

Abstract
Antimicrobial peptides with amphipathic β-hairpin-like structures have potent antimicrobial properties and low cytotoxicity. The effect of VR or RV motifs on β-hairpin-like antimicrobial peptides has not been investigated. In this study, a series of β-hairpin-like peptides, Ac-C(VR)nDPG (RV)nC-NH2 (n = 1, 2, 3, 4, or 5), were synthesized, and the effect of chain length on antimicrobial activity was evaluated. The antimicrobial activity of the peptides initially increased and then decreased with chain length. Longer peptides stimulated the toxicity to mammalian cells. VR3, a 16-mer peptide with seven amino acids in the strand, displayed the highest therapeutic index and represents the optimal chain length. VR3 reduced bacterial counts in the mouse peritoneum and increased the survival rate of mice at 7 days after Salmonella enterica serovar Typhimurium infection in vivo. The circular dichroism (CD) spectra demonstrated that the secondary structure of the peptides was a β-hairpin or β-sheet in the presence of an aqueous and membrane-mimicking environment. VR3 had the same degree of penetration into the outer and inner membranes as melittin. Experiments simulating the membrane environment showed that Trp-containing VRW3 (a VR3 analog) tends to interact preferentially with negatively charged vesicles in comparison to zwitterionic vesicles, which supports the biological activity data. Additionally, VR3 resulted in greater membrane damage than melittin as determined using a flow cytometry-based membrane integrity assay. Collectively, the data for synthetic lipid vesicles and whole bacteria demonstrated that the VR3 peptide killed bacteria via targeting the cell membrane. This assay could be an effective pathway to screen novel candidates for antibiotic development.
INTRODUCTION
There is an increasing need for alternatives to antibiotics due to the increasing problems associated with bacterial resistance to antibiotics. Antimicrobial peptides (AMPs) are major components of the innate immune system, and they have a broad spectrum of activities against bacteria, fungi, viruses, and parasites (12). AMPs are cationic and amphipathic with low molecular masses, ranging from approximately 1 to 5 kDa. The mechanisms of action of AMPs have been studied extensively and were proven to be different from those of antibiotics (2, 27). AMPs accumulate on the membrane surface and insert into the membrane bilayer to form pores through mechanisms that include “barrel stave,” “carpet,” or “toroidal pore” models (2). Additionally, AMPs may penetrate into the cell to bind intracellular molecules (27).
The β-sheet motif occurs frequently in known AMPs. As basic and simplified β-sheet structures, β-hairpins are composed of two primary structural units, strands and β-turns. Generally, a β-hairpin contains one or two disulfide bonds to cyclize the peptide chain. Previous studies have reported that amphipathic β-hairpin-like structures or two-strand β-sheet conformations had potent antimicrobial properties and high cell selectivity (9, 24, 35).
The α-helix has been widely investigated for the design of synthetic peptides with improved antibacterial activity (4, 15, 31). We have recently designed a series of α-helical peptides that are rich in valine (V) and arginine (R) residues and have potent antimicrobial activity (23). The effect of the VR or RV motif on the biological activities of β-hairpin-like antimicrobial peptides has not been studied. Electrostatic interactions occur between positively charged residues and negatively charged membrane surfaces (18). R-containing peptides may have stronger electrostatic interactions than those rich in lysine (45). Moreover, R can drag the lipid phosphate groups, which causes toroidal pore defects in the anionic membrane (43). Additionally, tight R-phosphate associations are important for the strong antimicrobial activities of the cationic β-hairpin antimicrobial peptide PG-1 (42). Val-rich arms are prone to extended strand conformations in β-sheets (37). Therefore, it is essential to investigate the antimicrobial properties of V- and R-rich AMPs with β-sheet structures. The biological function of these AMPs is usually modulated by their length (7, 22, 25). It is feasible to form an antiparallel β-sheet with different strand lengths by incorporating multiple alternating VR or RV dipeptide residues.
In this study, we designed a series of cyclic β-hairpin-like antimicrobial peptides by positioning hydrophobic or positively charged residues in the sequence template Ac-C(VR)nDPG (RV)nC-NH2 (n = 1, 2, 3, 4, or 5). A short two-residue loop segment (DPG) was used to link the two-strand antiparallel β-sheet. DPG stabilizes the type II′ β-turn and promotes the formation of β-hairpin conformations (13, 30). The β-hairpin was further stabilized by a disulfide bridge. The C terminus was aminated and the N terminus was protected by acetylation (Ac), which improve peptide stabilization (21, 26). The effect of the peptide chain length on antimicrobial activity was evaluated in vitro, and the peptide with the greatest cell selectivity was identified. The antimicrobial activity of the peptides was further investigated in vivo. To elucidate the peptide-membrane interaction mechanism, the tryptophan (W) fluorescence spectra of W-containing peptides were measured in the presence of synthetic lipid vesicles. The peptide-membrane interactions were further evaluated using the outer membrane (OM) permeabilization assay, the membrane depolarization assay, and flow cytometry analysis.
MATERIALS AND METHODS
Peptide design and synthesis.
We designed a series of cyclic amphipathic β-hairpin-like antimicrobial peptides according to the sequence template Ac-C(VR)nDPG (RV)nC-NH2 (n = 1, 2, 3, 4, or 5) (Table 1; Fig. 1). The peptide VRW3 was derived from the peptide VR3 by adding W to its C terminus to monitor lipid-peptide interactions using tryptophan fluorescence. The peptides were synthesized by GL Biochem Corporation (Shanghai, China) by solid-phase methods using N-(9-fluorenyl)methoxycarbonyl (Fmoc) chemistry. The C terminus was amine protected, and the N terminus was Ac protected. Electrospray mass spectrometry was used to identify the peptides. The purity of peptides was determined to be >95% using reverse-phase high-performance liquid chromatography.
Table 1.
Amino acid sequences, formulas, and molecular masses of the peptides
| Peptide | Sequence | Formula | Molecular mass (Da) |
|
|---|---|---|---|---|
| Calculated | Observed | |||
| VR1 | Ac-CVRDPGRVC-NH2 | C37H67N16O10S2 | 928.10 | 928.12 |
| VR2 | Ac-C(VR)2DPG(RV)2C-NH2 | C59H109N26O14S2 | 1,438.70 | 1,438.76 |
| VR3 | Ac-C(VR)3DPG(RV)3C-NH2 | C81H151N36O18S2 | 1,949.30 | 1,949.41 |
| VR4 | Ac-C(VR)4DPG(RV)4C-NH2 | C103H193N46O22S2 | 2,460.00 | 2,460.05 |
| VR5 | Ac-C(VR)5DPG(RV)5C-NH2 | C125H235N56O26S2 | 2,970.60 | 2,970.69 |
| VRW3 | Ac-C(VR)3DPG(RV)3CW-NH2 | C92H161N38O19S2 | 2,135.53 | 2,135.60 |
Fig 1.
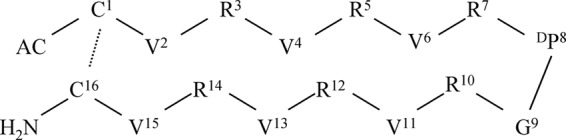
Sequence and chemical structure of the VR3 cyclic β-hairpin cationic peptide. VR3 is composed of two symmetric amphipathic R- and V-rich β-sheet antimicrobial peptides, which form two strands of a β-hairpin joined by a DP8G9 turn on one side and a disulfide bond between C1 and C16 bridging the N- and C-terminal residues on the other side. Prediction of the secondary structure preferences of the peptide sequences was performed using the PHD program (32). Dashed lines indicate disulfide bridges in VR3. Single-letter abbreviations represent the amino acid residues.
Antimicrobial assays.
The MICs of the peptides were measured using a modified version of the Clinical Laboratory and Standards Institute broth microdilution method to determine the in vitro antimicrobial activities of the peptides (46). Escherichia coli ATCC 25922, Salmonella enterica serovar Typhimurium C77-31, Bacillus subtilis CMCC 63501, and Staphylococcus epidermidis ATCC 12228 were obtained from the School of Veterinary Medicine, Northeast Agricultural University (Harbin, China). Bacteria were grown overnight at 37°C to mid-log phase and then diluted in Mueller-Hinton broth (MHB) (Sigma) to give a final concentration ranging from 2 × 105 to 7 × 105 CFU/ml. Peptides were dissolved and diluted in 0.01% acetic acid and 0.2% bovine serum albumin (BSA) (Sigma). Bacterial aliquots of 100 μl were incubated for 18 to 24 h at 37°C with 100 μl peptide in MHB. The tests were performed in triplicate. The MICs were calculated as the lowest concentration of peptide that prevented visible turbidity.
Quantification of hemolytic activity.
The hemolytic activities of the peptides were determined using a previously described method (40). Briefly, fresh human red blood cells (hRBCs) were collected and then centrifuged at 1,000 × g for 5 min at 4°C. The erythrocytes obtained were washed three times with phosphate-buffered saline (PBS) (pH 7.2), and resuspended in PBS. A 100-μl hRBC solution was incubated with 100 μl of the respective peptide dissolved in PBS for 1 h at 37°C. Intact erythrocytes were centrifuged at 1,000 × g for 5 min at 4°C, and the supernatant was transferred to a 96-well microtiter plate. The release of hemoglobin was monitored by measuring the optical density at 570 nm (OD570). As negative and positive controls, hRBCs in PBS and 0.1% Triton X-100 were employed, respectively. Minimum hemolytic concentrations (MHCs) are defined as the peptide concentrations resulting in 10% hemolysis. The therapeutic index (TI) is the ratio of the MHC to the geometric mean of the MIC (GM). Larger values indicate greater cell specificity.
Cytotoxicity assay.
The colorimetric 3-(4,5-dimethylthiozol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye reduction assay was used to determine the cytotoxicity of each peptide on MRC-5 cells, which were provided by the College of Veterinary Medicine, Northeast Agricultural University, according to a previously described method (16). Briefly, 1.0 × 104 cells/well in Dulbecco modified Eagle medium (DMEM) supplemented with l-glutamine (Gibco) and 10% fetal calf serum (Eurobio) were placed into 96-well plates and then incubated under a fully humidified atmosphere of 95% air and 5% CO2 at 37°C overnight. The next day, the peptides were added to cell cultures at final concentrations of 1 to 128 μg/ml. After incubation for 24 h, cell cultures were incubated with MTT (50 μl, 0.5 mg/ml) for 4 h at 37°C. The cell cultures were centrifuged at 1,000 × g for 5 min, and the supernatants were discarded. Subsequently, 150 μl of dimethyl sulfoxide was added to dissolve the formazan crystals formed, and the OD was measured using a microplate reader (Tecan GENios F129004; Tecan, Austria) at 492 nm.
Tryptophan fluorescence and quenching.
Small unilamellar vesicles (SUVs) were prepared for tryptophan fluorescence experiments as described previously (20). SUVs, including egg yolk l-α-phosphatidylcholine (PC), egg yolk l-α-phosphatidyl-dl-glycerol (PG), egg yolk l-α-phosphatidylethanolamine (PE), and cholesterol, were obtained from Sigma-Aldrich Corporation (St. Louis, MO). Following chloroform evaporation, the PE-PG (7:3, wt/wt) or PC-cholesterol (10:1, wt/wt) lipids were resuspended in 10 mM Tris-HCl buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 0.1 mM EDTA) by vortexing. The lipid dispersions were sonicated in ice water for 20 min using an ultrasonic cleaner until the solutions clarified. Tryptophan fluorescence spectra were measured using an F-4500 fluorescence spectrophotometer (Hitachi, Japan). The procedure was performed for each peptide in 10 mM Tris-HCl buffer (pH 7.4) with 500 μM PE-PG or PC-cholesterol lipids. The peptide/lipid molar ratio was 1:50, and the peptide-liposome mixture was allowed to interact for 2 min at 25°C. An excitation wavelength of 280 nm was used, and emission was scanned at wavelengths ranging from 300 to 400 nm. Spectra were baseline corrected by subtracting blank spectra of the corresponding solutions without the peptide. Quenching of fluorescence was accomplished using acrylamide (Sigma). To reduce the absorbance of acrylamide, Trp was excited at 295 nm instead of 280 nm (47). The final concentration of acrylamide was 0.4 M as determined by titrating the 4 M stock solution with liposomes at a lipid/peptide molar ratio of 50:1. The effects of the quenching reagent on peptide fluorescence intensities were assessed by the quenching constant (KSV), which was estimated using the Stern-Volmer equation, F0/F = 1 + KSV(Q), where F0 and F are the fluorescence values of the peptide in the absence or the presence of acrylamide, respectively, KSV represents the Stern-Volmer quenching constant, and Q represents the concentration of acrylamide.
Outer membrane permeabilization assay.
Outer membrane permeabilization activity was determined using the fluorescent dye N-phenyl-1-napthylamine (NPN) as described by Lee et al. (19). E. coli UB1005 cells were obtained from the State Key Laboratory of Microbial Technology, Shandong University. Briefly, E. coli cells were suspended in 5 mM sodium HEPES buffer (pH 7.4) containing 5 mM glucose. NPN was added to 2 ml of cells in a quartz cuvette to give a final concentration of 10 μM, and the background fluorescence was recorded (excitation wavelength = 350 nm, emission wavelength = 420 nm). Changes in fluorescence were recorded with an F-4500 fluorescence spectrophotometer (Hitachi, Japan). Peptide aliquots were added to the cuvette, and fluorescence was recorded as a function of time until no further increase in fluorescence was observed. As the outer membrane permeability increased due to the addition of peptide, NPN incorporated into the membrane resulted in an increase in fluorescence. Values were converted to percent NPN uptake using the equation % NPN uptake = (Fobs − F0)/(F100 − F0) × 100, where Fobs is the observed fluorescence at a given peptide concentration, F0 is the initial fluorescence of NPN with E. coli cells in the absence of peptide, and F100 is the fluorescence of NPN with E. coli cells upon addition of 10 μg/ml polymyxin B (Sigma). Polymyxin B is used as a positive control because of its strong outer membrane-permeabilizing properties.
Membrane depolarization.
The membrane depolarization activity of the peptides was determined with intact E. coli UB1005 cells and the membrane potential-sensitive fluorescent dye diSC3-5 (Sigma), according to the method described by Friedrich et al. (10). Briefly, E. coli were grown at 37°C with agitation to the mid-log phase and harvested by centrifugation. Cells were washed twice with washing buffer (5 mM HEPES, 20 mM glucose, pH 7.4) and resuspended to an OD600 of 0.05 in the same buffer. The cell suspension was incubated with 0.4 μM diSC3-5 until maximal dye uptake was reached. KCl was added to equilibrate the cytoplasm to a final concentration of 0.1 M and incubated at room temperature for 10 min. Two milliliters of the cell suspension was placed in a 1-cm cuvette, and the peptides were added. Changes in fluorescence were recorded using an F-4500 fluorescence spectrophotometer (Hitachi, Japan) at an excitation wavelength of 622 nm and an emission wavelength of 670 nm.
FACScan analysis.
The membrane integrities of the peptides were determined using a previously described method (28). In brief, E. coli was grown to log phase and harvested. The peptides were added and incubated for 30 min at 28°C with constant shaking at 140 rpm. The cells were harvested by centrifugation, washed three times, and incubated with propidium iodide (PI) (10-μg/ml final concentration, Sigma) at 4°C for 30 min, followed by removal of the unbound dye through washing with an excess of PBS. Flow cytometry was performed using a FACScan instrument (Becton Dickinson, San Jose, CA).
CD analysis.
To investigate the secondary structures of the peptides in different environments, circular dichroism (CD) spectra were measured under inert conditions with 10 mM sodium phosphate buffer (pH 7.4) (mimicking an aqueous environment), 50% trifluoroethanol (TFE) (mimicking the hydrophobic environment of the microbial membrane) (Sigma), and 30 mM SDS micelles (giving an environment comparable to a negatively charged prokaryotic membrane) (Sigma). The circular dichroism spectra of peptides were measured at 25°C with a J-820 spectropolarimeter (Jasco, Tokyo, Japan) equipped with a rectangular quartz cell with a path length of 0.1 cm. Spectra were recorded at a scanning speed of 10 nm/min over a wavelength range of 190 to 250 nm. An average of five to eight scans were collected for each peptide. The final concentration of peptides was 60 μM.
In vivo assay.
Male KM mice (4 weeks of age, 20.2 ± 1.5 g) were purchased from the Animal Center, Harbin Medical University (Harbin, China) and acclimatized for 1 week. Subsequently, the mice were infected with S. Typhimurium C77-31. Exponential-phase bacteria were resuspended in sterile PBS to achieve a final concentration of ∼1 × 108 CFU/ml. Animals were infected by intraperitoneal (i.p.) injection with 0.2 ml of the bacterial suspensions. Ten mice per group received a 0.2-ml i.p. injection of vehicle (PBS) or peptides approximately 60 min after the bacterial challenge. Peptides without bacteria were injected alone into each mouse as a control. The mice were monitored for 7 days. Two milliliters of sterile PBS was injected intraperitoneally into each mouse at 24 h, and peritoneal fluids (approximately 2 ml) were serially diluted. The colony counts of viable bacteria were determined by plating the fluids on Mueller-Hinton agar plates.
Statistical analysis.
Data were analyzed by analysis of variance (ANOVA) using SPSS 16.0 software. Quantitative data are presented as the mean ± standard deviation of the mean. Significant differences were identified at a P value of less than 0.05.
RESULTS
Antimicrobial activity.
The MICs of synthetic peptides against Gram-negative and Gram-positive bacteria are presented in Table 2. The potency of antimicrobial activity was ranked according to the geometric means of the MICs as follows: VR3 > VR2 > VR4 > VR5 > VR1. The MICs of VR3 ranged from 4 to 8 μg/ml. VR4 showed antimicrobial activity against Gram-positive bacteria, with MICs ranging from 8 to 16 μg/ml, but showed no antibacterial activity against Gram-negative bacteria at the tested concentrations. VR2 displayed moderate antibacterial activity, with MICs ranging from 16 to 128 μg/ml. The strand lengths of the peptides were quadratically correlated with antibacterial activity (Fig. 2). The 16-residue-long VR3 had the strongest antimicrobial activity, which was similar to those of VRW3 and the well-known peptide melittin.
Table 2.
MICs, minimum hemolytic concentrations, and therapeutic indices of the peptides against Gram-negative and Gram-positive bacterial strains
| Peptide | MICa (μg/ml) against: |
GMb (μg/ml) | MHCc (μg/ml) | TId | |||
|---|---|---|---|---|---|---|---|
| Gram-negative strains |
Gram-positive strains |
||||||
| E. coli | S. Typhimurium | B. subtilis | S. epidermidis | ||||
| VR1 | >256 | >256 | >256 | >256 | 512.0 | >128 | 0.5 |
| VR2 | 32 | 32 | 16 | 128 | 38.1 | >128 | 6.7 |
| VR3 | 8 | 8 | 4 | 4 | 5.7 | >128 | 44.9 |
| VR4 | >256 | >256 | 8 | 16 | 76.1 | 65.1 | 0.9 |
| VR5 | >256 | >256 | 64 | >256 | 304.4 | 27.4 | 0.1 |
| VRW3 | 8 | 16 | 2 | 4 | 5.7 | >128 | 44.9 |
| Melittin | 8 | 16 | 1 | 4 | 4.8 | 1 | 0.2 |
MICs were determined as the lowest concentration of peptide that prevented visible turbidity.
The geometric mean of the peptide MICs (GM) against all four bacterial strains was calculated. When no detectable antimicrobial activity was observed at 256 μg/ml, a value of 512 μg/ml was used to calculate the therapeutic index.
The MHC is the minimum hemolytic concentration that caused 10% hemolysis of human red blood cells (hRBCs). When no detectable hemolytic activity was observed at 128 μg/ml, a value of 256 μg/ml was used to calculate the therapeutic index.
The therapeutic index (TI) is the ratio of the MHC to the geometric mean of the MIC (GM). Larger values indicate greater cell selectivity.
Fig 2.

Correlation between antimicrobial activity and the chain lengths of Ac-C(VR)nDPG (RV)nC-NH2 peptides. The data can be fit to a power law as log GM = a(log n)2 − b(log n) + c (n = 1, 2, 3, 4, or 5), with a, b, and c equal to 11.97, 8.90, and 2.79 (R2 = 0.82), respectively.
Hemolytic activity.
The ability of these peptides to induce hemolysis of human erythrocytes was examined as a measure of their toxicity to mammalian cells (Table 2). The VR1, VR2, and VR3 peptides had no hemolytic activity at concentrations of as high as 128 μg/ml. The longer peptides VR4 and VR5 had 10% hemolytic activity at 65.1 μg/ml and 27.4 μg/ml, respectively. The MHCs of VR3 and its analog VRW3 were greater than 128 μg/ml, which is approximately 45 times greater than the MICs.
The therapeutic index is defined as the ratio of the MHC to the geometric mean of the MIC (GM) (Table 2) and was used to evaluate cell selectivity. VR3 had the highest TI at 44.9, whereas VR4 and VR5 had lower TIs of 0.9 and 0.1, respectively. VR2 had a slightly higher TI than VR4 and VR5.
Cytotoxicity.
The five peptides were tested for cytotoxic activity on MRC-5 cells, and the results are depicted in Fig. 3. The cytotoxic activities of the peptides were determined by the colorimetric MTT viability assay. Dose-response studies revealed that longer peptides (VR4 and VR5) displayed significantly higher cytotoxic activities on MRC-5 cells than the shorter peptides (VR1, VR2, and VR3). At the highest concentration of 128 μg/ml, the cell survival rates of VR1, VR2, and VR3 were 100%, 87%, and 82%, respectively, while VR4 and VR5 had greater cytotoxic activity, with cell viability being 36% and 15%, respectively.
Fig 3.

Cytotoxicities of the peptides. MRC-5 cells were used to evaluate the toxicities of the peptides to mammalian cells.
Binding of peptides to model membranes.
The fluorescence emission spectra for the tryptophan-containing peptides are shown in Table 3. VRW3 was used to evaluate the VR3 peptide because of their similar biological activity. VRW3 had maximal fluorescence emission at approximately 350 nm. The blue shifts observed with PE-PG phospholipid vesicles were larger than those observed with PC-cholesterol SUVs, which was consistent with their antimicrobial and hemolytic activities. In particular, a very small blue shift was observed in PC-cholesterol phospholipid vesicles in the presence of the VRW3 peptide, which was consistent with its low hemolytic activity. As a reference, melittin induced larger blue shifts in both PE-PG and PC-cholesterol vesicles than VRW3, which was consistent with the stronger biological activity of melittin.
Table 3.
Fluorescence spectroscopy parameters measured for the peptides in the presence and absence of PE-PG and PC-cholesterol vesicles
| Peptide | Fluorescence emission maximum (nm)a |
KSVb (M−1) |
||||
|---|---|---|---|---|---|---|
| Buffer | PE-PG | PC-cholesterol | Buffer | PE-PG | PC-cholesterol | |
| VRW3 | 351 | 334 (17) | 347 (4) | 6.12 | 0.51 | 2.88 |
| Melittin | 351 | 333(18) | 335(16) | 11.8 | 0.9 | 1.7 |
Values in parentheses are the blue shift of the emission maximum compared to Tris buffer.
The Stern-Vollmer constant KSV was calculated by the Stern-Vollmer equation, F0/F = 1 + KSV(Q), where Q is the concentration of the quencher (acrylamide). Concentrations of the quencher were increased from 0.01 to 0.40 M. A smaller KSV value reflects a more protected Trp residue.
Quenching of tryptophan fluorescence with acrylamide.
The relative extent to which the tryptophan residues of the peptides were buried into the phospholipid layer was examined by tryptophan fluorescence quenching (Table 3). The KSV value of VRW3 in PE-PG vesicles was less than that in PC-cholesterol, suggesting that the peptides were more protected in bacterium-mimicking cell membranes. This was in agreement with the observation that the antimicrobial activities of the peptides were stronger than their hemolytic activities.
Permeabilization of OMs.
Outer membranes (OMs) play an important role as a drug barrier in Gram-negative bacteria (14). NPN has been used to examine the permeabilization of the outer membranes of Gram-negative bacteria (34). The addition of VR3 to E. coli suspensions in the presence of NPN caused a rapid increase in fluorescence (Fig. 4). One hundred percent NPN uptake was produced at 2 μg/ml for VR3 and 0.5 μg/ml for melittin. The concentration of VR3 causing 100% NPN uptake was 4 times less than its MIC.
Fig 4.

Uptake of NPN by E. coli in the presence of different concentrations of VR3 and melittin. Outer membrane permeabilization activity was determined using the fluorescent dye (NPN) assay. NPN uptake was monitored at an excitation wavelength of 350 nm and an emission wavelength of 420 nm. As outer membrane permeability was increased by peptide addition, increased NPN incorporation into the membrane results in an increase in fluorescence. ▲, VR3; ■, melittin.
Membrane depolarization.
To assess bacterial membrane depolarization, the membrane potential-sensitive dye diSC3-5 was used. The chemical potential of K+ inside and outside the cells was balanced by adding 0.1 M KCl to the buffer. Depolarization by different concentrations of VR3 was monitored over a period of 1,200 s (Fig. 5). VR3 depolarized the bacterial cytoplasmic membrane in a dose- and time-dependent manner. Melittin caused depolarization of the cytoplasmic membrane faster than VR3, but VR3 and melittin displayed similar peak fluorescence values at 8 μg/ml after 1,200 s.
Fig 5.

Measurement of E. coli UB1005 cytoplasmic membrane depolarization by VR3 using the membrane potential-sensitive dye diSC3-5. Dye release was monitored at an excitation wavelength of 622 nm and an emission wavelength of 670 nm. A, VR3, 16 μg/ml; B, VR3, 8 μg/ml; C, melittin, 8 μg/ml; D, VR3, 4 μg/ml.
FACScan analysis.
Propidium iodide staining of nucleic acids in cells is indicative of compromised cell membrane permeability and cell death (5). Therefore, flow cytometric analysis was used to determine membrane integrity (Fig. 6). The analysis demonstrated that the control (no peptide) resulted in only 4.6% PI-positive cells. The percentage of PI-positive cells with membranes damaged by VR3 was 27.4% (MIC), 67.6% (2× MIC), and 78.0% (5× MIC). Melittin treatment resulted in positive nucleic acid staining of 31.8% (MIC), 48.3% (2× MIC), and 56.8% (5× MIC). Both peptides damaged the cell membrane in a dose-dependent manner, but VR3 caused a higher accumulation of PI than melittin at concentrations greater than 2× the MIC.
Fig 6.

Flow cytometric analysis. Exponential-phase E. coli cells were treated with VR3, and cellular fluorescence was analyzed by flow cytometry. The increments of the log fluorescence signal represent PI uptake resulting from peptide treatment. (A) No peptide, negative control; (B) VR3 (1× MIC, 8 μg/ml); (C) VR3 (2× MIC, 16 μg/ml); (D) VR3 (5× MIC, 40 μg/ml); (E) melittin (1× MIC,8 μg/ml); (F) melittin (2× MIC, 8 μg/ml); (G) melittin (5× MIC, 40 μg/ml).
CD analysis.
Circular dichroism (CD) spectroscopy was performed for all five peptides in sodium phosphate buffer, 50% TFE, and 30 mM SDS (Fig. 7). The VR1 spectra demonstrated significantly more β-turns in 30 mM SDS micelles, with minima between 200 and 205 nm and maxima between 225 and 230 nm (11). In TFE, VR1 and VR2 exhibited a population having a β-hairpin conformation characterized by a negative ellipticity near 205 nm and a crossover at 200 nm (44). In sodium phosphate buffer, VR1, VR2, and VR3 showed a range of secondary structures, but the spectra suggested a population with β-sheet structure. The spectra of VR3 showed an increase in β-sheet content in TFE and SDS. VR4 and VR5 displayed a maximum near 200 nm and a minimum just below 220 nm, suggesting a β-sheet structure (17).
Fig 7.
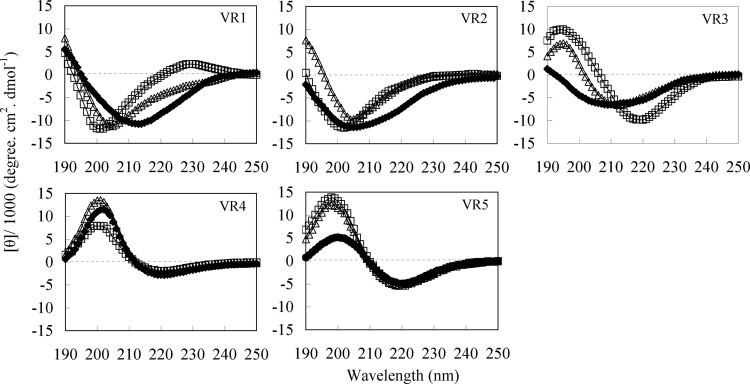
CD spectra of VR1, VR2, VR3, VR4, and VR5 in aqueous solution (♦), 50% TFE (△), and 30 mM SDS micelles (□). Data are expressed as the mean residue ellipticities.
In vivo activity.
The ability of VR3 to protect mice from a lethal challenge of bacteria from infections with S. Typhimurium C77-31 was determined (Fig. 8). By the i.p. infection model, we compared the efficacies of VR1 and VR3. At i.p. doses of 1.25 mg/kg, 2.5 mg/kg, and 5 mg/kg, the survival rates were 14%, 57%, and 71% with VR3, respectively (Fig. 8B). However, mice were not protected from bacterial challenge when VR1 was administered i.p. (Fig. 8A). As shown in Fig. 8A and B, 100% survival after 7 days was found in the groups that had been injected alone with 5 mg/kg of VR3 or VR1. Similar results were observed with exponentially growing cultures of S. Typhimurium from peritoneal fluids. VR3 decreased the bacterial load by 103-fold at 24 h postinjection in treated mice compared to control mice (P < 0.05). In contrast, VR1 was relatively ineffective at reducing the CFU of S. Typhimurium (Fig. 8C).
Fig 8.
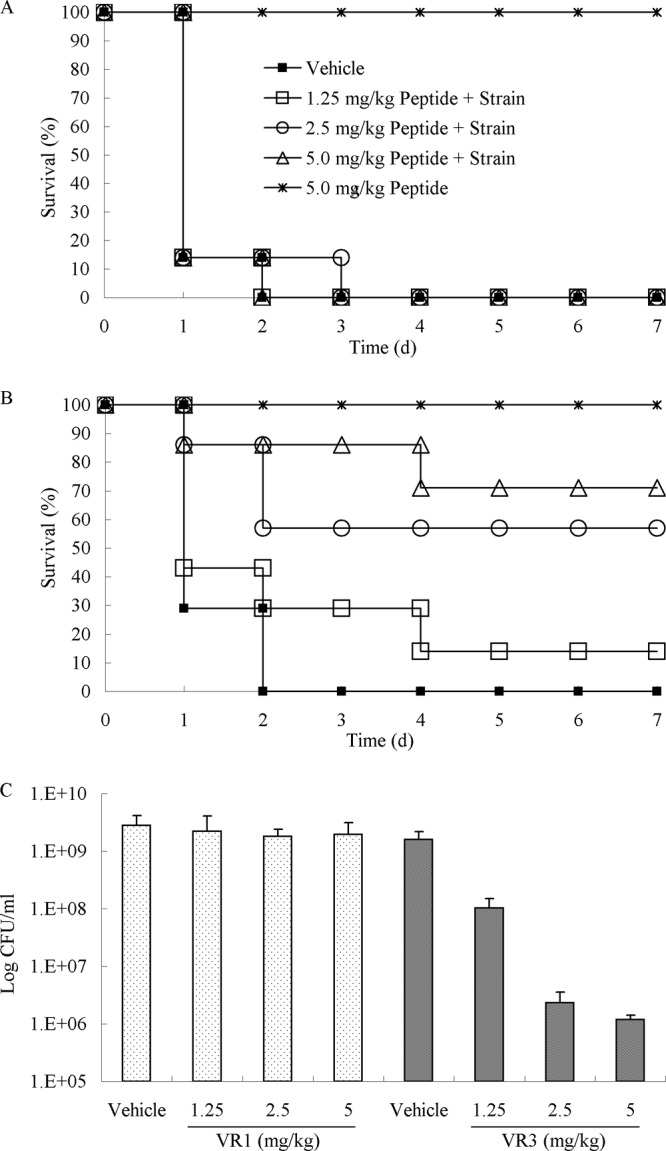
Evaluation of VR3 against S. Typhimurium in a bacterial infection model. S. Typhimurium was administered to mice by i.p. injection. (A and B) Survival analysis comparing various doses of VR1 (A) and VR3 (B) versus vehicle or peptide alone as control groups. The numbers of surviving mice were determined daily for 7 days. (C) Peritoneal bacterial counts were determined at 24 h for the VR1- and VR3-treated groups.
DISCUSSION
The relationship between the chain length and the antibacterial activities of peptides has been widely studied; however, poor agreement between these studies has been observed (7, 22, 25). Niidome et al. (25) designed a series of peptides with simple repeats, H-(LARL)3-(LRAL)n-NH2 (n = 0, 1, 2, or 3). Antibacterial activity decreased while hemolytic activity increased with increasing chain length of the peptides (25). However, another study indicated that longer-chain linear peptides had effective antibacterial activity and stimulated hemolytic activity as well (22). In the current study, the relationship between the lengths of five β-hairpin-like antimicrobial peptides and antibacterial activity was described by a quadratic function. We determined that the VR3 peptide was a potent killer of both Gram-negative and Gram-positive bacteria, with low cytotoxicity to mammalian cells. To investigate the interaction of peptides with membranes, VRW3 was synthesized by adding W to the C terminus of VR3. Compared with VR3, VRW3 had similar antimicrobial and hemolytic activities. We chose melittin as a reference to evaluate the cell selectivity of VR3. Melittin, derived from the venom of the European honeybee Apis mellifera, is commonly used as a cell-lysing and membrane-active peptide against both bacterial and eukaryotic cells (1, 6). Our results demonstrated that VR3 had higher cell selectivity than melittin. The reduction in both bacterial counts and mortality of infected mice suggest that VR3 effectively protects mice against microbial infection in vivo.
Stanger et al. found that two-stranded β-sheets (“β-hairpins”) became more stable with strand lengths of seven residues (38). In this study, the VR3 peptide containing 3 VR units in the strand showed the highest antibacterial activity, suggesting that optimal strand length improved the antibacterial activity of the peptides. The results from the analysis of secondary structure by CD provided more information regarding the quantitative structure-activity relationships (QSARs) of the peptides. Although VR1 and VR2 displayed greater β-hairpin content in a membrane-mimetic environment, they are also prone to fold into β-structures in aqueous environments. We used a DPro-Gly segment to achieve cyclization of the backbone because the DPro-Gly segment was a very strong inducer of β-sheet formation (3, 39). Furthermore, disulfide cyclization induced the formation of a superimposable β-turn in aqueous solution (41). The peptides form a β-hairpin conformation when the strand is not enough long; however, chain lengths longer than 12 amino acids may result in increased β-sheet content. VR3 underwent conformational transitions from an aqueous environment to a membrane-mimicking environment, which suggests that VR3 has an increase in β-sheet content upon binding to membranes.
To evaluate the antimicrobial mechanism of VR3, VRW3 was designed and synthesized to implement the tryptophan fluorescence experiment. PC-cholesterol (10:1, wt/wt) or PE-PG (7:3, wt/wt) SUVs were prepared to mimic zwitterionic or negatively charged membranes. The blue shift induced by VRW3 was greater in the negatively charged PE/PG vesicles than in the zwitterionic PC-cholesterol vesicles. This result suggests that the Trp of VRW3 penetrates bacterium-mimicking membranes more deeply than eukaryote-mimicking membranes. Peptide-membrane interactions are usually the result of electrostatic interactions, which may be essential for peptides to interact tightly with the membrane interface. Subsequently, peptides integrate into cell membranes, resulting in depolarization and microbial death (36). The peptides with a net positive charge of 7, such as VRW3 and its analog VR3, have electrostatic interactions between the positively charged residues and the negatively charged membrane surfaces (33). Our results suggest that VRW3 preferentially bound to model membranes containing a negatively charged head group rather than those containing a zwitterionic head group.
Furthermore, the tryptophan quenching assay performed with the neutral fluorescence quencher acrylamide was used to examine the relative extent of peptide penetration into model membranes. The Trp residue in VRW3 penetrated more efficiently into negatively charged PE-PG vesicles than into zwitterionic vesicles. This observation suggests that the selective antibacterial activity of VR3 and VRW3 correlated with the targeted cell membrane components.
To further investigate the interaction of peptides with membranes, membrane permeability was assayed to detect the target site of the VR3 peptide. VR3 permeabilized the outer membrane at a low concentration. This process involves the displacement of divalent cations that stabilize adjacent lipopolysaccharide (LPS) molecules (8, 29). Antimicrobial peptides with amphipathic β-hairpin-like structures have binding affinities to LPS and lipid A (LA) in the low micromolar range (9). This suggests that increased outer membrane permeability is due to the high binding affinity of the β-hairpin-like peptide with LPS in E. coli. VR3 induced a modest membrane depolarization in a dose- and time-dependent manner, indicating that the antibacterial target of VR3 was at the cytoplasmic membrane. To assess whether VR3 damaged the bacterial cell membrane, we determined PI staining of nucleic acids as an indicator of cell death. The results indicate that VR3 caused a higher accumulation of PI than melittin, suggesting that VR3 killed bacterial cells by disrupting their membranes. Initially, the peptides aggregate on the surface of the membrane. When a threshold is reached, they insert into the membrane bilayer to form pores or informal aqueous channels that allow the passage of ions and possibly larger molecules.
In summary, the 16-residue-long peptide VR3 had optimal cell selectivity and effectively protected mice against microbial infection. The relationship between antimicrobial activity and the chain lengths of the peptides was described by a quadratic function. The CD spectra demonstrated that the secondary structure of the peptides was a β-hairpin and/or β-sheet in aqueous and membrane-mimicking environments. The peptide killed bacteria by a membrane-active mechanism, which was evaluated by employing synthetic lipid vesicles and whole bacteria. These novel antimicrobial peptides can be designed by linking two-stranded antiparallel β-sheets with a short loop segment (DPG) and a disulfide bridge. The VR3 peptide could be developed as a promising antibiotic candidate.
ACKNOWLEDGMENTS
This work was supported by the National Basic Research Program (2012CB124703), the National Natural Science Foundation of China (31072046), and the China Agriculture Research System (CARS-36).
We thank Qi Qingsheng (Shandong University) for providing strains. We thank Ping Wei and Xin Yin (Northeast Agricultural University) for technical assistance.
Footnotes
Published ahead of print 5 March 2012
REFERENCES
- 1. Blondelle SE, Houghten RA. 1991. Hemolytic and antimicrobial activities of the twenty-four individual omission analogues of melittin. Biochemistry 30:4671–4678 [DOI] [PubMed] [Google Scholar]
- 2. Brogden KA. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Mol. 3:238–250 [DOI] [PubMed] [Google Scholar]
- 3. Chakraborty TK, Rao KS, Kiran MU, Jagadeesh B. 2008. Nucleation of the beta-hairpin structure in a linear hybrid peptide containing alpha-, beta- and gamma-amino acids. Tetrahedron Lett. 49:2228–2231 [Google Scholar]
- 4. Chen Y, et al. 2005. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 280:12316–12329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davey HM, Kell DB, Weichart DH, Kaprelyants AS. 2004. Estimation of microbial viability using flow cytometry. Curr. Protoc. Cytom. 11:11.3. [DOI] [PubMed] [Google Scholar]
- 6. Dempsey CE. 1990. The actions of melittin on membranes. Biochim. Biophys. Acta 1031:143–161 [DOI] [PubMed] [Google Scholar]
- 7. Deslouches B, et al. 2005. De novo generation of cationic antimicrobial peptides: influence of length and tryptophan substitution on antimicrobial activity. Antimicrob. Agents Chemother. 49:316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Falla TJ, Karunaratne DN, Hancock REW. 1996. Mode of action of the antimicrobial peptide indolicidin. J. Biol. Chem. 271:19298–19303 [DOI] [PubMed] [Google Scholar]
- 9. Frecer V, Ho B, Ding JL. 2004. De novo design of potent antimicrobial peptides. Antimicrob. Agents Chemother. 48:3349–3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Friedrich CL, Rozek A, Patrzykat A, Hancock REW. 2001. Structure and mechanism of action of an indolicidin peptide derivative with improved activity against gram-positive bacteria. J. Biol. Chem. 276:24015–24022 [DOI] [PubMed] [Google Scholar]
- 11. Gottler LM, de la Salud Bea R, Shelburne CE, Ramamoorthy A, Marsh ENG. 2008. Using fluorous amino acids to probe the effects of changing hydrophobicity on the physical and biological properties of the β-hairpin antimicrobial peptide protegrin-1. Biochemistry 47:9243–9250 [DOI] [PubMed] [Google Scholar]
- 12. Hancock REW. 1999. Host defence (cationic) peptides: what is their future clinical potential? Drugs 57:469–473 [DOI] [PubMed] [Google Scholar]
- 13. Haque TS, Little JC, Gellman SH. 1994. ‘Mirror-image’ reverse turns promote β-hairpin formation. J. Am. Chem. Soc. 116:4105–4106 [Google Scholar]
- 14. Helander IM, Nurmiaho-Lassila E-L, Ahvenainen R, Rhoades J, Roller S. 2001. Chitosan disrupts the barrier properties of the outer membrane of gram-negative bacteria, Int. J. Food Microbiol. 71:235–244 [DOI] [PubMed] [Google Scholar]
- 15. Jiang Z, Vasil AI, Gera L, Vasil ML, Hodges RS. 2011. Rational design of α-helical antimicrobial peptides to target gram-negative pathogens, Acinetobacter baumannii and Pseudomonas aeruginosa: utilization of charge, ‘specificity determinants,’ total hydrophobicity, hydrophobe type and location as design parameters to improve the therapeutic ratio. Chem. Biol. Drug Des. 77:225–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jin X, et al. 2010. Apoptosis-inducing activity of the antimicrobial peptide cecropin of Musca domestica in human hepatocellular carcinoma cell line BEL-7402 and the possible mechanism. Acta Biochim. Biophys. Sin. 42:259–265 [DOI] [PubMed] [Google Scholar]
- 17. Jin Y, et al. 2005. Antimicrobial activities and structures of two linear cationic peptide families with various amphipathic β-sheet and α-helical potentials. Antimicrob. Agents Chemother. 49:4957–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Juffer AH, Shepherd CM, Vogel HJ. 2001. Protein-membrane electrostatic interactions: application of the Lekner summation technique. J. Chem. Phys. 114:1892–1905 [Google Scholar]
- 19. Lee DL, et al. 2004. Effects of single d-amino acid substitutions on disruption of β-sheet structure and hydrophobicity in cyclic 14-residue antimicrobial peptide analogs related to gramicidin S. J. Pept. Res. 63:69–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee KH, et al. 2006. Interactions between the plasma membrane and the antimicrobial peptide HP (2-20) and its analogues derived from Helicobacter pylori. Biochem. J. 394:105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu LP, Deber CM. 1998. Uncoupling hydrophobicity and helicity in transmembrane segments. J. Biol. Chem. 273:23645–23648 [DOI] [PubMed] [Google Scholar]
- 22. Liu Z, et al. 2007. Length effects in antimicrobial peptides of the (RW)n series. Antimicrob. Agents Chemother. 51:597–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma Q-Q, et al. 2011. Cell selectivity and interaction with model membranes of Val/Arg-rich peptides. J. Pept. Sci. 17:520–526 [DOI] [PubMed] [Google Scholar]
- 24. Mani R, Cady SD, Tang M, AJ-Waring Lehrer RI, Hong M. 2006. Membrane-dependent oligomeric structure and pore formation of a β-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. U. S. A. 103:16242–16247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Niidome T, Matsuyama N, Kunihara M, Hatakeyama T, Aoyagi H. 2005. Effect of chain length of cationic model peptides on antibacterial activity. Bull. Chem. Soc. Jpn. 78:473–476 [Google Scholar]
- 26. Papanastasiou EA, et al. 2009. Role of acetylation and charge in antimicrobial peptides based on human β-defensin-3. APMIS 117:492–499 [DOI] [PubMed] [Google Scholar]
- 27. Park CB, Kim HS, Kim SC. 1998. Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 244:253–257 [DOI] [PubMed] [Google Scholar]
- 28. Park Y, Kim HJ, Hahm K-S. 2004. Antibacterial synergism of novel antibiotic peptides with chloramphenicol. Biochem. Biophys. Res. Commun. 321:109–115 [DOI] [PubMed] [Google Scholar]
- 29. Piers KL, Brown MH, Hancock REW. 1994. Improvement of outer membrane-permeabilizing and lipopolysaccharide-binding activities of an antimicrobial cationic peptide by C-terminal modification. Antimicrob. Agents Chemother. 38:2311–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ragothama SR, Awasthi SK, Balaram P. 1998. β-Hairpin nucleation by Pro-Gly β-turns. Comparison of d-Pro-Gly and l-Pro-Gly sequences in an apolar octapeptide. J. Chem. Soc. Perk. 2:137–143 [Google Scholar]
- 31. Rodríguez A, Villegas E, Satake H, Possani LD, Corzo G. 2011. Amino acid substitutions in an alpha-helical antimicrobial arachnid peptide affect its chemical properties and biological activity towards pathogenic bacteria but improves its therapeutic index. Amino Acids 40:61–68 [DOI] [PubMed] [Google Scholar]
- 32. Rost B, Sander C. 1994. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins 19:55–72 [DOI] [PubMed] [Google Scholar]
- 33. Schmidt NW, et al. 2011. Criterion for amino acid composition of defensins and antimicrobial peptides based on geometry of membrane destabilization. J. Am. Chem. Soc. 133:6720–6727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sedgwick EG, Bragg PD. 1987. Distinct phases of the fluorescence response of the lipophilic probe N-phenyl-1-naphthylamine in intact cells and membrane vesicles of Escherichia coli. Biochim. Biophys. Acta 894:499–506 [DOI] [PubMed] [Google Scholar]
- 35. Shenkarev ZO, et al. 2011. Molecular mechanism of action of β-hairpin antimicrobial peptide arenicin: oligomeric structure in DPC micelles and pore formation in planar lipid bilayers. Biochemistry 50:6255–6265 [DOI] [PubMed] [Google Scholar]
- 36. Shepherd CM, Schaus KA, Vogel HJ, Juffer AH. 2001. Molecular dynamics study of peptide-bilayer adsorption. Biophys. J. 80:579–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith CK, Withka JM, Regan L. 1994. A thermodynamic scale for the beta-sheet forming tendencies of the amino acids. Biochemistry 33:5510–5517 [DOI] [PubMed] [Google Scholar]
- 38. Stanger HE, et al. 2001. Length-dependent stability and strand length limits in antiparallel β-sheet secondary structure. Proc. Natl. Acad. Sci. U. S. A. 98:12015–12020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stanger HE, Gellman SH. 1998. Rules for antiparallel β-sheet design: d-Pro-Gly is superior to l-Asn-Gly for β-hairpin nucleation. J. Am. Chem. Soc. 120:4236–4237 [Google Scholar]
- 40. Stark M, Liu L, Deber CM. 2002. Cationic hydrophobic peptides with antimicrobial activity. Antimicrob. Agents Chemother. 46:3585–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Syud FA, Espinosa JF, Gellman SH. 1999. NMR-based quantification of β-sheet populations in aqueous solution through use of reference peptides for the folded and unfolded states. J. Am. Chem. Soc. 121:11577–11578 [Google Scholar]
- 42. Tang M, Waring AJ, Hong M. 2009. Effects of arginine density on the membrane-bound structure of a cationic antimicrobial peptide from solid-state NMR. Biochim. Biophys. Acta 1788:514–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tang M, Hong M. 2009. Structure and mechanism of beta-hairpin antimicrobial peptides in lipid bilayers from solid-state NMR spectroscopy. Mol. Biosyst. 5:317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Varkey J, Nagaraj R. 2005. Antibacterial activity of human neutrophil defensin HNP-1 analogs without cysteines. Antimicrob. Agents Chemother. 49:4561–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vogel HJ, et al. 2002. Towards a structure-function analysis of bovine lactoferricin and related tryptophan-and arginine-containing peptides. Biochem. Cell Biol. 80:49–63 [DOI] [PubMed] [Google Scholar]
- 46. Yang L, Harroun TA, Weiss TM, Ding L, Huang HW. 2001. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 81:1475–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhu WL, et al. 2006. Design and mechanism of action of a novel bacteria-selective antimicrobial peptide from the cell-penetrating peptide Pep-1. Biochem. Bioph. Res. Co. 349:769–774 [DOI] [PubMed] [Google Scholar]