

Abstract
Plasmodium falciparum has developed resistance to most available treatments, underscoring the need for novel antimalarial drugs. Fibrates are lipid-modifying agents used to reduce morbidity and mortality associated with cardiovascular disease. They may have antimalarial activity through modulation of P-glycoprotein and ATP-binding cassette subfamily A member (ABC-1)-mediated nutrient transport and/or via a putative peroxisome proliferator-activated receptor alpha-like protein. We therefore examined in vitro antimalarial activities of fibrates and their interactions with chloroquine and dihydroartemisinin in chloroquine-sensitive (3D7) and chloroquine-resistant (W2mef) strains of P. falciparum using the conventional isotopic assay microtechnique. A bioassay was used to assess inhibition activities of human plasma after therapeutic fenofibrate doses. Fenofibric acid, the main metabolite of fenofibrate, was the most potent of the fibrates tested, with mean 50% inhibitory concentrations of 152 nM and 1,120 nM for chloroquine-sensitive and -resistant strains, respectively. No synergistic interaction between fibrates and chloroquine or dihydroartemisinin was observed. Plasma fenofibric acid concentrations, quantified by high-performance liquid chromatography in seven healthy volunteers after treatment (mean, 15.3 mg/liter, or 48 μM), inhibited P. falciparum. BLAST analysis revealed the likely presence of an ABC-1 transporter homolog in P. falciparum. Our findings demonstrate that fenofibric acid has activity similar to the activities of conventional antimalarial drugs at concentrations well below those achieved after therapeutic doses. It may inhibit P. falciparum growth by inhibiting intracellular lipid transport.
INTRODUCTION
The progressive increase in drug-resistant malaria, including the recent emergence of delayed clearance of Plasmodium falciparum after treatment with artemisinin derivatives (8, 16, 36), highlights the need for novel effective antimalarial drugs. One possible source is compounds which have been approved for other indications but which are found to have parasiticidal or disease-modifying effects in malaria infection. Given prior detailed knowledge of their pharmacokinetic properties, safety, and tolerability, it is likely that such compounds can be fast-tracked through the usual drug development process. Examples that may prove to be pertinent to the treatment of human malaria are the glitazone drug rosiglitazone (4) and the 3-hydroxy-3-methyl-glutaryl coenzyme A reductase inhibitor atorvastatin (30, 31, 34).
Fibrates such as gemfibrozil and fenofibrate are agonists of peroxisome proliferator-activated receptor alpha (PPARα) that are in relatively widespread clinical use as potent lipid-modifying drugs (13). The first study of fibrates in malaria found that clofibrate directly inhibited the development of parasitemia in P. berghei-infected mice (27). Subsequent studies have provided indirect evidence that fibrates might have therapeutic potential in malaria. One study showed that fenofibrate was the only member of the fibrate class to inhibit P-glycoprotein-mediated transport (19). This finding suggests that fenofibrate might also inhibit the P-glycoprotein homologue 1 (Pgh1) in P. falciparum and, thus, through reduced efflux of chloroquine (CQ) and mefloquine, reverse resistance (32). In addition, the beneficial anti-inflammatory properties of fibrates, such as those seen in a study of gemfibrozil in murine influenza (5), might attenuate host inflammatory responses that have been proposed to be part of the pathophysiology of complications of malaria, including coma (9–11). This mechanism is thought to underlie the disease-modifying effects of rosiglitazone (4).
In the light of these observations, we have examined the in vitro activity of gemfibrozil, clofibrate, fenofibrate, and fenofibric acid (the main metabolite of fenofibrate, which is also marketed as a lipid-modifying drug) against CQ-sensitive and CQ-resistant P. falciparum, alone and in combination with either CQ or dihydroartemisinin (DHA).
MATERIALS AND METHODS
P. falciparum isolates and culture.
The laboratory-adapted P. falciparum strains 3D7 (Africa; CQ sensitive), Dd2, and W2mef (Indochina; CQ resistant) were maintained in continuous culture in RPMI 1640-HEPES supplemented with 10% (vol/vol) human plasma, 92.6 mg/liter l-glutamine, 500 μg/liter gentamicin, 50 mg/liter hypoxanthine, all obtained from Sigma-Aldrich, St. Louis, MO. Cultures were maintained in a low-oxygen atmosphere (5% O2, 5% CO2, 90% N2; BOC Gas, Perth, Australia) with daily changes of culture medium at 5% hematocrit and diluted with noninfected erythrocytes when parasitemia exceeded 5%. Synchronous cultures were prepared by sorbitol lysis (26).
In vitro parasite growth inhibition.
Stock solutions of chloroquine diphosphate (Sigma Chemicals, St. Louis, MO), dihydroartemisinin (Sigma), atorvastatin (Waterstonetech, Carmel, IN), fenofibrate (Sigma), clofibrate (Sigma), gemfibrozil (Sigma-Aldrich), and fenofibric acid (Tyger Scientific Inc., NJ) were prepared in distilled water (CQ) and dimethyl sulfoxide (fibrates and atorvastatin). For in vitro susceptibility, serial dilutions of each drug were prepared in RPMI 1640 and added in triplicate to 96-well plates at final concentrations of 6.25 to 1,600 nM (CQ), 0.78 to 51.2 nM (DHA), 0.3 μM to 200 μM (atorvastatin), 6.2 to 800 μM (gemfibrozil, fenofibrate, clofibrate), and 39 to 5,000 nM (fenofibric acid). Synchronous parasite suspensions (≥90% rings) were adjusted to 0.5% parasitemia and a final hematocrit of 1.5% in the drug-parasite mixture and were incubated for 48 h. Parasite growth was measured by a modification of the [3H]hypoxanthine incorporation assay as previously described (15, 35). Fifty percent inhibitory concentrations (IC50s) were determined by nonlinear regression analysis.
Drug interaction studies.
A modified fixed-ratio isobologram method was used to assess drug interactions (20, 35). Inhibition assays incorporating 1.5% parasitemia and 1.5% hematocrit were first carried out to determine IC50s for individual fibrates, atorvastatin, CQ, and DHA. These were used to establish test concentration ranges in the combination assays. Briefly, a total of 11 solutions containing fixed-ratio mixtures of fibrates with either atorvastatin, CQ, or DHA were prepared in the following ratios: 1:0, 0:1, 1:1, 1:3, 1:30, 1:300, 1:3,000, 3:1, 30:1, 300:1, and 3,000:1. The agent at a ratio of 1 within the pair would have its concentration fixed at 1,600 μM (fibrates), 200 μM (atorvastatin), 1,600 nM (CQ), and 100 nM (DHA), while the concentration of the other agent is determined according to the combination's ratio factor (i.e., for the combination gemfibrozil-DHA at the ratio 1:3, the concentrations of each component are 533 μM and 100 nM, respectively).
These initial mixtures (200 μl/well) were dispensed into the bottom row of 96-well plates in duplicate (1:0 and 0:1 mixtures were dispensed in triplicate) and serially diluted with 100 μl of assay medium six times, leaving the top row as a drug-free control. Fractional inhibitory concentrations (FICs) of each drug in each combination determined from dose-response curves were used to construct isobolograms from which the sum of each FIC was calculated (2). The interaction value (I) is calculated by fitting the data to the function Yi = 1 − {XI/[XI + e(−I)(1 − Xi)]}, where Yi is the IC50 of drug A combined with drug B and Xi is the IC50 of drug B when combined with drug A (7).
Dosed-plasma bioassay.
Due to the possibility that active metabolites of fenofibrate and other in vivo factors might contribute to enhanced antimalarial activity such as those observed for atovaquone (6, 18), a bioassay was performed. Pretreatment heparinized blood samples were taken from seven consenting healthy adults on no regular medication who were given fenofibrate at 145 mg as nanoparticles (Lipidil; Abbott Pharmaceuticals, Sydney, Australia) once daily for six consecutive days. A second blood sample was drawn on the last day of dosing at the time of the predicted maximal plasma concentration (Cmax) of fenofibric acid at steady state (about 6 h after the final fenofibrate dose) (23). Approval for these procedures was obtained from the South Metropolitan Area Health Service Human Research Ethics Committee.
All blood samples were promptly centrifuged, and aliquots of separated plasma were stored at −20°C. After extraction in hexane-chloroform-isopropanol (18:80:2, vol/vol/vol), plasma fenofibric acid concentrations in pre- and posttreatment plasma were measured by validated high-performance liquid chromatographic (HPLC) assay with tandem mass spectrometric detection and 2-(2,4,5-trichlorophenoxy)-propionic acid as the internal standard (Laboratoires Fournier SA, Daix, France) (37). The lower limit of quantitation (LLOQ) was 0.030 μg/ml, and the between-run accuracy (percent deviation) and precision (coefficient of variation [CV]) were <4% and <8%, respectively (37).
A modified microdilution isotopic technique (15, 34, 35) was used to determine the antimalarial activities of pre- and posttreatment plasma using serial dilution in drug-free RPMI. In triplicate experiments, aliquots of 100 μl of plasma were added to 90 μl of parasite suspension (1% parasitemia, 1.5% hematocrit) and 10 μl [3H]hypoxanthine (0.5 μCi) in 96-well plates, and the mixture was incubated, harvested, and counted (22).
BLAST analysis.
To elucidate possible mechanisms underlying the activity of fenofibric acid against malaria parasites, similarity searches were conducted between mRNA and protein sequences of human PPARα and P. falciparum using the Basic Local Alignment Search Tool (BLAST; NCBI).
RESULTS
In vitro antimalarial activity.
The in vitro inhibitory effects of the four fibrates and CQ are summarized in Table 1. All fibrates showed antimalarial activity, with fenofibric acid being the most potent. With the exception of fenofibric acid, the IC50s of the other fibrates did not differ between CQ-sensitive and CQ-resistant strains and were well above those for CQ.
Table 1.
In vitro activity of fibrates against CQ-sensitive (3D7) and CQ-resistant (W2mef) strains of P. falciparuma
| Drug | 3D7 |
W2mef |
||
|---|---|---|---|---|
| IC50 | IC90 | IC50 | IC90 | |
| Clofibrate | 184 (98–345) | 441 (264–736) | 256 (197–332) | 708 (514–976) |
| Gemfibrozil | 311 (245–395) | 541 (446–657) | 245 (181–332) | 511 (445–587) |
| Fenofibrate | 69 (54–88) | 228 (171–304) | 72 (65–80) | 178 (152–210) |
| Fenofibric acid | 152 (109–212) | 250 (191–326) | 1,120 (916–1,369) | 2,436 (1,866–3,179) |
| Chloroquine | 23 (15–34) | 48 (23–99) | 230 (202–261) | 495 (334–732) |
Data represent at least six experiments performed in triplicate. The 95% confidence intervals of geometric mean IC50 and IC90 values are given in parentheses. Data for clofibrate, gemfibrozil, and fenofibrate are in micromolar, and data for fenofibric acid and chloroquine are in nanomolar.
Interactions with conventional antimalarial drugs.
The results of drug interaction analysis are shown in Table 2. There were no synergistic combinations identified from the drug interaction studies involving fibrates, CQ, DHA, and atorvastatin. For fenofibric acid-CQ, there was no interaction for 3D7 but there was antagonism for W2mef. In the case of gemfibrozil-CQ, there was antagonism against 3D7 and an indifferent interaction for W2mef. The remaining combinations, including the combination fenofibric acid-atorvastatin, showed indifferent interactions.
Table 2.
In vitro efficacy of antimalarial drug combinations against P. falciparum clones 3D7, W2mef, and Dd2 assessed by isobolographic analysisa
| Drug combination | 3D7 |
W2mef/Dd2 |
||||
|---|---|---|---|---|---|---|
| I (95% CI) | ΣFIC (95% CI) | Interpretation | I (95% CI) | ΣFIC (95% CI) | Interpretation | |
| Fenofibric acid-chloroquine | 0.23 (−0.06 to 0.51) | 0.99 (0.95 to 1.02) | Indifferent | −1.07 (−2.08 to −0.07) | 1.19 (1.02 to 1.36) | Antagonistic |
| Fenofibric acid-dihydroartemisinin | −0.26 (−1.02 to 0.50) | 1.11 (0.99 to 1.21) | Indifferent | −0.48 (−1.31 to 0.36) | 1.11 (1.02 to 1.21) | Indifferent |
| Fenofibric acid-atorvastatin | −0.33 (−1.92 to 1.26) | 1.19 (1.02 to 1.35) | Indifferent | −0.81 (−1.52 to −0.09) | 1.08 (0.96 to 1.19) | Indifferent |
| Fenofibrate-chloroquine | −0.67 (−1.40 to 0.05) | 1.08 (0.98 to 1.12) | Indifferent | −0.41 (−1.01 to 0.19) | 1.01 (0.93 to 1.10) | Indifferent |
| Fenofibrate-dihydroartemisinin | 0.36 (−0.83 to 1.55) | 1.16 (1.03 to 1.30) | Indifferent | ND | ND | ND |
| Gemfibrozil-chloroquine | −1.53 (−2.44 to −0.62) | 1.12 (1.01 to 1.23) | Antagonistic | −0.55 (−1.80 to 0.73) | 1.19 (0.99 to 1.40) | Indifferent |
| Gemfibrozil-dihydroartemisinin | −1.34 (−3.34 to 0.66) | 1.20 (0.95 to 1.45) | Indifferent | −0.25 (−2.10 to 1.61) | 1.15 (0.95 to 1.40) | Indifferent |
Data are the interaction factor (I) and the summed fractional inhibitory concentration (ΣFIC) with 95% confidence intervals (CIs). The assessment of interaction is based on both I and ΣFIC data (see the text). Data for CQ-resistant strains are presented together. ND, not determined.
Bioassay of fenofibric acid.
Fenofibrate is a prodrug that is hydrolyzed by tissue and plasma esterases to fenofibric acid. No fenofibric acid was detected by HPLC in pretreatment plasma from any of the seven healthy volunteers. Fenofibric acid was detectable in all the volunteers in the posttreatment sample with a mean steady-state concentration of 15.3 mg/liter (48 μM). The antimalarial activity of fenofibric acid generated in vivo after fenofibrate dosing was assessed by bioassay (Fig. 1). Although undiluted pretreatment plasma inhibited parasite growth as effectively as when drug was present, posttreatment plasma inhibited growth of 3D7 at dilutions between 8- and 64-fold.
Fig 1.
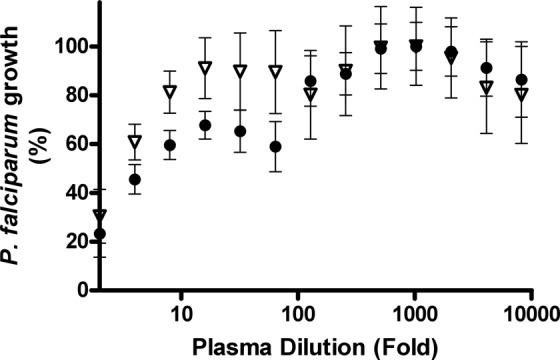
Bioassay of plasma before and after fenofibrate treatment against laboratory-adapted clone 3D7. Growth response to plasma dilutions was assessed in triplicate in two independent experiments. Data points represent pretreatment plasma (triangles) and posttreatment plasma (circles).
BLAST analysis for PPAR-like region and ATP-binding cassette subfamily A member (ABC-1) transporter in P. falciparum.
Nucleic acid alignments between human and Plasmodium mRNA were generally weak. Comparisons between the human PPARα protein (NCBI reference sequence no. NP_001001928.1) and reference proteins for P. falciparum taxid 5833 resulted in three BLAST hits. Although the alignments were low, two of these were located at the PPARα DNA binding domain and one was located at the ligand binding domain. The latter, identified to be a conserved Plasmodium protein (GenBank accession no. XP_001350490.2), is of particular interest since it aligns with both the ligand binding site and the heterodimer interface of human PPARα in a similar fashion to protein homologs found in Babesia bovis and Toxoplasma gondii. The region (positions 959 to 1030) of the Plasmodium protein identified as Plasmodium 1 showed similarity with hypothetical proteins in other Plasmodium species, including P. knowlesi, P. vivax, P. yoelii, P. chabaudi, and P. berghei.
Proteins within ABC-1 mediate cholesterol and lipid transport and are within the same superfamily of ATP-binding cassette (ABC) transporters as the multidrug-resistant P-glycoprotein. Nucleic acid alignments between human and Plasmodium mRNA produced 103 BLAST hits of short sequences (up to 50 bp). Similarity searches between reference protein sequences of human ABC-1 and P. falciparum revealed the presence of a parasite homolog of this lipid efflux pump. There were 49 protein alignment hits, and 13 of the P. falciparum nucleotide sequences spanned across two regions (positions 900 to 1116 and 1927 to 2110) with a coverage of up to 19% (E < 0.006) of the human ABC-1 query sequence. Two significant alignments were found in the P. falciparum ABC transporter protein (GenBank accession no. XP_001350233.1) and P. falciparum multidrug resistance protein 2 (GenBank accession no. XP_001348629.1) with E values of 8 to 16 and 1 to 12, respectively.
DISCUSSION
The present study demonstrates that gemfibrozil, fenofibrate, and clofibrate have weak activity relative to conventional antimalarial drugs in vitro. Nonetheless, their IC50s are similar to those of antibiotics that are used for malaria prophylaxis and adjunctive therapy (29). This level of activity may have accounted for the lower infection rate in the clofibrate-treated group of animals observed previously (27), although inflammation-modulating effects similar to those of gemfibrozil (5) may have also contributed. Fenofibric acid, the major metabolite of fenofibrate in vivo (25), had the greatest activity of all the fibrate compounds that we tested, with activity at the nM medium concentrations seen with conventional antimalarial agents. As might have been predicted from this observation and consistent with the hypothesis that in vivo metabolism of therapeutic doses of fenofibrate generates fenofibric acid with useful antimalarial activity, the bioassay experiments showed that therapeutic plasma fenofibric acid concentrations inhibited CQ-sensitive and CQ-resistant strains of cultured P. falciparum.
For a drug to be clinically useful, the plasma concentration achievable in vivo should be at least an order of magnitude higher than the in vitro inhibitory concentration. The steady-state Cmax of fenofibric acid was 15.3 mg/liter (48 μM) in the present study, consistent with a mean steady-state Cmax of 23 mg/liter (72 μM) following 5 days of treatment with a micronized capsule formulation of 200 mg fenofibrate in six healthy volunteers (14). Given that the present IC50s were 0.15 μM and 1.12 μM for CQ-sensitive and CQ-resistant P. falciparum strains, respectively, the present data suggest that steady-state in vivo concentrations were between 40 and 320 times these IC50s. When the respective IC90 values were used, these ratios were still ≥20. Using published mean values for fenofibric acid pharmacokinetic parameters, including an elimination half-life of 20 h (23, 37), and utilizing a simple one-compartment model, we estimate that the mean in vivo concentrations would be >20 times the IC50 and >10 times the IC90 values 4 to 6 h after the first dose. Use of fenofibric acid may further enhance bioavailability (37) and thus potential in vivo antimalarial efficacy.
A second consideration for clinical application is toxicity. Fenofibrate is generally well tolerated, with the most frequent acute adverse effects being mild gastrointestinal symptoms (nausea, vomiting, diarrhea, and abdominal pain) and dermatological complaints (rash, pruritus, and photosensitivity), while elevated hepatic transaminase concentrations can occasionally be observed (23). An increase in serum creatinine is reversible and not a marker of renal toxicity (12). Thromboembolic disease and pancreatitis have been reported with long-term use (24), but their potential significance in patients treated with short courses for malaria is uncertain. Because of evidence of embryofetal toxicity in animal studies, fenofibrate is not recommended in pregnancy.
Under current recommendations regarding artemisinin combination therapies, fenofibric acid could be partnered with a compound such as DHA as, from the present study, their interactions are not antagonistic. Unless sustained-release formulations are developed, the half-life of fenofibric acid suggests that fenofibrate (or fenofibric acid itself) would need to be administered daily for 7 days in combination with an initial 3-day artemisinin regimen to ensure therapeutic concentrations over at least three P. falciparum asexual life cycles. Although 7-day treatment regimens have previously been used for uncomplicated malaria (e.g., quinine-tetracycline), this has implications for compliance. Its usefulness as a prophylactic drug is yet to be elucidated. Although the initial use of fenofibrate may be limited to developed countries due to cost of treatment, wider future deployment as an antimalarial could be possible as economies develop and costs of established medications fall.
Although its mode of action remains to be elucidated, fenofibric acid may act by interfering with Pgh1 (19) and ABC-1-mediated transport and/or via a putative PPARα-like protein. BLAST analysis revealed partial similarities between a conserved Plasmodium protein sequence and that of the PPARα ligand binding domain in humans. Despite the low alignment scores, there may be sufficient similarities at key amino acid positions for the tertiary protein conformation to interact with fenofibric acid, resulting in subsequent metabolic interference. In human and murine cells, fenofibric acid interferes with the expression of ABC-1 (1, 21), thus altering lipid accumulation. Fenofibric acid effects on the Plasmodium ABC-1 homolog may disturb the development of P. falciparum by similar mechanisms, depriving the growing parasite of lipid components of membranes and other cellular structures.
There was a mildly antagonistic interaction between fenofibric acid and CQ in the W2mef strain in the present experiments. If fibrates act through inhibition of Pgh1, as might be predicted (19), a synergistic interaction with reversal of CQ resistance would have occurred. Fenofibric acid and clofibrate appeared to be less active against W2mef than 3D7, suggesting that they, like CQ, are exerting their effect within the food vacuole and are at risk of drug efflux mechanisms in CQ-resistant parasites. Indeed, the present BLAST analysis suggests that it may inhibit P. falciparum growth by inhibiting intracellular lipid transport. The present observations must, however, be viewed as preliminary.
An inhibitory effect against P. falciparum was observed in pretreatment human plasma. Plasma from untreated healthy individuals can interfere with parasite survival via complement-mediated cell lysis. Non-heat-treated serum has been shown to reduce parasite growth as much as 25% compared to heat-treated control sera (33). Therefore, heat inactivation of pooled human plasma is required to reduce this growth-inhibitory activity prior to supplementation in culture medium. Plasma fenofibric acid is stable for up to 8 h at room temperature (17). Its stability in plasma at 50°C is, however, unknown. Therefore, to minimize the risk of drug degradation, plasma samples in the bioassays were not heat treated. This may explain the apparent dose-response of pretreatment plasma against the parasite. Alternative non-heat treatment of bioassay plasma, such as with Affingel protein A, may reduce inhibition of parasite growth by removal of human immunoglobulin G (33). Toward the other end of the curve, a 1-in-64 dilution was the last to show inhibitory activity against 3D7. The equivalent medium fenofibric acid concentration (0.8 μmol/liter at this dilution) was also the last above the 3D7 IC90 (median, 0.25 μmol/liter) in the initial in vitro experiments with added fenofibric acid rather than plasma from treated volunteers.
The present study has revealed fenofibric acid, the principal metabolite of fenofibrate, to be the most active lipid-modifying agent against P. falciparum in vitro, including statin drugs such as atorvastatin (30, 31, 34). The favorable pharmacokinetic properties of fenofibrate as well as its known safety and tolerability (3, 28) suggest that it might have future clinical application. Its use may be restricted to combination with other established drugs such as artemisinin derivatives. Indeed, there was no evidence of an unfavorable interaction in our isobolographic analyses. Further in vitro and animal studies are warranted.
ACKNOWLEDGMENTS
We thank Jean-Claude Ansquer, Delphine Chevillon, and Colette Prevost from Abbott Laboratories Fournier SA for performing the HPLC analysis and Jenny Wong for technical assistance with in vitro cultures.
This study was supported by the National Health and Medical Research Council of Australia (grant 513782).
Footnotes
Published ahead of print 19 March 2012
REFERENCES
- 1. Arakawa R, Tamehiro N, Nishimaki-Mogami T, Ueda K, Yokoyama S. 2005. Fenofibric acid, an active form of fenofibrate, increases apolipoprotein A-I-mediated high-density lipoprotein biogenesis by enhancing transcription of ATP-binding cassette transporter A1 gene in a liver X receptor-dependent manner. Arterioscler. Thromb. Vasc. Biol. 25:1193–1197 [DOI] [PubMed] [Google Scholar]
- 2. Berenbaum MC. 1978. A method for testing for synergy with any number of agents. J. Infect. Dis. 137:122–130 [DOI] [PubMed] [Google Scholar]
- 3. Bhavesh D, Shah S. 2009. Determination of fenofibric acid in human plasma by ultra performance liquid chromatography-electrospray ionization mass spectrometry: application to a bioequivalence study. Biomed. Chromatogr. 23:922–928 [DOI] [PubMed] [Google Scholar]
- 4. Boggild AK, et al. 2009. Use of peroxisome proliferator-activated receptor gamma agonists as adjunctive treatment for Plasmodium falciparum malaria: a randomized, double-blind, placebo-controlled trial. Clin. Infect. Dis. 49:841–849 [DOI] [PubMed] [Google Scholar]
- 5. Budd A, et al. 2007. Increased survival after gemfibrozil treatment of severe mouse influenza. Antimicrob. Agents Chemother. 51:2965–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Butcher GA, Sinden RE. 2003. Persistence of atovaquone in human sera following treatment: inhibition of Plasmodium falciparum development in vivo and in vitro. Am. J. Trop. Med. Hyg. 68:111–114 [PubMed] [Google Scholar]
- 7. Canfield CJ, Pudney M, Gutteridge WE. 1995. Interactions of atovaquone with other antimalarial drugs against Plasmodium falciparum in vitro. Exp. Parasitol. 80:373–381 [DOI] [PubMed] [Google Scholar]
- 8. Carrara VI, et al. 2009. Changes in the treatment responses to artesunate-mefloquine on the northwestern border of Thailand during 13 years of continuous deployment. PLoS One 4:e4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clark CJ, et al. 2005. Prolonged survival of a murine model of cerebral malaria by kynurenine pathway inhibition. Infect. Immun. 73:5249–5251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clark IA, Alleva LM. 2009. Is human malarial coma caused, or merely deepened, by sequestration? Trends Parasitol. 25:314–318 [DOI] [PubMed] [Google Scholar]
- 11. Clark IA, Alleva LM, Budd AC, Cowden WB. 2008. Understanding the role of inflammatory cytokines in malaria and related diseases. Travel Med. Infect. Dis. 6:67–81 [DOI] [PubMed] [Google Scholar]
- 12. Davis TM, et al. 2011. Effects of fenofibrate on renal function in patients with type 2 diabetes mellitus: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study. Diabetologia 54:280–290 [DOI] [PubMed] [Google Scholar]
- 13. Derosa G, et al. 2009. Fenofibrate, simvastatin and their combination in the management of dyslipidaemia in type 2 diabetic patients. Curr. Med. Res. Opin. 25:1973–1983 [DOI] [PubMed] [Google Scholar]
- 14. Desager JP, Horsmans Y, Vandenplas C, Harvengt C. 1996. Pharmacodynamic activity of lipoprotein lipase and hepatic lipase, and pharmacokinetic parameters measured in normolipidaemic subjects receiving ciprofibrate (100 or 200 mg/day) or micronised fenofibrate (200 mg/day) therapy for 23 days. Atherosclerosis 124(Suppl):S65–S73 [DOI] [PubMed] [Google Scholar]
- 15. Desjardin RE, Canfield Haynes CJ, Chulay DJD. 1979. Quantitative assessment of antimalarial activity in vitro by semiautomated microdilution technique. Antimicrob. Agents Chemother. 16:710–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dondorp AM, et al. 2010. Artemisinin resistance: current status and scenarios for containment. Nat. Rev. Microbiol. 8:272–280 [DOI] [PubMed] [Google Scholar]
- 17. Dubey S, et al. 2010. Rapid, sensitive and validated ultra-performance liquid chromatography/mass spectrometric method for the determination of fenofibric acid and its application to human pharmacokinetic study. E-J. Chem. 7:25–36 [Google Scholar]
- 18. Edstein MD, et al. 2005. Lengthy antimalarial activity of atovaquone in human plasma following atovaquone-proguanil administration. Antimicrob. Agents Chemother. 49:4421–4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ehrhardt M, Lindenmaier H, Burhenne J, Haefeli WE, Weiss J. 2004. Influence of lipid lowering fibrates on P-glycoprotein activity in vitro. Biochem. Pharmacol. 67:285–292 [DOI] [PubMed] [Google Scholar]
- 20. Fivelman QL, Adagu IS, Warhurst DC. 2004. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob. Agents Chemother. 48:4097–4102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jaye M, Duverger N, Searfoss G, Minnich A. November 2003. Therapeutic uses of PPAR mediators. U.S. patent 20030220373A1
- 22. Karl S, Wong RP, St Pierre TG, Davis TM. 2009. A comparative study of a flow-cytometry-based assessment of in vitro Plasmodium falciparum drug sensitivity. Malar. J. 8:294–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keating GM, Ormrod D. 2002. Micronised fenofibrate: an updated review of its clinical efficacy in the management of dyslipidaemia. Drugs 62:1909–1944 [DOI] [PubMed] [Google Scholar]
- 24. Keech A, et al. 2005. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 366:1849–1861 [DOI] [PubMed] [Google Scholar]
- 25. Kirchgassler KU, Schmitz H, Bach G. 1998. Effectiveness and tolerability of 12-week treatment with micronised fenofibrate 200 mg in a drug monitoring programme involving 9884 patients with dyslipidaemia. Clin. Drug Invest. 15:197–204 [Google Scholar]
- 26. Lambros C, Vanderberg JP. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65:418–420 [PubMed] [Google Scholar]
- 27. McQuistion TE. 1979. Effect of temperature and clofibrate on Plasmodium berghei infection in mice. Am. J. Trop. Med. Hyg. 28:12–14 [DOI] [PubMed] [Google Scholar]
- 28. Miller DB, Spence JD. 1998. Clinical pharmacokinetics of fibric acid derivatives (fibrates). Clin. Pharmacokinet. 34:155–162 [DOI] [PubMed] [Google Scholar]
- 29. Nakornchai S, Konthiang P. 2006. Activity of azithromycin or erythromycin in combination with antimalarial drugs against multidrug-resistant Plasmodium falciparum in vitro. Acta Trop. 100:185–191 [DOI] [PubMed] [Google Scholar]
- 30. Parquet V, et al. 2009. Atorvastatin is a promising partner for antimalarial drugs in treatment of Plasmodium falciparum malaria. Antimicrob. Agents Chemother. 53:2248–2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pradines B, et al. 2007. Atorvastatin is 10-fold more active in vitro than other statins against Plasmodium falciparum. Antimicrob. Agents Chemother. 51:2654–2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF. 2000. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature 403:906–909 [DOI] [PubMed] [Google Scholar]
- 33. Teja-Isavadharm P, et al. 2004. Plasmodium falciparum-based bioassay for measurement of artemisinin derivatives in plasma or serum. Antimicrob. Agents Chemother. 48:954–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wong R, Davis TM. 2009. Statins as potential antimalarial drugs: low relative potency and lack of synergy with conventional antimalarial drugs. Antimicrob. Agents Chemother. 53:2212–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wong RP, et al. 2011. Desbutyl-lumefantrine is a metabolite of lumefantrine with potent in vitro antimalarial activity that may influence artemether-lumefantrine treatment outcome. Antimicrob. Agents Chemother. 55:1194–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wongsrichanalai C, Meshnick SR. 2008. Declining artesunate-mefloquine efficacy against falciparum malaria on the Cambodia-Thailand border. Emerg. Infect. Dis. 14:716–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhu T, Ansquer JC, Kelly MT, Sleep DJ, Pradhan RS. 2010. Comparison of the gastrointestinal absorption and bioavailability of fenofibrate and fenofibric acid in humans. J. Clin. Pharmacol. 50:914–921 [DOI] [PubMed] [Google Scholar]