

Abstract
The direct α, β-dehydrogenation of aldehydes and ketones represents an efficient alternative to stepwise methods to prepare enal and enone products. Here, we describe a new Pd(TFA)2/4,5-diazafluorenone dehydrogenation catalyst that overcomes key limitations of previous catalyst systems. The scope includes successful reactivity with pharmaceutically important cyclopentanone and flavanone substrates, as well as acyclic ketones. Preliminary mechanistic studies compare the reactivity of this catalyst to previously reported dehydrogenation catalysts and reveal that cleavage of the α-C–H bond of the ketone is the turnover-limiting step of the catalytic mechanism.
Enones and other α,β-unsaturated carbonyl compounds are important intermediates in the synthesis of pharmaceuticals and other complex organic molecules.1 Such compounds are frequently prepared via stepwise protocols, including α-bromination-dehydrobromination,2 α-selenylation followed by oxidation to a selenoxide and elimination, 3,4 and formation of a silyl enol ether, followed by PdII-mediated dehydrosilylation (“Saegusa” oxidation). 5 Direct α,β-dehydrogenation of ketones and aldehydes has also been achieved using stoichiometric reagents, such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)6 and 2-iodoxybenzoic acid (IBX).7,8 PdII-catalyzed direct dehydrogenation of carbonyl compounds with O2 as the oxidant represents an appealing atom-economical alternative to these methods.9,10 Here, we report the discovery of a new Pd(TFA)2/4,5-diazafluorenone (TFA = trifluoroacetate) catalyst system that overcomes key limitations of previously reported catalysts for these reactions. Comparison between this catalyst system and other catalysts, as well as preliminary mechanistic insights into these reactions are described below.
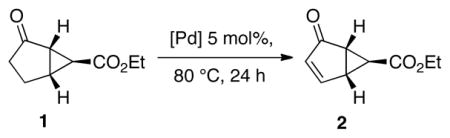
Early studies of Pd-catalyzed dehydrogenation reactions focused on cyclohexanone derivatives;11 however, low yields and/or limited substrate scope restricted the synthetic utility of these methods. We recently reported a Pd(DMSO)2(TFA)2 catalyst system that overcomes many of these limitations and enables aerobic dehydrogenation of a variety of substituted cyclohexanones and other cyclic ketones, including heterocycles (Scheme 1). 12 Independently, we discovered a different PdII catalyst system, Pd(TFA)2/2-Me2Npy (2-Me2Npy = 2-(N,N-dimethylamino)pyridine), that affords substituted phenols via aerobic dehydrogenation of cyclohexanone and cyclohexenone derivatives (Scheme 1).13 Subsequent efforts in our lab to apply these methods to specific target molecules of interest have revealed limitations. In particular, the bicyclic cyclopentanone derivative 2 is a key precursor to a pharmaceutically important anatagonist of the metabotropic glutamate receptor (mGluR) (Scheme 2).14 This molecule was previously prepared from 1 using IBX as the oxidant.14b An aerobic dehydrogenation method could provide a scalable route to this molecule,15 but efforts to apply the Pd(DMSO)2(TFA)2 and Pd(TFA)2/2-Me2Npy catalyst systems to this reaction resulted in unsatisfactory yields (≤37%, Scheme 2). Dehydrogenation of acyclic aldehydes and ketones have less precedent than reactions of cyclic ketones. 3-Arylpropanal (i.e., hydrocinnamaldehyde) derivatives were recently shown to undergo Pd(OAc)2-catalyzed aerobic dehydrogenation in the presence of an amine cocatalyst, possibly via in situ formation of an enamine intermediate (Scheme 3).16 Analogous ketones (e.g., R = Me) were unreactive, however.
Scheme 1.

Palladium-catalyzed dehydrogenation of cyclohexanones
Scheme 2.

Attempted dehydrogenation of a pharmaceutically important cyclopentanone derivative
Scheme 3.

Dehydrogenation of β-aryl carbonyl compounds
Efforts to address these limitations were initiated by exploring possible Pd catalysts for aerobic dehydrogenation of cyclopentanone 1 at 80 °C under 1 atm of O2. As noted above, the previously reported Pd(DMSO)2(TFA)2 catalyst exhibited poor reactivity (Table 1, entry 1). Use of the Pd(TFA)2/2-Me2Npy catalyst system in DMSO or Pd(TFA)2 in the absence of an added ligand led to a slightly improved result (37% and 32% yield, respectively; entries 2 and 3). Other PdII sources were less effective than Pd(TFA)2 (entries 4–7), and no reactivity was observed with traditional heterogeneous Pd catalysts (entries 8 and 9). A number of ancillary ligands were tested in combination with Pd(TFA)2 (entries 10–19). In general, the ancillary ligands had little benefit or were deleterious relative to the reactivity of Pd(TFA)2 alone. The best yield was observed with 4,5-diazafluorenone as a ligand; however, the yield maximized at 40% when the reaction was performed under 1 atm O2 (entry 20). We reasoned that catalyst stability and turnover numbers could be improved by increasing the pressure of O2 (used as a mixture of 9% O2 in N2 to minimize safety risks). Upon testing this hypothesis, a 61% yield of cyclopentenone product 2 was obtained with the Pd(TFA)2/2-Me2Npy catalyst system when the reaction was carried out under 7.2 atm O2 (entry 21). The optimal yield, however, was achieved with the 4,5-diazafluorenone ligand (85% GC, 79% isolated yield; entry 22). The beneficial effect of diazafluorenone in this reaction complements our recent discovery of the utility of this ligand in other Pd-catalyzed aerobic oxidation reactions (allylic C–H oxidation and direct biaryl coupling).17
Table 1.
Catalyst optimization for the aerobic dehydrogenation of 1a
 | ||||
|---|---|---|---|---|
| entry | Pd source | ligand | solvent | yield (%)b |
| 1 | Pd(TFA)2 | DMSO | HOAc | 13 |
| 2 | Pd(TFA)2 | 2-Me2Npy/TsOH | DMSO | 37 |
| 3 | Pd(TFA)2 | none | DMSO | 32 |
| 4 | Pd(OAc)2 | none | DMSO | 21 |
| 5 | Pd(BF4)2(CH3CN)4 | none | DMSO | 21 |
| 6 | PdCl2 | none | DMSO | 6 |
| 7 | Pd2(dba)3 | none | DMSO | 8 |
| 8 | Pd/C | none | DMSO | 0 |
| 9 | Pd(OH)2/charcoal | none | DMSO | 0 |
| 10 | Pd(TFA)2 | pyridine | DMSO | 27 |
| 11 | Pd(TFA)2 | 2-F pyridine | DMSO | 31 |
| 12 | Pd(TFA)2 | 2-MeO-pyridine | DMSO | 32 |
| 13 | Pd(TFA)2 | 3-NO2-pyridine | DMSO | 21 |
| 14 | Pd(TFA)2 | 2,2′-bipyridine (bpy) | DMSO | 33 |
| 15 | Pd(TFA)2 | 6,6′-Me2bpy | DMSO | 15 |
| 16 | Pd(TFA)2 | 5,5′-Me2bpy | DMSO | 12 |
| 17 | Pd(TFA)2 | phenanthroline | DMSO | 36 |
| 18 | Pd(TFA)2 | 2,9-Me2phenanthroline | DMSO | 14 |
| 19 | Pd(TFA)2 | bipyrimidine | DMSO | 19 |
| 20 | Pd(TFA)2 | 4,5-diazafluorenone | DMSO | 40 |
| 21 | Pd(TFA)2 | 2-Me2Npy/TsOH | DMSO | 61c |
| 22 | Pd(TFA)2 | 4,5-diazafluorenone | DMSO | 85c[79]d |
Conditions: [1] = 0.2 M (16.8 mg, 0.1 mmol), 5% catalyst (0.005 mmol), solvent (0.5 mL), 1 atm O2, 80 °C, 24 h.
Determined by GC.
7.2 atm O2 (9% in N2), 48 h.
Isolated yield.
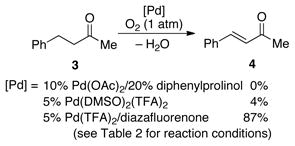
The success of Pd(TFA)2/4,5-diazafluorenone in the dehydrogenation of 1 prompted us to explore the utility of this catalyst system in reactions of acyclic ketones. As noted above, previously reported catalyst systems were essentially unreactive in the dehydrogenation of benzylacetone (3, Eqn. (1)): 0% yield of 4 with Pd(OAc)2/diphenylprolinol16a and 4% yield with Pd(DMSO)2(TFA)2.12 The poor reactivity of ketone 3 relative to hydrocinnamaldehyde (cf. Scheme 3) probably reflects the higher pKa of the α-C–H bond of ketones relative to aldehydes (see further discussion below).18 In contrast, use of the optimized Pd(TFA)2/diazafluorenone catalyst system to the dehydrogenation of 3 afforded enone 4 in 87% isolated yield (eqn (1)). With this substrate, the reaction was compatible with 1 atm O2. To the best of our knowledge, this reaction represents the first example of aerobic dehydrogenation of acyclic ketones.
 |
(1) |
Apigenin is a flavone natural product that has attracted considerable interest as a cancer chemopreventative agent.19 The saturated precursor, naringenin, and related analogs can be readily obtained via condensation of simple benzaldehyde and o-acetylphenol precursors (Scheme 4).20 An apigenin analog was recently prepared in 66% yield by DDQ-promoted dehydrogenation of the corresponding naringenin derivative.21 The Pd(DMSO)2(TFA)2 catalyst system was completely unreactive in an attempted dehydrogenation of naringenin, whereas the Pd(TFA)2/4,5-diazafluorenone catalyst afforded the desired product in 81% yield (Scheme 4).
Scheme 4.

Aerobic dehydrogenation of naringenin with the Pd(TFA)2/diazafluorenone catalyst system.
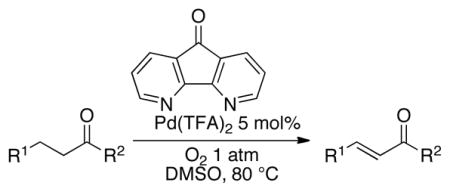
A number of related acyclic β-arylaldehyde and -ketone substrates and chroman-4-one and flavanone derivatives underwent successful aerobic dehydrogenation with this catalyst system (Table 2). Hydrocinnamaldehyde underwent facile dehydrogenation to afford cinnamaldehyde in 91% yield (entry 1). A small amount of benzaldehyde (8%) was formed as a byproduct in this reaction. Other benzyl acetone derivatives, including those with electron-donating and electron-withdrawing substituents, and a phenyl ketone derivative underwent successful dehydrogenation (Table 2, entries 2–5). Dehydrogenation of methyl 3-benzoylpropanoate afforded the expected alkene in a 13:1 trans:cis isomeric ratio (entry 6). Chromones 22 and flavones 23 have important biological activities, and the Pd(TFA)2/4,5-diazafluorenone catalyst exhibits excellent reactivity in the dehydrogenative synthesis of these compounds (Table 2, entries 7–10), including chloro- and fluoro-substituted derivatives.
Table 2.
Pd-catalyzed aerobic dehydrogenation of aldehydes and ketonesa
 | |||||
|---|---|---|---|---|---|
| entry | substrate | time (h) | temp (°C) | enone | yield (%)b |
| 1 | 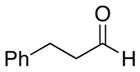 |
8 | 80 | 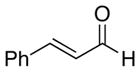 |
91 |
| 2 | 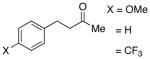 |
48 | 80 | 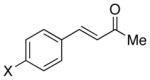 |
83 |
| 3 | 48 | 80 | 87 | ||
| 4 | 48 | 100 | 86 | ||
| 5 | 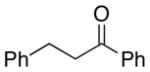 |
36 | 100 | 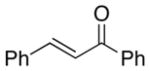 |
87 |
| 6 | 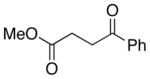 |
48 | 100 |
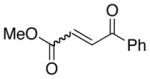 trans : cis = 13 : 1 |
74 |
| 7 | 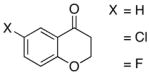 |
32 | 80 | 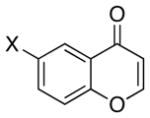 |
96 |
| 8 | 32 | 80 | 94 | ||
| 9 | 32 | 80 | 94 | ||
| 10 | 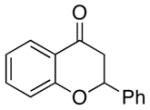 |
24 | 100 |
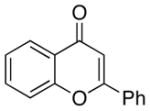 Flavone |
88 |
Conditions: [substrate] = 0.8 M (0.4 mmol), Pd(TFA)2 (6.6 mg, 0.02 mmol), 4,5-diazafluorenone (3.6 mg, 0.02 mmol), DMSO (0.5 mL), 1 atm O2.
Isolated yield.
Preliminary efforts to expand the substrate scope beyond that indicated above have revealed some limitations. Acyclic substrates lacking an aryl group in the β position are susceptible to further dehydrogenation at the γ,δ-position, resulting in a mixture of enone and dienone products. For example, an unoptimized reaction of 2-octanone afforded oct-3-en-2-one in 17% yield and octa-3,5-dien-2-one in 6% yield after 18 h, with 35% recovered starting material. The reaction of propyl phenyl ketone, which cannot undergo a second dehydrogenation step, afforded the enone in only low yield (20%) after 48 h. The low substrate conversion in this reaction possibly reflects deactivation of the catalyst by formation of an inactive Pd-π-allyl species. Finally, esters such as ethyl hydrocinnamate were unreactive under the optimized conditions and increasing the reaction temperature to 100 °C failed to promote reactivity. This lack of reaction probably reflects the reduced acidity of the α-C–H bond of esters relative to ketones and aldehydes.18
Future efforts to expand the scope of these reactions will benefit from mechanistic insights, and a thorough study of the different aerobic dehydrogenation catalysts has been initiated. A few preliminary results are worth noting. The Pd(TFA)2/4,5-diazafluorenone catalyst was evaluated in the dehydrogenation of 4-tert-butylcyclohexanone (Table 3) in order to compare its reactivity with the recently reported Pd(DMSO)2(TFA)212 and Pd(TFA)2/2-Me2Npy catalyst systems.13 Analysis of the reaction time courses for each of these reactions shows that the Pd(TFA)2/4,5-diazafluorenone catalyst behaves similarly to the Pd(TFA)2/2-Me2Npy catalyst.24 Both of these catalysts show a preference for formation of the phenol product, owing to faster dehydrogenation of the cyclohexenone to the phenol relative to the initial dehydrogenation step to afford the enone. In contrast, the first dehydrogenation step (k1) is much faster than the second (k2) when Pd(DMSO)2(TFA)2 is used as the catalyst, resulting in high selectivity for the enone product. The basis for these reactivity differences remains to be elucidated, but the results are consistent with the observation of single and double dehydrogenation products in the reaction of 2-octanone with the Pd(TFA)2/4,5-diazafluorenone catalyst system (see above).
Table 3.
Comparison of Pd catalysts in the oxidation of 4-tert-butylcyclohexanonea
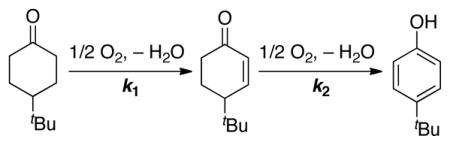 | |||
|---|---|---|---|
| Catalyst | k1 (h−1) | k2 (h−1) | k1/k2 |
| Pd(TFA)2/2-Me2Npy/TsOH | 0.086 | 0.14 | 0.61 |
| Pd(DMSO)2(TFA)2 | 0.19 | 0.0057 | 33 |
| Pd(TFA)2/4,5-diazafluorenone | 0.036 | 0.099 | 0.36 |
Conditions: [substrate] = 0.2 M (15.4 mg, 0.1 mmol), [catalyst] = 0.05 M (0.005 mmol), solvent (0.5 mL), 1 atm O2, 80 °C.
Deuterium kinetic isotope effects in the reaction of benzylacetone were evaluated by comparing the reaction rate of the parent substrate 3 with those of the α- and β-deuterated derivatives 3-d5 and 3-d2 (Scheme 5).25 A primary kinetic isotope effect (kH/kD = 2.6) was observed with the α-deuterated substrate, whereas a negligible isotope effect was observed from deuteration of the β position. This observation suggests that cleavage of the α-C–H bond is the turnover-limiting step of the catalytic reaction while β-hydride elimination from a presumed Pd-enolate intermediate is comparatively fast. These results are consistent with the correlation between the acidity of the α-C–H bond of the substrate and catalytic reactivity: aldehydes > ketones ≫ esters. A proposed catalytic cycle is shown in Scheme 6. The reaction is initiated by turnover-limiting “activation” (i.e., deprotonation) of the α-C–H bond. Subsequent fast β-hydride elimination from a PdII-enolate intermediate affords the enone product and a PdII–hydride intermediate.26 The latter species can be oxidized by O2 to regenerate the PdII catalyst (Scheme 6).27
Scheme 5.

Kinetic isotope effects based on independent initial-rate measurements
Scheme 6.

Proposed catalytic cycle for Pd-catalyzed α,β-dehydrogenation of carbonyl compounds
In summary, we have identified a new catalyst, Pd(TFA)2/4,5-diazafluorenone, that significantly expands the utility of direct aerobic dehydrogenation of carbonyl compounds. Noteworthy results include the first demonstration of aerobic dehydrogenation of acyclic ketones and the application of this catalyst to pharmaceutically important target molecules that were unreactive with previously reported catalysts.
Supplementary Material
Acknowledgments
We thank Eli Lilly for providing us with a sample of compound 1. We are grateful to NIH for financial support of this work (RC1-GM091161). We thank the UW-Madison Department of Chemistry for an undergraduate scholarship to TJW. High-pressure instrumentation was partially supported by the NSF (CHE-0946901).
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures and compound characterization data.
References
- 1.For reviews of methods for α,β-dehydrogenation of carbonyl compounds, see: Buckle DR, Pinto IL. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 7. Oxford; 1991. p. 119.Larock RC. Comprehensive Organic Transformations. John Wiley & Sons; New York: 1999. p. 251.
- 2.(a) Miller B, Wong H. Tetrahedron. 1972;28:2369. [Google Scholar]; (b) Stotter PL, Hill KA. J Org Chem. 1973;38:2576. [Google Scholar]
- 3.For a leading reference and review of selenium based reagents, see: Sharpless KB, Lauer RF, Teranishi AY. J Am Chem Soc. 1973;95:6137.Reich HJ. Acc Chem Res. 1979;12:22.
- 4.Related sulfoxide elimination reactions are also used. For a leading reference, see: Trost BM, Salzmann TN, Hiroi K. J Am Chem Soc. 1976;98:4887.
- 5.The original report and many subsequent applications of the Saegusa oxidation protocol use 0.5–1 equiv of PdII, but catalytic examples have also been reported. See the following leading references: Ito Y, Hirao T, Saegusa T. J Org Chem. 1978;43:1011.Larock RC, Hightower TR, Kraus GA, Hahn P, Zheng D. Tetrahedron Lett. 1995;36:2423.Yu JQ, Wu HC, Corey EJ. Org Lett. 2005;7:1415. doi: 10.1021/ol050284y.
- 6.(a) Walker D, Hiebert JD. Chem Rev. 1967;67:153. doi: 10.1021/cr60246a002. [DOI] [PubMed] [Google Scholar]; (b) Buckle DR. Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Inc; New York: 2010. 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone. [Google Scholar]
- 7.(a) Nicolaou KC, Zhong YL, Baran PS. J Am Chem Soc. 2000;122:7596. [Google Scholar]; (b) Nicolaou KC, Montagnon T, Baran PS, Zhong YL. J Am Chem Soc. 2002;124:2245. doi: 10.1021/ja012127+. [DOI] [PubMed] [Google Scholar]; (c) Nicolaou KC, Montagnon T, Baran PS. Angew Chem Int Ed. 2002;41:993. doi: 10.1002/1521-3773(20020315)41:6<993::aid-anie993>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; (d) Nicolaou KC, Gray DLF, Montagnon T, Harrison ST. Angew Chem Int Ed. 2002;41:996. doi: 10.1002/1521-3773(20020315)41:6<996::aid-anie996>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 8.For a method employing catalytic 2-iodoxybenzene sulfonate (IBS) in combination with stoichiometric Oxone®, see: Uyanik M, Akakura M, Ishihara K. J Am Chem Soc. 2009;131:251. doi: 10.1021/ja807110n.
- 9.For a recent review of efforts toward this goal, see: Muzart J. Eur J Org Chem. 2010:3779.
- 10.For recent advances in Pd-catalyzed aerobic oxidation reactions, see the following references: Stahl SS. Angew Chem Int Ed. 2004;43:3400. doi: 10.1002/anie.200300630.Gligorich KM, Sigman MS. Chem Commun. 2009:3854. doi: 10.1039/b902868d.Chen X, Engle KM, Wang DH, Yu JQ. Angew Chem Int Ed. 2009;48:5094. doi: 10.1002/anie.200806273.
- 11.For leading references, see ref. 9 and the following: Theissen RJ. J Org Chem. 1971;36:752.Muzart J, Pete JP. J Mol Catal. 1982;15:373.Wenzel TT. J Chem Soc, Chem Commun. 1989:932.Park YW, Oh HH. Bull Korean Chem Soc. 1997;18:1123.Tokunaga M, Harada S, Iwasawa T, Obora Y, Tsuji Y. Tetrahedron Lett. 2007;48:6860.
- 12.Diao T, Stahl SS. J Am Chem Soc. 2011;133:14566. doi: 10.1021/ja206575j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izawa Y, Pun D, Stahl SS. Science. 2011;333:209. doi: 10.1126/science.1204183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Massey SM, Monn JA, Valli MJ. 878463. Eur Patent. 1998; (b) Zhang F, Moher ED, Zhang TY. Tetrahedron Lett. 2007;48:3277. [Google Scholar]
- 15.For progress in the development of a safe and scalable flow process for aerobic oxidation, see: Ye X, Johnson MD, Diao T, Yates MH, Stahl SS. Green Chemistry. 2010;12:1180. doi: 10.1039/c0gc00106f.
- 16.(a) Zhu J, Liu J, Ma RQ, Xie HX, Li J, Jiang HL, Wang W. Adv Synth Catal. 2009;351:1229. [Google Scholar]; (b) Liu J, Zhu J, Jiang HL, Wang W, Li J. Chem Asian J. 2009;4:1712. doi: 10.1002/asia.200900238. [DOI] [PubMed] [Google Scholar]
- 17.(a) Campbell AN, White PB, Guzei IA, Stahl SS. J Am Chem Soc. 2010;132:15116. doi: 10.1021/ja105829t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Campbell AN, Meyer EB, Stahl SS. Chem Commun. 2011;47:10257. doi: 10.1039/c1cc13632a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.For pKa values relevant to the present study, see: Matthews WS, Bares JE, Bartmess JE, Bordwell FG, Cornforth FJ, Drucker GE, Margolin Z, McCallum RJ, McCollum GJ, Vanier NR. J Am Chem Soc. 1975;97:7006.Bordwell FG, Bares JE, Bartmess JE, Drucker GE, Gerhold J, McCollum GJ, Van der Puy M, Vanier NR, Matthews WS. J Org Chem. 1977;42:326.Bordwell FG, Harrelson JA., Jr Can J Chem. 1990;68:1714.For a useful table of Bordwell pKa values, see: (d)http://www.chem.wisc.edu/areas/reich/pkatable/
- 19.Patel D, Shukla S, Gupta S. Int J Oncol. 2007;30:233. [PubMed] [Google Scholar]
- 20.Choudary BM, Ranganath KVS, Yadav J, Lakshmi Kantam M. Tetrahedron Lett. 2005;46:1369. [Google Scholar]
- 21.Kondo T, Oyama K, Yoshida K. Angew Chem Int Ed. 2001;40:894. doi: 10.1002/1521-3773(20010302)40:5<894::AID-ANIE894>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 22.Si D, Wang Y, Zhou Y, Guo Y, Wang J, Zhou H, Li Z, Fawcett JP. Drug Metab Dispos. 2009;37:629. doi: 10.1124/dmd.108.023416. [DOI] [PubMed] [Google Scholar]
- 23.Cermak R, Wolffram S. Curr Drug Metab. 2006;7:729. doi: 10.2174/138920006778520570. [DOI] [PubMed] [Google Scholar]
- 24.The reactions were monitored for 24 h, during which > 50% conversion was achieved for each reaction. The rate constants were obtained by kinetic simulation of the reaction timecourses to a simple A → B → C sequential kinetic model. See Supplementary Information for kinetic profiles.
- 25.Oxidation of the 3-d5 was carried out in DMSO-d6 to avoid background α–H/D exchange.
- 26.A Pd0 species, formed from a PdII–H intermediate, has been isolated from a “Saegusa”-type oxidation reaction: Porth S, Bats JW, Trauner D, Giester G, Mulzer J. Angew Chem Int Ed. 1999;38:2015. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<2015::AID-ANIE2015>3.0.CO;2-#.
- 27.Recent studies suggest that aerobic oxidation of PdII–hydrides proceed via Pd0, as shown in Scheme 1. See: Konnick MM, Stahl SS. J Am Chem Soc. 2008;130:5753. doi: 10.1021/ja7112504.Popp BV, Stahl SS. Chem Eur J. 2009;15:2915. doi: 10.1002/chem.200802311.Decharin N, Popp BV, Stahl SS. J Am Chem Soc. 2011;133:13268. doi: 10.1021/ja204989p.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.