeting control siRNA using Lipofectamine 2000 (Invitrogen/LifeTechnologies Austria, Vienna, Austria). Briefly, for each well 0.5 μL Lipofectamine 2000 was diluted with 24.5 μL OptiMEM medium (Invitrogen/LifeTechnologies Austria, Vienna, Austria), and after 5 min of incubation, 25 μL diluted Lipofectamine 2000 was mixed with 25 μL of a specific siRNA/reporter vector mix (diluted in OptiMEM). After 20 min of incubation, the mixes were pipetted directly into the wells of a 96-well plate and freshly harvested cells were added. After 24 h of incubation, medium was exchanged and cells were incubated for another 24 h. Culture conditions were as described above. Firefly and Renilla luciferase activities were determined at 48 h post-transfection using the Dual-Glo luciferase assay (Promega), according to the manufacturer’s instructions. Briefly, 75 μL of Dual-Glo Reagent was added to cells grown in 75 μL medium, and after 10 min of incubation at room temperature, firefly luciferase activity was measured. Next, one volume of Dual-Glo Stop & Glo reagent was added to each well, plates were incubated for an additional 10 min at room temperature, and eventually, Renilla luciferase activity was determined. Luminescence was measured on a Wallac Victor 1420 Multilabel Counter (Perkin Elmer Austria, Brunn am Gebirge, Austria). Knockdown rates were calculated by normalizing Renilla luciferase activities to firefly luciferase activities, and comparing dual-luciferase ratios between targeting and non-targeting control siRNAs.
2.6. Determination of mRNA levels
1.25e+05 A549 cells were seeded into the wells of 24-well plates and reverse transfected with siRNAs at a concentration of 10 nM. Volumes of transfection mixes were adjusted to the 24-well plate format. Briefly, for each well 1 μL Lipofectamine 2000 was diluted with 49 μL OptiMEM medium (Invitrogen/LifeTechnologies Austria, Vienna, Austria), and after 5 min of incubation, 50 μL diluted Lipofectamine 2000 was mixed with 50 μL of a specific siRNA diluted in OptiMEM. Transfection conditions were otherwise as described under 2.5. After 24 h of incubation, medium was exchanged and cells were infected with Ad5 at an multiplicity of infection (MOI) of 0.01 TCID50/cell, and total RNA was isolated at 24 h post-infection using an RNeasy Mini® Kit (QIAGEN). Residual DNA was removed with RQ1 DNase (Promega), and reverse transcription was carried out using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Expression levels of the E1A-12S, E1A-13S, DNA polymerase, pTP, IVa2, hexon, and protease genes were determined by TaqMan real-time quantitative PCR (qPCR), using the LightCycler 480 Probes master mix (Roche Diagnostics) and primer/probe sets specific for E1A-13S (E1A 289R-cDNA-f1 5′-GCATGTTTGTCTACAGTCCTGTGTC-3′, E1A 289R-cDNA-r1 5′-GGCGTCTCAGGATAGCAGGC-3′, and E1A 289R-cDNA-p1 5′-AGGCTCCGGTTCTGGCTCGGG-3′), E1A-12S (E1A 12S-cDNA-f1 5′-AGGATGAAGAGGGTCCTGTGTCT-3′, E1A 289R-cDNA-r1 5′-GGCGTCTCAGGATAGCAGGC-3′, and E1A 289R-cDNA-p1 5′-AGGCTCCGGTTCTGGCTCGGG-3′), DNA polymerase (Pol-cDNA-f1 5′-ATGGCCTTGGCTCAAGCTC-3′, Pol-cDNA-r1 5′-GCGTAGGTTGCTGGCGAAC-3′, and Pol-cDNA-p1 5′-CGCCTCTGCGTGAAGACGACGG-3′), pTP (pTP-cDNA-f2 5′-AAACCAACGCTCGGTGCC-3′, pTP-cDNA-r2 5′-GGACGCGGTTCCAGATGTT-3′, and pTP-cDNA-p2 5′-CGCGCGCAATCGTTGACGCT-3′), IVa2 (IVa2-cDNA-f1 5′-GGAAACCAGAGGGCGAAGA-3′, IVa2-cDNA-r1 5′-AGGGTCCTCGTCAGCGTAGTC-3′, and IVa2-cDNA-p1 5′-CTTGAGGCTGGTCCTGCTGGT-3′), hexon (Hex-cDNA-f1 5′-CAGTCACAGTCGCAAGAGGAGC-3′, Hex-cDNA-r1 5′-AGGTACTCCGAGGCGTCCTG-3′, and Hex-cDNA-p1 5′-ACCACTGCGGCATCATCGAAGGG-3′), and protease (Prot-cDNA-f1 5′-TCACAGTCGCAAGTCTTTGACG-3′, Prot-cDNA-r1 5′-GCGGCAGCTGTTGTTGATG-3′, and Prot-cDNA-p1 5′-CCGAGAAGGGCGTGCGCAGGTA-3′). The specificity of the primers employed (i.e., covered exon–exon junctions) enabled them to discriminate between overlapping transcripts or transcripts originating from the (+) or (−) strand, respectively. Ad5 gene expression levels were normalized to GAPDH expression levels, which were previously proven to remain unchanged upon Ad5 infection under the selected experimental conditions. GAPDH expression was determined with the primer/probe set GAPDH-f1 5′-TGCACCACCAACTGCTTAGC-3′, GAPDH-r1 5′-GGCATGGACTGTGGTCATGAG-3′, and GAPDH-p1 5′-CCTGGCCAAGGTCATCCATGACAACTT-3′. All q PCR assays were set up in 96-well plates and contained 1× LightCycler 480 Probes master mix (Roche Diagnostics, Vienna, Austria), 500 nM of forward and reverse primers, each, 100 nM of probe, and 1 μL of cDNA in a total volume of 20 μL. For the detection of GAPDH cDNA, 300 nM of forward and reverse primers, each, and 1 μL of 1:10 diluted template cDNA were used per reaction. All assays were performed in duplicates using a LightCycler 480 system (Roche Diagnostics, Vienna, Austria) with the following cycling parameters: heating to 95 °C for 1 min followed by 50 cycles at 95 °C for 15 s and 60 °C for 1 min. Data were analyzed using the LightCycler 480 software. Control included with every assay consisted of a ‘no template control’ (no DNA added).
2.7. Determination of adenovirus genome copy numbers
3e+04 A549 cells were seeded into the wells of 96-well plates, and reverse transfected with siRNAs at concentrations ranging from 0.04 nM to 30 nM. Transfection conditions were as described under 2.5., except that reporter plasmid DNA was omitted. After 24 h, cells were infected with Ad1, Ad2, Ad5, or Ad6 at an MOI of 0.01 TCID50/cell. Samples were collected at 2, 4, and 6 days post-infection. Viral DNA was isolated using a QIAamp DNA Blood Mini Kit (QIAGEN). Ad5 genome copy numbers were determined by qPCR, using the following TaqMan primer/probe set directed against the viral E1A region: E1A-fwd 5′-GACGGCCCCCGAAGATC-3′, E1A-rev 5′-TCCTGCACCGCCAACATT-3′, and E1A-p 5′-CGAGGAGGCGGTTTCGCAGA-3′. The setup of qPCR assays and the cycling parameters were the same as described above. For each reaction, 1 μL of isolated DNA was used. Adenovirus genome copy numbers were calculated by using serial dilutions of an adenoviral reference DNA as a standard.
2.8. Determination of numbers of infectious virus particles
To liberate the viruses from the cells, 96-well plates containing cells and viruses were subjected to three freeze–thaw cycles. Crude lysates were cleared by centrifugation of the plates for 15 min at 2800 rpm. The numbers of infectious virions were determined on A549 cells by TCID50 assays.
2.9. Cell viability
The experimental setup for the determination of the viability of infected cells was as described for other virus inhibition experiments, except that A549 cells were infected at higher MOIs of 2 TCID50/cell, 4 TCID50/cell, or 6 TCID50/cell. Metabolic activity as a measure of cell viability was determined at 6 days post-infection by performing an MTS assay (CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay), according to the manufacturer’s instructions (Promega). Absorbance was determined at 490 nm on a Wallac Victor 1420 Multilabel Counter (Perkin Elmer).
2.10. Statistical analysis
All the data are expressed as mean ± standard deviation (SD). To test for statistical significance, one-way ANOVA corrected with Bonferroni’s post hoc test was applied. A p value of <0.05 was considered statistically significant.
3. Results
3.1. Identification of best-performing siRNAs directed against E1A, DNA polymerase, pTP, IVa2, hexon, and protease
To analyze which adenoviral processes may constitute useful targets for RNAi-mediated inhibition of adenovirus multiplication, we designed a set of siRNAs targeting the E1A, DNA polymerase, pTP, IVa2, hexon, and protease mRNAs (Table 1). E1A siRNAs were designed to target E1A-12S and also E1A-13S splice isoforms. With the exception of pTP-si1 to pTP-si4, all siRNAs were 25-mer, blunt-ended siRNAs carrying the Invitrogen “Stealth” modification. pTP-si1 to pTP-si4 were 21-mer, unmodified siRNAs carrying 2 nt TT overhangs at their 3′ ends. To screen for the best-performing siRNA of each group, we employed a dual-luciferase assay-based system. The respective target sequences were individually inserted into the 3′ UTR of a plasmid-located Renilla luciferase gene. The DNA polymerase, pTP, IVa2, hexon, and protease siRNAs, together with the respective reporter vectors, were used to co-transfect HEK293 cells. Knockdown of Renilla luciferase expression in relation to the expression of a firefly luciferase gene located on the same plasmid was determined in dual-luciferase assays. The silencing capacity of the E1A siRNAs was assessed in A549 cells because promoter activities of the reporter vectors turned out to be altered upon silencing of the endogenous E1A gene present in HEK293 cells (Graham et al., 1977) (data not shown). For all target mRNAs, we identified siRNAs enabling a knockdown of ⩾78% at a concentration of 30 nM (Fig. 1). The best-performing siRNAs of each group, i.e., pTP-si8, Pol-si2, Hex-si2, E1A-si3, Iva2-si2, and Prot-si1, were selected for further characterization.
Fig. 1.

Screening for functional siRNAs. SiRNAs and their respective dual-luciferase target vectors were used to co-transfect HEK293 cells (DNA polymerase, pTP, IVa2, hexon, and protease siRNAs) or A549 cells (E1A siRNAs). A non-targeting control siRNA served as a negative control (neg. ctrl.). All siRNAs were employed at a concentration of 30 nM. Renilla luciferase activities in relation to firefly luciferase activities were determined 48 h post-transfection. Relative light units (RLU; mean ± SD, n = 3) in comparison to a non-targeting control siRNA are shown. Differences between neg. ctrl. siRNA and targeting siRNAs were statistically significant in all cases (p < 0.05).
3.2. SiRNAs decrease mRNA levels directly or indirectly
The dual-luciferase assay-based screening system was employed to select the best-performing siRNAs of each group. Next, we investigated whether the selected siRNAs were able to knockdown gene expression during an adenovirus infection of A549 cells. Cells were transfected with the siRNAs at a concentration of 10 nM, and then infected with Ad5 at an MOI of 0.01 TCID50/cell. Target mRNA levels were determined by RT-qPCR, using primers specific for the individual mRNAs (Fig. 2A). The highest silencing rates (93–97%) were observed for the E1A-, DNA polymerase-, pTP-, and IVa2-targeting siRNAs. Silencing of the hexon and protease genes was less pronounced (79–87%). Except for the difference in residual hexon and pTP mRNA levels, the differences between hexon or protease mRNA levels and those of all other early genes were statistically significant.
Fig. 2.

siRNAs decrease mRNA levels directly and indirectly. A549 cells were transfected with siRNAs directed against the E1A, DNA polymerase (Pol), pTP, IVa2, hexon (Hex), and protease (Prot) genes, or a non-targeting control siRNA (neg. ctrl.) at a concentration of 10 nM, and then infected with Ad5 at an MOI of 0.01 TCID50/cell. Levels of direct targets (A) or indirect targets such as the E1A (B), DNA polymerase (C), pTP (D), and IVa2 (E) mRNAs were determined by RT-qPCR. Relative mRNA levels in comparison to a non-targeting siRNA are shown. Values represent mean ± SD of three independent experiments, each performed in triplicate. For each experiment, real-time qPCR quantification was performed in duplicate. *p < 0.05, ***p < 0.001.
As the pTP, DNA polymerase, and IVa2 mRNAs share a common 3′ part (Supplementary Fig. 1), and the DNA polymerase target site is also part of the pTP mRNA, IVa2- and DNA polymerase-directed siRNAs were therefore expected to concomitantly silence pTP/DNA polymerase/IVa2 and pTP/DNA polymerase, respectively. Furthermore, siRNA-mediated silencing of early genes was expected indirectly to affect the expression of those middle or late genes for which expression is known to depend on early viral gene products. Thus, we also determined the effect of the E1A-, pTP-, DNA polymerase-, and IVa2-targeting siRNAs on the expression of the other genes. Silencing of E1A resulted in a marked reduction in the expression of all other genes (Fig. 2B). This can be attributed to the central role of E1A in activating the expression of downstream genes.
Silencing of the E1A gene actually resulted in a greater reduction in the expression of hexon and protease than did direct silencing of these genes by the hexon and protease siRNAs (compare Fig. 2A and B). As expected, the DNA polymerase-directed siRNA also silenced the pTP gene, and indirectly also affected the expression of all other genes (Fig. 2C). This finding is in accordance with the dependency of IVa2 and ML transcription on the replication of the adenoviral genome, for which DNA polymerase expression is mandatory (Flint, 1986; Iftode and Flint, 2004; Shaw and Ziff, 1980). The same holds true for silencing of pTP (Fig. 2D), which is also essential for virus DNA replication, and consequently activation of transcription from the other promoters. Although the pTP siRNA target site is absent from DNA polymerase mRNA, pTP silencing also decreased DNA polymerase mRNA levels, albeit to a lesser extent than DNA polymerase silencing did. This reduction can be attributed to the inhibition of DNA replication by the pTP siRNA, and consequently decreased DNA polymerase gene copy numbers. As expected, the IVa2 siRNA led to a reduction not only in IVa2, but also in pTP and DNA polymerase mRNA levels (Fig. 2E). Since transcription from the MLP is highly activated by the IVa2 protein (Tribouley et al., 1994), ML transcript levels were also indirectly decreased.
3.3. SiRNAs decrease Ad5 genome copy numbers, and also the output of infectious virus progeny
In order to investigate the gene silencing effect of the individual siRNAs on adenovirus replication, A549 cells were transfected with the siRNAs at a concentration of 10 nM and infected as before. At 2 days post-infection, Ad5 genome copy numbers were determined by qPCR, using primers directed against the E1A gene (Fig. 3A). With the exception of the hexon and protease siRNAs, all siRNAs effectively inhibited adenovirus replication. The highest inhibition rate was achieved with the DNA polymerase siRNA, which decreased Ad5 genome copy numbers on average by approximately 2.5 orders of magnitude (99.6%). The failure of the hexon and protease siRNAs to decrease virus genome copy numbers was not surprising, because a reduction in hexon and protease levels is not expected to affect viral DNA replication.
Fig. 3.

Impact of siRNAs on viral DNA replication and virus spreading. (A) A549 cells were transfected with siRNAs directed against the E1A, DNA polymerase (Pol), pTP, IVa2, hexon (Hex), and protease (Prot) genes, or a non-targeting control siRNA (neg. ctrl.) at a concentration of 10 nM, and then infected with Ad5 at an MOI of 0.01 TCID50/cell. Virus genome copy numbers were determined at 48 h post-infection by qPCR, using E1A-specific primers. Values represent mean ± SD of three independent experiments, each performed in triplicate. For each experiment, real-time qPCR quantification was performed in duplicate. *p < 0.001. (B) Long-term infection of A549 cells with Ad5 at an MOI of 0.01 TCID50/cell, and treatment with 10 nM of the indicated siRNAs. Virus genome copy numbers were determined at time points 0, 2, 4, and 6 days post-infection by qPCR. Representative data from at least three independent experiments, each performed in triplicates, are shown (mean ± SD; n = 3). For each experiment, real-time qPCR quantification was performed in duplicate. **p < 0.01.
Next, we evaluated the performance of those siRNAs that were expected directly or indirectly to affect the output of viral DNA (i.e., E1A, DNA polymerase, pTP, and IVa2 siRNAs) in a time-course experiment spanning 6 days in which Ad5 was allowed to spread throughout the cultures (Fig. 3B). As expected, viral genome copy numbers were also decreased at later time points. We repeated the experiments with higher siRNA concentrations (30 nM and 90 nM) and obtained comparable results (data not shown). The inhibition rate at late time points may be generally underestimated; although the cells were infected with Ad5 at a low MOI of 0.01 TCID50/cell, the high burst size of adenovirus rapidly leads to infection of the entire culture. This prevents an exponential increase in virus multiplication at later time points, in those cultures in which replication is not attenuated by siRNAs.
The impact of siRNAs on viral processes other than DNA replication is not fully elucidated by the measurement of virus genome copy numbers. Thus, we also determined the output of infectious virus progeny. Cells were transfected and infected as described above, and the numbers of infectious virus particles at 48 h post-infection were determined (Fig. 4). We observed that hexon and protease siRNAs inhibited the production of infectious virus progeny by approximately 1.3 and 0.8 orders of magnitude (94.9% and 83.1%), respectively. However, the other siRNAs led to an even higher decrease in virus titers of up to 2.8 orders of magnitude (99.8%).
Fig. 4.

siRNAs decrease the numbers of infectious virus particles. A549 cells were transfected with siRNAs directed against the E1A, DNA polymerase (Pol), pTP, IVa2, hexon (Hex), and protease (Prot) genes, or a non-targeting control siRNA (neg. ctrl.) at a concentration of 10 nM, and then infected with Ad5 at an MOI of 0.01 TCID50/cell. Numbers of infectious virus particles at 48 h post-infection were determined on A549 cells by TCID50 assays. Representative data from three independent experiments, each performed in triplicate, are shown (mean ± SD; n = 3). **p < 0.01, ***p < 0.001.
Taken together, our data indicate that silencing of early or intermediate genes seems to be more effective in terms of reducing the output of viral DNA, and also the number of infectious virus progeny, than is silencing of late genes. Computational calculation of the target site accessibility of the DNA polymerase siRNA, using the RNAxs software tool, suggested high accessibility of the entire region embedding the Pol-si2 target site. Target site accessibility has been reported to correlate with high effectiveness of siRNAs (Tafer et al., 2008; Westerhout and Berkhout, 2007). Thus, we speculated that siRNAs capable of binding to target sites in the immediate vicinity of, or overlapping, the target site of the Pol-si2 siRNA may allow simlar or even better knockdown of DNA polymerase gene expression than Pol-si2. Thus we designed three more such siRNAs (Fig. 5A). However, none of them proved superior to the Pol-si2 siRNA (Fig. 5B). The functionality of Pol-si2 was also validated by comparing its activity not only to that of a universal non-targeting control siRNA but also to that of a scrambled version. No change in the inhibition rate was observed (Supplementary Fig. 2). The inhibitory effect of Pol-si2 was also shown to be dose-dependent (Fig. 6). The silencing capacity of low siRNA concentrations may even be underestimated in some experiments; in control experiments employing fluorescence-labeled siRNAs, the transfection efficiency decreased significantly at concentrations of <5 nM (data not shown). Thus, low siRNA concentrations do not truly reflect the silencing capacity, because significant numbers of cells contain no siRNA.
Fig. 5.

Differential inhibition of Ad5 replication by DNA polymerase siRNAs binding in the immediate vicinity of, or overlapping, the Pol-si2 target sequence. (A) Region of the DNA polymerase open reading frame (indicated as DNA pol) targeted by siRNAs Pol-si2, Pol-si4, Pol-si5, and Pol-si6. The DNA sequences corresponding to the individual siRNA target sites on the target mRNAs are given below. The nucleotides corresponding to the seed regions of the respective siRNAs are shaded in grey. (B) A549 cells were transfected with the viral DNA polymerase-directed siRNAs or a non-targeting control siRNA (neg. ctrl.) at a concentration of 10 nM, and then infected with Ad5 at an MOI of 0.01 TCID50/cell. Virus genome copy numbers from triplicate infections (mean ± SD; n = 3) were determined at 48 h post-infection by qPCR using E1A-specific primers. ***p < 0.001.
Fig. 6.

Dose-dependent decrease in Ad5 genome copy numbers mediated by the DNA polymerase siRNA. A549 cells were transfected with the DNA polymerase siRNA or a non-targeting control siRNA (neg. ctrl.) in decreasing concentrations as indicated, and then infected with Ad5 at an MOI of 0.01 TCID50/cell. Virus genome copy numbers from triplicate infections (mean ± SD; n = 3) were determined at 48 h post-infection by qPCR, using E1A-specific primers. For each experiment, real-time qPCR quantification was performed in duplicate. *p < 0.05, **p < 0.01, ***p < 0.001.
The target sequence of the DNA polymerase siRNA is also present in the mRNAs of the other members of adenovirus species C (i.e., Ad1, Ad2, and Ad6), all of which commonly account for life-threatening disseminated adenovirus disease. Consequently, the inhibitory effect of the DNA polymerase siRNA was not restricted to Ad5. Replication of Ad1, Ad2, and Ad6 was also efficiently inhibited (Supplementary Fig. 3).
3.4. Treatment with individual siRNAs or combinations of siRNAs results in a comparable decrease in the numbers of infectious virus particles
Given the dependency of intermediate or late adenoviral gene expression on certain early viral gene products, simultaneous silencing of different adenoviral genes may have synergistic effects on the inhibition of virus multiplication. We therefore performed virus inhibition experiments using combinations of siRNAs. In all of these experiments, we used a total siRNA concentration of 10 nM, i.e., combined siRNAs were employed at a concentration of 5 nM each. As a control, cells were transfected with the individual siRNAs at a concentration of 10 nM. To correct for potential saturation effects (e.g., during transfection and/or RISC loading of siRNAs), cells were also transfected with a combination of 5 nM individual targeting siRNA and 5 nM non-targeting control siRNA. The numbers of infectious virus particles were determined at 48 h post-infection by TCID50 assay (Fig. 7). As shown in Fig. 7B, the superior anti-adenoviral effect mediated by the DNA polymerase siRNA was not enhanced by simultaneous targeting of those mRNAs whose generation depends on the function of the DNA polymerase, e.g., the IVa2 or hexon genes. Similarly, combined E1A and DNA polymerase silencing did not further decrease virus titers (Fig. 7A). The same held true for all other siRNA combinations. In general, combining a highly effective siRNA with a less well-performing siRNA led to an intermediate inhibition rate, or an inhibition rate equal to the one caused by the individual better-performing siRNA. Moreover, the anti-adenoviral effect of an individual siRNA was not reduced by halving its concentration upon combination with an equal concentration of non-targeting negative control siRNA.
Fig. 7.

Effect of combinatorial silencing of Ad5 genes on virus output. A549 cells were transfected with the indicated siRNAs either alone or in combination. SiRNA combinations for E1A (A), DNA polymerase (B), pTP (C), IVa2 (D), hexon (E), and protease (F) are depicted. For all transfections, the total concentration of siRNA was 10 nM. For combinations, each siRNA was employed at a concentration of 5 nM. As a control, cells were transfected with the individual siRNAs alone at a concentration of 10 nM, or as a mix of 5 nM targeting siRNA and 5 nM non-targeting negative control siRNA. Subsequently, cells were infected with Ad5 at an MOI of 0.01 TCID50/cell, and cells and supernatants were harvested at 48 h post-infection. Numbers of infectious Ad5 particles of triplicate infections were determined on A549 cells by TCID50 assays (mean ± SD; n = 3).
We speculated that possible synergistic effects may have been undetectable, because the cells were harvested at a relatively early time point (48 h post-infection). However, they might become detectable at later time points, when the virus was allowed to spread throughout the culture. We hypothesized that combinations comprising the E1A siRNA on the one hand, and siRNAs targeting mRNAs originating from other early/middle genes on the other, would be most likely to cause a synergistic effect. We therefore repeated the virus inhibition experiment using the respective siRNA combinations, and determined Ad5 genome copy numbers at 6 days post-infection. However, we did not detect any synergistic effects at this late time point (Supplementary Fig. 4). We also repeated the experiment using lower concentrations of siRNAs. Although there was a slight trend toward somewhat increased inhibition for some combinations, none of these differences were statistically significant, and under no conditions did any combinations of siRNAs result in a higher inhibition rate than the inhibition rate caused by Pol-si2 when applied alone (Supplementary Fig. 5).
3.5. SiRNAs increase the viability of infected cells to different extents
Next, we quantitatively assessed the impact of Ad5 gene silencing on the viability of infected cultures. We transfected A549 cells with the siRNAs at a concentration of 10 nM as before, and then infected them with Ad5 at a higher MOI (4 TCID50/cell) to ensure pronounced cell killing. We determined the metabolic activity as a measure of cell viability at 6 days post-infection, by means of an MTS assay (Fig. 8). As expected, the siRNAs, although greatly decreasing the output of virus progeny, were not capable of preventing already infected cells from cell death. This was also clearly deducible from experiments in which the overall appearance of infected cultures was assessed by crystal violet staining (data not shown).
Fig. 8.
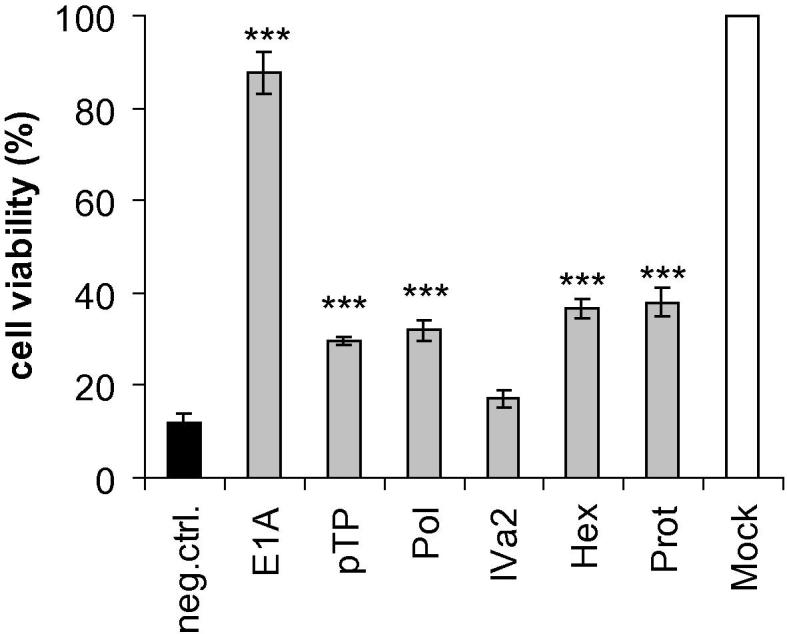
SiRNAs increase the viability of infected cells. A549 cells were transfected with siRNAs directed against the E1A, DNA polymerase (Pol), pTP, IVa2, hexon (Hex), and protease (Prot) genes, or a non-targeting control siRNA (neg. ctrl.) at a concentration of 10 nM, and then infected with Ad5 at an MOI of 4 TCID50/cell. An MTS assay was performed at 6 days post-infection. The viability of cells from triplicate infection experiments (mean ± SD; n = 3) was calculated in relation to the viability of mock-infected cells. ***p < 0.001.
However, all siRNAs were capable of prolonging cell survival, albeit to different extents. This protective effect was most pronounced for cells transfected with the E1A siRNA. Although such cells displayed severe cytopathic effects and were already partially detached from the culture vessels, the culture viability was remarkably high (>80%) at 6 days post-infection. We repeated the experiment using lower and higher MOIs (2 TCID50/cell and 6 TCID50/cell, respectively) and obtained comparable results with a tendency towards higher and lower protection at decreased and increased MOIs, respectively (data not shown). The observed protective effect of the E1A siRNA could not be attributed to a possible unspecific general increase in cellular metabolic activity, because neither the E1A siRNA nor any of the other siRNAs altered the viability of uninfected cells (Supplementary Fig. 6). Thus, although the E1A siRNA did not inhibit the output of infectious virus progeny as efficiently as did the DNA polymerase siRNA, it enhanced the viability of infected cells and kept them alive for a prolonged time period.
4. Discussion
In the present study, we evaluated a larger panel of potential targets, and also determined the inhibitory effect of siRNAs on wild-type adenovirus. SiRNAs directed against the E1A, DNA polymerase, pTP, and IVa2 transcripts were all capable of efficiently silencing the respective genes in the course of an adenovirus infection. By contrast, although having displayed a comparable silencing capacity in luciferase reporter assays, the hexon- and protease-directed siRNAs, showed only a limited capacity to reduce the number of ML transcripts. This observation can be attributed to the markedly higher amounts of hexon and protease mRNAs generated from the particularly strong MLP, in comparison with the mRNA levels of the other genes. This high number of MLP-derived late mRNAs may become even more problematic in RNAi-based attempts to inhibit adenovirus multiplication, because the virus-associated RNAs (VA-RNAs) I and II (non-coding RNAs produced in low amounts during the early stages of infection, but in vast amounts at later time points) appear to counteract RNAi. This effect is thought to be partially caused by the incorporation into and saturation of the RISC by VA-RNA subfragments, which behave like miRNAs (Andersson et al., 2005). Thus, siRNA-mediated inhibition of adenovirus gene expression during the early stages of infection may generally be more beneficial than inhibition of late-stage gene expression. In this regard, inhibition of viral DNA replication may be particularly advantageous, because a decrease in viral genome copy numbers should significantly lower VA-RNA gene copy numbers. In the present study, we observed such an indirect effect when measuring pTP mRNA levels following knockdown of viral DNA polymerase expression. Although not a direct target of the DNA polymerase siRNA, the pTP mRNA levels dropped significantly as a consequence of reduced genome (and pTP gene) copy numbers (Fig. 2D).
Effective knockdown of hexon gene expression may be even more complicated, because hexon mRNA-directed siRNAs target not only the hexon, but also the pVI mRNA. This is caused by the presence of the hexon-encoding sequence downstream of the pVI open reading frame on all pVI transcripts. Thus, hexon mRNA-targeting siRNAs may be partially sequestered away from their actual target by the pVI mRNA, thereby becoming limiting in hexon silencing. The same holds true for the protease siRNA (which concomitantly silences all other genes of the L3 region, i.e., pVI and hexon), the IVa2 siRNA (which additionally binds to the DNA polymerase and pTP mRNAs), and the DNA polymerase siRNA (which concomitantly silences the pTP gene). However, the mRNA levels of these genes, especially those coding for DNA polymerase and pTP, are far lower than those produced by the MLP, and siRNAs may less easily become limiting. Hexon gene silencing was previously demonstrated to be as effective in inhibiting adenovirus multiplication as was silencing of the early E1A gene (Eckstein et al., 2010). This may be attributed to the fact that the mutant virus used was deficient in the E1B-55K gene. E1B-55K has been reported to promote the export of MLP-derived transcripts from the nucleus (Woo and Berk, 2007). Thus, and consequently, lower amounts of ML mRNAs may accumulate in the cytoplasm of cells infected with this mutant virus.
In the present study, we speculated that silencing of early rather than late adenoviral genes would be more effective in inhibiting adenovirus multiplication. We observed that indirect inhibition of hexon and protease gene expression by silencing of genes for which expression activates ML transcription was more effective than was direct targeting of the hexon and protease transcripts (Fig. 2B–E). Importantly, this included E1A silencing. It was previously reported that E1A promotes adenoviral DNA replication, even when present at very low concentrations (Hitt and Graham, 1990). The rather disappointing anti-adenoviral effect obtained with an E1A-directed siRNA (Eckstein et al., 2010) was ascribed to this fact. In the present study, the E1A siRNA employed was obviously potent enough efficiently to decrease not only the E1A mRNA levels, but also, indirectly, the mRNA levels of E1A downstream targets such as the DNA polymerase, pTP, IVa2, hexon, and protease genes (Fig. 2B). Consequently, E1A silencing markedly inhibited the synthesis of viral DNA, and also the generation of infectious virus progeny (Figs. 3 and 4).
The E1A siRNA also substantially improved the viability of the infected cultures, as measured by MTS assay (Fig. 8). This effect was not immediately evident by visual inspection of the cultures, because the siRNA failed to prevent certain adenovirus-related cytopathic effects. The increase in cell viability may be derived from prevention of the well-known toxic effects caused by the main E1A splice isoforms, which eventually drive cells into apoptosis (Cuconati et al., 2002; Lowe and Ruley, 1993; White, 2001). Cell viability was only moderately improved upon silencing of the other early genes. This contradicts a possible indirect E1A siRNA-mediated protective effect (which may occur following blockage of viral DNA replication), and a consequent decrease in the copy numbers of other genes, such as the adenovirus death protein (ADP) gene, which is required for efficient cell lysis and virus release (Tollefson et al., 1996). The inability of the E1A siRNA used by Eckstein et al. (2010) to increase cell viability may also be partially related to the absence of the anti-apoptotic E1B genes from the mutant virus employed.
A reduction in infectious virus progeny was also achievable by knockdown of IVa2 gene expression. However, the fact that IVa2-directed siRNAs silenced not only the IVa2 gene, but also the DNA polymerase and pTP genes, makes it impossible to distinguish whether the main inhibitory effect was caused by blockage of IVa2-mediated viral processes (i.e., activation of late gene expression or DNA packaging), or by inhibition of viral DNA synthesis. The other 2 siRNAs targeting the viral DNA replication machinery (i.e., the pTP and DNA polymerase genes) were among the most effective in inhibiting adenovirus multiplication. This finding does not exclude IVa2-mediated viral processes as potential targets for RNAi-mediated intervention, but clearly establishes adenoviral DNA replication as a key target for the inhibition of adenovirus multiplication.
Combinatorial targeting of different viral transcripts has occasionally been reported to lead to synergistic effects (Chen et al., 2005; ter Brake et al., 2006). In the present study, combinatorial targeting of different adenoviral transcripts did not further decrease virion production. This observation is in accordance with similar findings of Eckstein et al. (2010). It is possible that, in some cases, targeting of 2 distinct transcripts may be redundant. For example, it is conceivable that reducing hexon protein, and also viral genome numbers, is of no additional benefit, because the output of DNA-containing virions will remain unchanged regardless of whether high or low amounts of structural proteins are produced. Nevertheless, synergistic effects are conceivable for other combinations. At least at high siRNA concentrations, competitive effects during lipofection or saturation of RISC are conceivable reasons for the failure to observe synergistic effects. To correct for these, we compared the inhibitory effects of combined siRNAs to those of individual siRNAs, and also to individual siRNAs combined with non-targeting negative control siRNA. Throughout these experiments, we maintained a constant concentration of each siRNA, regardless of whether it was combined with another targeting siRNA. However, we did not observe any synergistic effects. The repeatedly observed failure to produce synergistic effects upon combining siRNAs has been suspected to be attributable to the competition between siRNAs for RISC loading (Castanotto et al., 2007; Formstecher et al., 2006; Koller et al., 2006). It is possible that some of the siRNAs employed in the present study were more efficiently incorporated into the RISC, and were therefore able to outcompete the others.
Animal studies will eventually reveal how efficiently the siRNAs selected in this study can inhibit adenovirus multiplication in vivo. Delivery of siRNAs into living organisms is much more challenging than delivery into cells in vitro. However, a number of delivery vehicles have been developed over the past years which have continuously improved the delivery rates in vivo (Rettig and Behlke, 2011), and RNAi has successfully been applied to condemn virus replication in vivo (Arbuthnot, 2010; Haasnoot et al., 2007; Zhou and Rossi, 2011). The results reported here may also help to generate viral vectors for the efficient expression and delivery of anti-adenoviral siRNAs in the form of shRNAs or artificial miRNAs, a potential alternative way of eliciting anti-adenoviral RNAi in infected cells.
5. Conclusion
Taken together, our data indicate that: (i) highly potent siRNAs are able to inhibit adenovirus multiplication, making them attractive anti-adenoviral drug candidates; (ii) silencing of early adenoviral genes may be more beneficial than silencing of late genes; (iii) silencing of certain early genes can indirectly reduce late gene products more efficiently, or at least as well as, direct silencing of the late genes; (iv) adenoviral infections may be more effectively treated by reduction of adenoviral DNA than by reduction of the proteinaceous components of the virion; (v) the adenoviral DNA replication machinery, and in particular the DNA polymerase gene, constitutes a key target for RNAi-mediated inhibition of adenovirus multiplication; and (vi) silencing of the E1A gene (although less effective than silencing of the DNA polymerase gene in preventing the generation of virus progeny) should not be excluded as a potential strategy, because it may impair virus spread in vivo, by prolonging the survival of infected cells.
Acknowledgment
This work was supported by the Austrian Science Fund through Grant L665-B13.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.antiviral.2012.03.011.
Appendix A. Supplementary data
Supplementary Figure 1.

Schematical drawing of adenoviral transcripts of the E2B transcription unit targeted by siRNAs IVa2-si2, pTP-si8, and Pol-si2. Intronic sequences are indicated by dashed lines. Binding sites for siRNAs are indicated by vertical arrows.
Supplementary Figure 2.

Comparison of the activity two siRNAs that performed best in terms of inhibition of virus replication, i.e., Pol-si2 (A) or protecting infected cells from cell death, i.e., E1A-si3 (B) to that of their respective scrambled controls. A549 cells were transfected with siRNAs Pol-si2 (Pol), E1A-si3 (E1A), a universal non-targeting negative control siRNA (neg. ctrl.), or scrambled versions of the two targeting siRNAs (scrambled Pol, scrambled E1A) at a concentration of 10 nM. Control cells receiving only Lipofectamine 2000 (Lipo) or fresh medium instead of any components of the transfection mix (mock) were included as well. Transfected cells were infected with Ad5 at an MOI of 0.01 TCID50/cell and virus genome copy numbers were determined at 48 h post-infection by qPCR, using E1A-specific primers. Values represent mean ± SD of two independent experiments, each performed in triplicate. For each sample, real-time qPCR quantification was performed in duplicate. ***p < 0.001. Not significant (ns).
Supplementary Figure 3.

The DNA polymerase siRNA inhibits all members of adenovirus species C. A549 cells were transfected with the Pol-si2 siRNA (Pol) or a non-targeting control siRNA (neg. ctrl.) at a concentration of 10 nM, and then infected with Ad1, Ad2, Ad5, or Ad6 at an MOI of 0.01 TCID50/cell. Virus genome copy numbers from triplicate infections (mean ± SD; n = 3) were determined at 48 h post-infection by qPCR, using E1A-specific primers. For each sample, real-time qPCR quantification was performed in duplicate. **p < 0.01, ***p < 0.001.
Supplementary Figure 4.

Effect of long-term combinatorial silencing of early and middle Ad5 genes on Ad5 replication. A549 cells were transfected with siRNAs directed against the E1A, DNA polymerase (Pol), pTP, and IVa2 genes, or a non-targeting control siRNA (neg. ctrl.), either alone (at a concentration of 10 nM) or in combination (at a concentration of 5 nM each). At 24 h after transfection, cells were infected with Ad5 at an MOI of 0.01 TCID50/cell. Cells and supernatants from triplicate infection experiments were harvested at 6 days post-infection, and virus genome copy numbers (mean ± SD; n = 3) were determined by qPCR, using E1A-specific primers. For each smple, real-time qPCR quantification was performed in duplicate. **p < 0.01.
Supplementary Figure 5.

Effect of combinatorial silencing of early and middle Ad5 genes on Ad5 replication at different siRNA concentrations. A549 cells were transfected with the indicated siRNAs either alone or in combination. Subsequently, cells were infected with Ad5 at an MOI of 0.01 TCID50/cell, and cells and supernatants were harvested at 48 h post-infection. Numbers of infectious Ad5 particles were determined on A549 cells by qPCR, using E1A-specific primers. The means (± SD; n = 3) of one representative experiment out of three independent experiments performed in triplicates, each, are shown. For each sample, real-time qPCR quantification was performed in duplicate. (A) For combinations, each siRNA was employed at a concentration of 5 nM. As a control, cells were transfected with the individual siRNAs alone at a concentration of 10 nM, 5 nM, or as a mix of 5 nM targeting siRNA and 5 nM non-targeting negative control siRNA. (B) For combinations, each siRNA was employed at a concentration of 1 nM. As a control, cells were transfected with the individual siRNAs alone at a concentration of 2 nM, 1 nM, or as a mix of 1 nM targeting siRNA and 1 nM non-targeting negative control siRNA. (C) For combinations, each siRNA was employed at a concentration of 0.2 nM. As a control, cells were transfected with the individual siRNAs alone at a concentration of 0.4 nM, 0.2 nM, or as a mix of 0.2 nM targeting siRNA and 0.2 nM non-targeting negative control siRNA.
Supplementary Figure 6.

SiRNAs do not alter the viability of uninfected cells. A549 cells were transfected with siRNAs directed against the E1A, DNA polymerase (Pol), pTP, IVa2, hexon (Hex), and protease (Prot) genes, a universal non-targeting negative control siRNA (neg. ctrl.), and scrambled versions of the E1A and DNA polymerase siRNAs (scrambled E1A, scrambled Pol) at a concentration of 10 nM. At 6 days post-transfection, cells were subjected to MTS assay analysis. The viability of transfected cells was calculated in relation to the viability of cells treated only with Lipofectamine 2000. A representative example (mean ± SD; n = 3) of one out of several control experiments performed in triplicates, each, is shown.
References
- Andersson M.G., Haasnoot P.C., Xu N., Berenjian S., Berkhout B., Akusjarvi G. Suppression of RNA interference by adenovirus virus-associated RNA. J. Virol. 2005;79:9556–9565. doi: 10.1128/JVI.79.15.9556-9565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbuthnot P. Harnessing RNA interference for the treatment of viral infections. Drug News Perspect. 2010;23:341–350. doi: 10.1358/dnp.2010.23.6.1437713. [DOI] [PubMed] [Google Scholar]
- Berk A.J. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene. 2005;24:7673–7685. doi: 10.1038/sj.onc.1209040. [DOI] [PubMed] [Google Scholar]
- Blackford A.N., Grand R.J. Adenovirus E1B 55-kilodalton protein: multiple roles in viral infection and cell transformation. J. Virol. 2009;83:4000–4012. doi: 10.1128/JVI.02417-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanke C., Clark C., Broun E.R., Tricot G., Cunningham I., Cornetta K., Hedderman A., Hromas R. Evolving pathogens in allogeneic bone marrow transplantation: increased fatal adenoviral infections. Am. J. Med. 1995;99:326–328. doi: 10.1016/s0002-9343(99)80169-7. [DOI] [PubMed] [Google Scholar]
- Carthew R.W., Sontheimer E.J. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanotto D., Sakurai K., Lingeman R., Li H., Shively L., Aagaard L., Soifer H., Gatignol A., Riggs A., Rossi J.J. Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucl. Acids Res. 2007;35:5154–5164. doi: 10.1093/nar/gkm543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S., Mautner V., Osman H., Collingham K.E., Fegan C.D., Klapper P.E., Moss P.A., Milligan D.W. Adenovirus infections following allogeneic stem cell transplantation: incidence and outcome in relation to graft manipulation, immunosuppression, and immune recovery. Blood. 2002;100:1619–1627. doi: 10.1182/blood-2002-02-0377. [DOI] [PubMed] [Google Scholar]
- Chen Z., Xu Z.F., Ye J.J., Yao H.P., Zheng S., Ding J.Y. Combination of small interfering RNAs mediates greater inhibition of human hepatitis B virus replication and antigen expression. J. Zhejiang Univ. Sci. B. 2005;6:236–241. doi: 10.1631/jzus.2005.B0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y.S., Kim M.K., Lee W.J., Kang C. Silencing E1A mRNA by RNA interference inhibits adenovirus replication. Arch. Virol. 2007;152:1305–1314. doi: 10.1007/s00705-007-0951-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuconati A., Degenhardt K., Sundararajan R., Anschel A., White E. Bak and Bax function to limit adenovirus replication through apoptosis induction. J. Virol. 2002;76:4547–4558. doi: 10.1128/JVI.76.9.4547-4558.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson B.L., McCray P.B., Jr. Current prospects for RNA interference-based therapies. Nat. Rev. Genet. 2011;12:329–340. doi: 10.1038/nrg2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong R.N., van der Vliet P.C., Brenkman A.B. Adenovirus DNA replication: protein priming, jumping back and the role of the DNA binding protein DBP. Curr. Top. Microbiol. Immunol. 2003;272:187–211. doi: 10.1007/978-3-662-05597-7_7. [DOI] [PubMed] [Google Scholar]
- Ebner K., Rauch M., Preuner S., Lion T. Typing of human adenoviruses in specimens from immunosuppressed patients by PCR-fragment length analysis and real-time quantitative PCR. J. Clin. Microbiol. 2006;44:2808–2815. doi: 10.1128/JCM.00048-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echavarria M. Adenoviruses in immunocompromised hosts. Clin. Microbiol. Rev. 2008;21:704–715. doi: 10.1128/CMR.00052-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckstein A., Grossl T., Geisler A., Wang X., Pinkert S., Pozzuto T., Schwer C., Kurreck J., Weger S., Vetter R., Poller W., Fechner H. Inhibition of adenovirus infections by siRNA-mediated silencing of early and late adenoviral gene functions. Antiviral. Res. 2010;88:86–94. doi: 10.1016/j.antiviral.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Elbashir S.M., Harborth J., Lendeckel W., Yalcin A., Weber K., Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Fechner H., Wang X., Srour M., Siemetzki U., Seltmann H., Sutter A.P., Scherubl H., Zouboulis C.C., Schwaab R., Hillen W., Schultheiss H.P., Poller W. A novel tetracycline-controlled transactivator–transrepressor system enables external control of oncolytic adenovirus replication. Gene Ther. 2003;10:1680–1690. doi: 10.1038/sj.gt.3302051. [DOI] [PubMed] [Google Scholar]
- Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E., Mello C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Flint S.J. Regulation of adenovirus mRNA formation. Adv. Virus Res. 1986;31:169–228. doi: 10.1016/s0065-3527(08)60264-x. [DOI] [PubMed] [Google Scholar]
- Flint S.J., Gonzalez R.A. Regulation of mRNA production by the adenoviral E1B 55-kDa and E4 Orf6 proteins. Curr. Top. Microbiol. Immunol. 2003;272:287–330. doi: 10.1007/978-3-662-05597-7_10. [DOI] [PubMed] [Google Scholar]
- Formstecher E., Reverdy C., Cholay M., Planquette C., Trouplin V., Lehrmann H., Aresta S., Calabrese A., Arar K., Daviet L., Colland F. Combination of active and inactive siRNA targeting the mitotic kinesin Eg5 impairs silencing efficiency in several cancer cell lines. Oligonucleotides. 2006;16:387–394. doi: 10.1089/oli.2006.16.387. [DOI] [PubMed] [Google Scholar]
- Ghildiyal M., Zamore P.D. Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon Y.J., Aoki K., Kinchington P.R. Adenovirus keratoconjunctivitis. In: Pepose J.S., Holland G.N., Wilhelmus K.R., editors. Ocular Infection and Immunity. Mosby; St. Louis: 1996. pp. 877–894. [Google Scholar]
- Graham F.L., Smiley J., Russell W.C., Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- Greber U.F., Webster P., Weber J., Helenius A. The role of the adenovirus protease on virus entry into cells. EMBO J. 1996;15:1766–1777. [PMC free article] [PubMed] [Google Scholar]
- Haasnoot J., Westerhout E.M., Berkhout B. RNA interference against viruses: strike and counterstrike. Nat. Biotechnol. 2007;25:1435–1443. doi: 10.1038/nbt1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale G.A., Heslop H.E., Krance R.A., Brenner M.A., Jayawardene D., Srivastava D.K., Patrick C.C. Adenovirus infection after pediatric bone marrow transplantation. Bone Marrow Transpl. 1999;23:277–282. doi: 10.1038/sj.bmt.1701563. [DOI] [PubMed] [Google Scholar]
- Hartline C.B., Gustin K.M., Wan W.B., Ciesla S.L., Beadle J.R., Hostetler K.Y., Kern E.R. Ether lipid-ester prodrugs of acyclic nucleoside phosphonates: activity against adenovirus replication in vitro. J. Infect. Dis. 2005;191:396–399. doi: 10.1086/426831. [DOI] [PubMed] [Google Scholar]
- Heemskerk B., Lankester A.C., van Vreeswijk T., Beersma M.F., Claas E.C., Veltrop-Duits L.A., Kroes A.C., Vossen J.M., Schilham M.W., van Tol M.J. Immune reconstitution and clearance of human adenovirus viremia in pediatric stem-cell recipients. J. Infect. Dis. 2005;191:520–530. doi: 10.1086/427513. [DOI] [PubMed] [Google Scholar]
- Hitt M.M., Graham F.L. Adenovirus E1A under the control of heterologous promoters: wide variation in E1A expression levels has little effect on virus replication. Virology. 1990;179:667–678. doi: 10.1016/0042-6822(90)90134-d. [DOI] [PubMed] [Google Scholar]
- Howard D.S., Phillips I.G., Reece D.E., Munn R.K., Henslee-Downey J., Pittard M., Barker M., Pomeroy C. Adenovirus infections in hematopoietic stem cell transplant recipients. Clin. Infect. Dis. 1999;29:1494–1501. doi: 10.1086/313514. [DOI] [PubMed] [Google Scholar]
- Huntzinger E., Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- Hutvagner G., Simard M.J. Argonaute proteins: key players in RNA silencing. Nat. Rev. Mol. Cell. Biol. 2008;9:22–32. doi: 10.1038/nrm2321. [DOI] [PubMed] [Google Scholar]
- Iftode C., Flint S.J. Viral DNA synthesis-dependent titration of a cellular repressor activates transcription of the human adenovirus type 2 IVa2 gene. Proc. Natl. Acad. Sci. USA. 2004;101:17831–17836. doi: 10.1073/pnas.0407786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ison M.G. Adenovirus infections in transplant recipients. Clin. Infect. Dis. 2006;43:331–339. doi: 10.1086/505498. [DOI] [PubMed] [Google Scholar]
- Kawamata T., Tomari Y. Making RISC. Trends Biochem. Sci. 2010;35:368–376. doi: 10.1016/j.tibs.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Kojaoghlanian T., Flomenberg P., Horwitz M.S. The impact of adenovirus infection on the immunocompromised host. Rev. Med. Virol. 2003;13:155–171. doi: 10.1002/rmv.386. [DOI] [PubMed] [Google Scholar]
- Koller E., Propp S., Murray H., Lima W., Bhat B., Prakash T.P., Allerson C.R., Swayze E.E., Marcusson E.G., Dean N.M. Competition for RISC binding predicts in vitro potency of siRNA. Nucl. Acids Res. 2006;34:4467–4476. doi: 10.1093/nar/gkl589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaerts L., De Clercq E., Naesens L. Clinical features and treatment of adenovirus infections. Rev. Med. Virol. 2008;18:357–374. doi: 10.1002/rmv.589. [DOI] [PubMed] [Google Scholar]
- Lindemans C.A., Leen A.M., Boelens J.J. How I treat adenovirus in hematopoietic stem cell transplant recipients. Blood. 2010;116:5476–5485. doi: 10.1182/blood-2010-04-259291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lion T., Baumgartinger R., Watzinger F., Matthes-Martin S., Suda M., Preuner S., Futterknecht B., Lawitschka A., Peters C., Potschger U., Gadner H. Molecular monitoring of adenovirus in peripheral blood after allogeneic bone marrow transplantation permits early diagnosis of disseminated disease. Blood. 2003;102:1114–1120. doi: 10.1182/blood-2002-07-2152. [DOI] [PubMed] [Google Scholar]
- Lion T., Kosulin K., Landlinger C., Rauch M., Preuner S., Jugovic D., Potschger U., Lawitschka A., Peters C., Fritsch G., Matthes-Martin S. Monitoring of adenovirus load in stool by real-time PCR permits early detection of impending invasive infection in patients after allogeneic stem cell transplantation. Leukemia. 2010;24:706–714. doi: 10.1038/leu.2010.4. [DOI] [PubMed] [Google Scholar]
- Ljungman P., Ribaud P., Eyrich M., Matthes-Martin S., Einsele H., Bleakley M., Machaczka M., Bierings M., Bosi A., Gratecos N., Cordonnier C. Cidofovir for adenovirus infections after allogeneic hematopoietic stem cell transplantation: a survey by the Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Bone Marrow Transpl. 2003;31:481–486. doi: 10.1038/sj.bmt.1703798. [DOI] [PubMed] [Google Scholar]
- Lowe S.W., Ruley H.E. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev. 1993;7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- Munoz F.M., Piedra P.A., Demmler G.J. Disseminated adenovirus disease in immunocompromised and immunocompetent children. Clin. Infect. Dis. 1998;27:1194–1200. doi: 10.1086/514978. [DOI] [PubMed] [Google Scholar]
- Paolino K., Sande J., Perez E., Loechelt B., Jantausch B., Painter W., Anderson M., Tippin T., Lanier E.R., Fry T., DeBiasi R.L. Eradication of disseminated adenovirus infection in a pediatric hematopoietic stem cell transplantation recipient using the novel antiviral agent CMX001. J. Clin. Virol. 2011;50:167–170. doi: 10.1016/j.jcv.2010.10.016. [DOI] [PubMed] [Google Scholar]
- Pelka P., Ablack J.N., Fonseca G.J., Yousef A.F., Mymryk J.S. Intrinsic structural disorder in adenovirus E1A: a viral molecular hub linking multiple diverse processes. J. Virol. 2008;82:7252–7263. doi: 10.1128/JVI.00104-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig, G.R., Behlke, M.A., 2011. Progress toward in vivo use of siRNAs-II. Mol. Ther. http://dx.doi.org/10.1038/mt.2011.263 (Epub ahead of print). [DOI] [PMC free article] [PubMed]
- Russell W.C. Adenoviruses: update on structure and function. J. Gen. Virol. 2009;90:1–20. doi: 10.1099/vir.0.003087-0. [DOI] [PubMed] [Google Scholar]
- Shaw A.R., Ziff E.B. Transcripts from the adenovirus-2 major late promoter yield a single early family of 3′ coterminal mRNAs and five late families. Cell. 1980;22:905–916. doi: 10.1016/0092-8674(80)90568-1. [DOI] [PubMed] [Google Scholar]
- Subramanian T., Tarodi B., Chinnadurai G. P53-independent apoptotic and necrotic cell deaths induced by adenovirus infection: suppression by E1B 19K and Bcl-2 proteins. Cell Growth Differ. 1995;6:131–137. [PubMed] [Google Scholar]
- Symeonidis N., Jakubowski A., Pierre-Louis S., Jaffe D., Pamer E., Sepkowitz K., O’Reilly R.J., Papanicolaou G.A. Invasive adenoviral infections in T-cell-depleted allogeneic hematopoietic stem cell transplantation: high mortality in the era of cidofovir. Transpl. Infect. Dis. 2007;9:108–113. doi: 10.1111/j.1399-3062.2006.00184.x. [DOI] [PubMed] [Google Scholar]
- Tafer H., Ameres S.L., Obernosterer G., Gebeshuber C.A., Schroeder R., Martinez J., Hofacker I.L. The impact of target site accessibility on the design of effective siRNAs. Nat. Biotechnol. 2008;26:578–583. doi: 10.1038/nbt1404. [DOI] [PubMed] [Google Scholar]
- ter Brake O., Konstantinova P., Ceylan M., Berkhout B. Silencing of HIV-1 with RNA interference: a multiple shRNA approach. Mol. Ther. 2006;14:883–892. doi: 10.1016/j.ymthe.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Tollefson A.E., Scaria A., Hermiston T.W., Ryerse J.S., Wold L.J., Wold W.S. The adenovirus death protein (E3-11.6K) is required at very late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J. Virol. 1996;70:2296–2306. doi: 10.1128/jvi.70.4.2296-2306.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribouley C., Lutz P., Staub A., Kedinger C. The product of the adenovirus intermediate gene IVa2 is a transcriptional activator of the major late promoter. J. Virol. 1994;68:4450–4457. doi: 10.1128/jvi.68.7.4450-4457.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster A., Russell S., Talbot P., Russell W.C., Kemp G.D. Characterization of the adenovirus proteinase: substrate specificity. J. Gen. Virol. 1989;70(Pt. 12):3225–3234. doi: 10.1099/0022-1317-70-12-3225. [DOI] [PubMed] [Google Scholar]
- Westerhout E.M., Berkhout B. A systematic analysis of the effect of target RNA structure on RNA interference. Nucl. Acids Res. 2007;35:4322–4330. doi: 10.1093/nar/gkm437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. Regulation of the cell cycle and apoptosis by the oncogenes of adenovirus. Oncogene. 2001;20:7836–7846. doi: 10.1038/sj.onc.1204861. [DOI] [PubMed] [Google Scholar]
- Woo J.L., Berk A.J. Adenovirus ubiquitin-protein ligase stimulates viral late mRNA nuclear export. J. Virol. 2007;81:575–587. doi: 10.1128/JVI.01725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf U., Hale G.A., Carr J., Gu Z., Benaim E., Woodard P., Kasow K.A., Horwitz E.M., Leung W., Srivastava D.K., Handgretinger R., Hayden R.T. Cidofovir for the treatment of adenoviral infection in pediatric hematopoietic stem cell transplant patients. Transplantation. 2006;81:1398–1404. doi: 10.1097/01.tp.0000209195.95115.8e. [DOI] [PubMed] [Google Scholar]
- Zhang W., Imperiale M.J. Requirement of the adenovirus IVa2 protein for virus assembly. J. Virol. 2003;77:3586–3594. doi: 10.1128/JVI.77.6.3586-3594.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Granberg F., Elfineh L., Pettersson U., Svensson C. Strategic attack on host cell gene expression during adenovirus infection. J. Virol. 2003;77:11006–11015. doi: 10.1128/JVI.77.20.11006-11015.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Rossi J.J. Progress in RNAi-based antiviral therapeutics. Methods Mol. Biol. 2011;721:67–75. doi: 10.1007/978-1-61779-037-9_4. [DOI] [PubMed] [Google Scholar]