

Abstract
In this review, based on my Garrod Lecture to the British Society for Antimicrobial Chemotherapy, I have given a brief outline of my career over the past 40 years, starting with research in the 1970s into the properties and functions of penicillin-binding proteins (PBPs), leading to the identification of the high molecular mass PBPs as the physiological targets of penicillin, and subsequent studies showing the emergence of low-affinity PBPs in penicillin-resistant clinical isolates by inter-species recombination and the generation of mosaic PBP genes. The studies of clinical isolates of gonococci, meningococci and pneumococci with PBP-mediated resistance to penicillin led to new interests in molecular epidemiology and the population and evolutionary biology of bacterial pathogens. The development (with colleagues) of multilocus sequence typing provided a method for the unambiguous characterization of bacterial strains that has proved to be very widely used, but the recent remarkable (and ongoing) developments in DNA sequencing technologies have provided the prospect of being able routinely to use whole genome sequences to characterize pathogen isolates. These developments will soon have major implications for diagnostic microbiology, outbreak investigations and our ability to follow the spread of strains of community-acquired and nosocomial pathogens at local, national and international levels. However, there are major barriers to be overcome, particularly with respect to how the avalanche of genome sequence data will be stored so that its transformative potential for molecular epidemiology and international public health are fully realized.
Keywords: penicillin resistance, mosaic genes, multilocus sequence typing, whole genome sequencing, sexual networks
Introduction
The theme of the meeting at which I presented my Garrod Lecture was the celebration of the first 40 years of the BSAC and in the spirit of the meeting, self-indulgently, I looked back at my own career, which also started about 40 years ago. In this article I will pass quickly by my early work and focus on a more recent interest, molecular epidemiology, an area that is undergoing a renaissance due to the emergence of new technology and new possibilities.
Penicillin-binding proteins
My PhD at University College London, with Robin Rowbury, was on the genetics of DNA synthesis and cell division in Salmonella typhimurium and, at the completion of my studies, I chose to move in 1973 to Princeton University to work with Arthur B. Pardee. Art gave me some papers to read from Jack Strominger at Harvard University, whose group had been studying proteins in the bacterial cell envelope that bound penicillin. Their studies had clearly shown that both Escherichia coli and Bacillus subtilis contained several proteins that covalently bound penicillin, which were believed (correctly) to be the penicillin-susceptible enzymes that catalysed the final steps in peptidoglycan biosynthesis.1 However, the method they used to detect these proteins was laborious and they had been struggling, without success, to develop a more convenient way of studying them. Since low concentrations of many penicillins and cephalosporins were well known to inhibit cell division in E. coli, leading to the formation of long filamentous cells, I decided to work on these proteins, with the original aim of identifying a protein whose inhibition by penicillin led to the block in cell division. I therefore set about developing a convenient method that could be used to study the individual ‘penicillin-binding proteins’ (PBPs) and to identify what would have been the first example of a cell division protein. At that time, Uli Laemmli was at Princeton and the SDS–polyacrylamide slab gel electrophoresis system that he had just developed was already being used in Pardee's laboratory. I therefore decided to develop a method that could detect the multiple PBPs of E. coli on SDS–polyacrylamide slab gels, using radioactive benzylpenicillin and autoradiography. Fairly rapidly I managed to detect seven PBPs with this approach and used a combination of biochemistry and genetics (Figure 1) to dissect the role of each of the PBPs in the elongation, shape determination and division of E. coli, and thereby identified the higher molecular mass PBPs as the primary ‘killing targets’ of this important group of antibiotics.2,3 The new ‘PBP assay’ became widely used to study the affinities of new β-lactam antibiotics for the individual killing targets and also allowed studies of the role of PBP alterations in resistance to β-lactam antibiotics.
Figure 1.
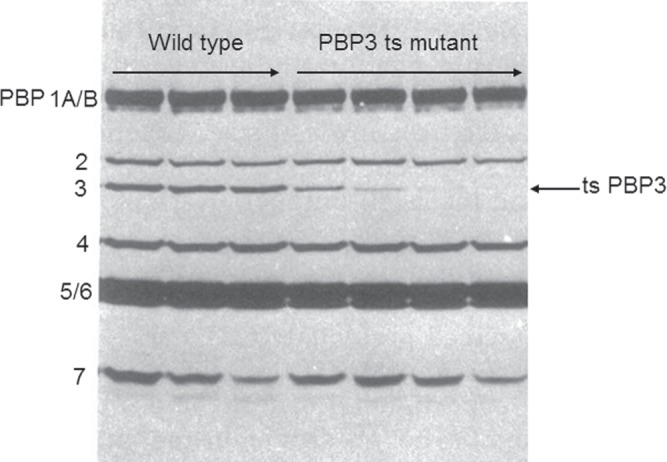
Identifying the function of an E. coli PBP using temperature-sensitive mutants. Most penicillins and cephalosporins inhibit E. coli cell division at low concentrations and one of the PBPs should be essential for this process. Mutants that could not divide at 42°C were isolated and screened for the production of a thermolabile PBP, by pre-incubating cell envelopes for increasing times at 42°C and examining their subsequent ability to bind radioactive penicillin when returned to 30°C. Whereas pre-incubation at 42°C does not reduce penicillin binding in the wild type (left three lanes), in this cell division mutant pre-incubation at 42°C rapidly reduces the ability of PBP 3 to bind penicillin at 30°C (right four lanes). This PBP was therefore assigned an essential role in peptidoglycan synthesis at cell division. Reproduced from reference 36 with kind permission from the American Society for Microbiology.
PBP-mediated resistance to β-lactam antibiotics
Our laboratory studies with E. coli showed that reductions in the affinities of PBPs for penicillins and cephalosporins clearly could lead to increased resistance to these antibiotics,4,5 but the real utility of the PBP assay was demonstrated by studies, published in 1980, that showed reductions in the affinity for penicillin of high molecular mass PBPs in penicillin-resistant isolates of Streptococcus pneumoniae6,7 and Neisseria gonorrhoeae (Figure 2)8 and, remarkably, the presence of a novel low-affinity PBP in methicillin-resistant isolates of Staphylococcus aureus.9
Figure 2.

Alterations of PBP 1 and PBP 2 in a non-β-lactamase-producing penicillin-resistant clinical isolate of N. gonorrhoeae. In a penicillin-susceptible strain (S) radioactive penicillin binds to all three gonococcal PBPs at concentrations ≥50 ng/mL (A), whereas in the resistant clinical isolate no binding to PBP 1 or PBP 2 is observed even at 1 μg/mL (B, C). Reproduced from reference 8 with kind permission from the American Society for Microbiology.
In those days most academic microbiologists worked on E. coli, and there were few links with clinical microbiologists. Consequently (and still with some lingering regrets), I started to work on pathogens too late to have been involved in these important discoveries of PBP changes in clinical isolates of major pathogens. However, by the late 1980s my laboratory started to work on pathogens and, having missed out on the discovery of PBP changes in clinical isolates, we looked at the molecular basis of PBP-mediated resistance to β-lactams in gonococci,10 meningococci11 and pneumococci,12 by cloning the PBP genes and comparing the nucleotide sequences of the genes in penicillin-susceptible and penicillin-resistant clinical isolates. The results were very surprising as, in each of these species, the sequences of the PBP genes of susceptible isolates were virtually identical, whereas those from the resistant isolates were very different in sequence, particularly in the region encoding the protein domain that interacted with penicillin. The large numbers of nucleotide differences clearly ruled out the expected process of point mutation and led to the concept of hybrid or mosaic PBP genes and the idea that increased resistance had arisen by the replacement, by homologous recombination, of parts of the genes of susceptible isolates with the corresponding parts from the PBP genes of very closely related species.10
This work led to an interest in the extent and consequences of homologous recombination in bacterial species, since the prevailing view at the time, derived from the analysis of pathogen populations by Bob Selander's group using multilocus enzyme electrophoresis (MLEE; see below), was that bacterial populations were highly clonal and that recombination was rare in nature. With the late John Maynard Smith, we re-evaluated the evidence for this view, and showed that recombination was in fact quite common in some species, and that pathogen populations ranged from clonal to nearly non-clonal,13 a view strikingly confirmed by recent studies on the genome sequences from multiple isolates of individual S. aureus14 and S. pneumoniae strains.15
Multilocus sequence typing
The studies of clinical isolates of several pathogens with reduced susceptibility or resistance to β-lactam antibiotics owing to PBP changes, and particularly the emergence of multiply antibiotic-resistant strains of S. pneumoniae, resulted in my laboratory entering the field of molecular typing and molecular epidemiology. At this time (the early 1990s) many methods for strain characterization were available and some of these worked well for outbreak investigation and are still used, notably PFGE, but the only method that was suitable for studying the global or long-term spread of strains was MLEE. This method identifies variants of the gene products of 10–20 housekeeping genes (genes encoding basic metabolic functions) using electrophoresis of cell extracts on starch gels, followed by detection using specific enzyme stains. In populations of most bacterial species there are a number of variants of each enzyme that differ in charge, reflecting slight differences in the amino acid sequences of the proteins, and thus of their corresponding gene sequences, and these variants are assigned as different alleles. Isolates with the same alleles at each housekeeping locus are clearly very closely related and are assigned to the same clone (strain).
Although MLEE is based on sound population genetic principles, and its use by Bob Selander's laboratory in the 1980s laid the foundations of the population biology of many bacterial pathogens,16 it had major deficiencies, notably the great difficulty in comparing results from one laboratory with those from another. An obvious step was to convert the method into a DNA sequence-based procedure so that the different DNA sequences at each housekeeping gene (locus) in a pathogen were assigned directly as different alleles, rather than assigning alleles indirectly using differences in the electrophoretic mobilities of their gene products on starch gels. This simple modification has huge advantages as sequence data are unambiguous and easily compared between laboratories, and the alleles at each locus, and the allelic profiles and isolate information for each pathogen, could be stored in online databases that can be interrogated via the internet. It also required the use of fewer loci than MLEE as sequencing identifies more alleles per locus, and provides a simple nomenclature as each different allelic profile can be assigned as a different sequence type (ST), which provides a convenient strain descriptor.
The desirability of this approach had been discussed for several years by Martin Maiden and myself, and undoubtedly by others, but the prospect of asking microbiologists to sequence several genes for each clinical isolate they wished to type, at a time when sequencing was a laborious manual procedure, was unrealistic. Furthermore, the housekeeping genes would have to be cloned as, in the mid-1990s, very few housekeeping gene sequences were available and they could not simply be extracted from the genome sequences of each major bacterial pathogen as these were not then available. However, by 1998, automated DNA sequencers were starting to appear in UK laboratories and the sequence-based modification of MLEE that we had discussed for several years became feasible. The resulting procedure, multilocus sequence typing (MLST), was introduced in 1998 and has proved to be a highly successful approach to molecular typing.17 The first paper involved a collaboration between the laboratories of Martin Maiden, Ian Feavers, Mark Achtman and myself, who had each just obtained ABI377 slab gel DNA sequencers, and typed 107 meningococci that Mark Achtman and Dominique Caugant had previously characterized using MLEE. We soon speeded up and the second MLST scheme (for S. pneumoniae), also published in 1998,18 involved Mark Enright, single-handedly, characterizing almost three times as many isolates as in the first paper. MLST schemes for most of the major bacterial pathogens were soon developed, with seven housekeeping loci becoming established as the norm.
MLST was designed for global or long-term epidemiology rather than outbreaks, where PFGE usually works well, and even today there is, I believe, no other simple procedure that provides the ability to unambiguously assign strains so that, for example, changes in the population of a pathogen can be followed for decades after the implementation of a new vaccine, such as the MenC19 and pneumococcal conjugate vaccines.20
MLST can distinguish extremely large numbers of strains within a pathogen species as long as there is sufficient sequence variation within their housekeeping genes to have many different alleles (sequences) at each locus. Most of the bacterial pathogens that are of major public health concern have sufficient sequence diversity to be able to distinguish billions of different genotypes (allelic profiles) using MLST, but there are some important exceptions, notably Mycobacterium tuberculosis, where there is too little sequence diversity, and a few cases where there is almost no genetic diversity as disease is caused by a distinctive clone or lineage of a species, such as typhoid fever or anthrax.21
One of the important features of MLST is the unambiguous and ‘digital’ nature of the typing data—assigning strains as a string of seven integers (the allele numbers assigned to each locus)—so that the typing data be stored in a publicly accessible online database, along with basic epidemiological data, and can be queried to allow the assignment of newly typed isolates as known or novel STs. In this way, isolates can readily be identified as, for example, one of the major clones causing meningococcal meningitis, or one of the internationally disseminated multidrug-resistant pneumococcal clones. The original MLST web site was set up in Oxford soon after MLST was developed22 and, following my move from Oxford to Imperial College, there were two websites (http://www.mlst.net and http://pubmlst.org), hosting databases for different pathogen species, and there are now additional sites for other species, for example, at University College Cork (http://mlst.ucc.ie) and the Pasteur Institute (http://www.pasteur.fr/mlst).
These websites have tools that allow the databases to be queried and the relationships between new isolates and those in the database to be visualized. The relatedness among isolates characterized by MLST and other molecular typing methods has normally been visualized by clustering approaches using, in the case of MLST, the differences in the allelic profiles of isolates, so that those most similar to a query isolate can be identified on a tree. As databases grow, the ability to visualize the relatedness of thousands of isolates using trees (particularly within the confines of a web page) becomes problematic. However, the relationships between distantly related isolates are almost always of no interest to public health and it is the isolates that are most closely related to a query isolate that are relevant.
A new way to look at the relatedness of all isolates in a large bacterial population was developed by Ed Feil when he was a postdoctoral fellow with me in Oxford. His ‘based upon related sequence types’ (BURST) algorithm allowed a database of any size to be divided into non-overlapping groups of STs that show some defined level of similarity in their allelic profiles.23 Using the default parameters, all STs in a group share alleles at six out of seven loci with at least one other ST in the group. This approach was incorporated as eBURST into our MLST websites (also available at http://eburst.mlst.net/) and identifies groups of STs (usually referred to as clonal complexes) that are similar and may be descended from a recent common ancestor. eBURST also provides a hypothesis about the evolutionary relationships among isolates within an eBURST group, by predicting its founding ST and the patterns of descent of the other STs in the group from this founder, and also the ability to display all isolates in a database as a ‘population snapshot’ (Figure 3). The current version of the MLST software at www.mlst.net (developed by David Aanensen) incorporates ‘comparative eBURST’, and uses a geographic interface, allowing users to analyse isolates from their own country but, using different colours in the eBURST diagram, to compare these with those from any other country, or to compare isolates returned from two different database searches (Figure 4).
Figure 3.

The use of an eBURST population snapshot to display the clonal complexes, and those isolates not closely related to others (singletons), within the current Haemophilus influenzae MLST database. The arrows identify the major serotype b ST6 clonal complex, a minor serotype b clonal complex and clonal complexes of serotype e and f strains (see reference 23 for details).
Figure 4.

Geographic eBURST available within the current version of the Imperial College MLST web site. Isolates of S. aureus from Spain were compared with those from Portugal. The population snapshot shows strains in the MLST database from Spain as black circles, those from Portugal as green circles, and those found in both countries as magenta. Some of the STs that correspond to major MRSA clones are indicated, all of which are found in both Spain and Portugal.
Although the eBURST approach has been very successful, it has its limitations, particularly for bacterial species in which evolutionary change in the housekeeping genes predominantly occurs by recombination (rather than point mutation), as the frequent replacement of chromosomal segments with the corresponding segments from other strains of the species can occasionally lead to STs that have very similar allelic profiles due to recombination, rather than recent shared ancestry. This is particularly seen with Enterococcus faecium and Burkholderia pseudomallei, which have very high rates of recombination; in each case, a large proportion of the STs are joined inappropriately into one large eBURST group.24 As databases get very large this problem is being observed, to a minor extent, in species with lower (S. pneumoniae; ∼7000 STs) or low rates of recombination (S. aureus; ∼1900 STs), where, in a couple of cases, the addition of a new ST has linked eBURST groups that previously had been distinct, although the reason in the latter case is less clear than in the previous examples.
Assigning species using MLST data
Besides its use in clinical microbiology and public health, MLST has been widely used for addressing questions in population and evolutionary biology and has provided one of the first and most general methods for estimating the relative contributions of homologous recombination and point mutation during the recent diversification of strains of different bacterial species.25 MLST data have also been used to assign species among populations of very similar bacteria using the concatenated sequences of the seven loci to construct trees, rather than the allelic profiles.26 A good example is provided by an analysis of the clustering patterns of over 400 viridans-group streptococci and the relationships between the observed multilocus sequence clusters and the named species.27 This approach (called multilocus sequence analysis; MLSA) has considerable advantages over classical methods of defining species as it allows the assignment of isolates to known species (and the recognition of possible new species) via the internet.27
From genes to genomes
MLST is still an important tool for molecular epidemiology (and is likely to remain so for the rest of the decade), and the MLST databases for individual pathogen species provide an invaluable resource. However, the development of new DNA sequencing methods that greatly decrease the cost and increase the speed of obtaining the complete genome sequences of bacterial isolates is certain to make an increasingly major impact on clinical microbiology and public health, as well as microbial population and evolutionary biology. Fortunately, the MLST ST can be obtained easily from the genome sequence, ensuring backwards compatibility such that the large MLST databases will retain their relevance.
The major advances in DNA sequencing technology are providing high-throughput bacterial genome sequencing, where projects that involve sequencing the genomes of many thousands of isolates of a pathogen species are now entirely feasible (and under way). With the sequencing platform that currently is most commonly used for high-throughput bacterial genome sequencing, the Illumina HiSeq, the length of individual sequence reads is fairly short (currently up to ∼100 nucleotides), but vast numbers (billions) of reads are generated—so much so that, by multiplexing, 96 isolates can be sequenced in each lane of this sequencer.
These sequence reads are mapped to a reference genome, which identifies the nucleotide positions that differ among a set of isolates (single-nucleotide polymorphisms; SNPs), and these differences within the core genome are used to construct a phylogenetic tree to show the likely evolutionary relationships among the isolates. Individual isolates of bacterial pathogens vary in the non-core genes that they possess and the set of these accessory genes in individual isolates cannot be obtained by mapping the sequence reads to the reference genome, since the latter will not possess many of the accessory genes found in other isolates of the species. These accessory genes are of crucial interest to clinical microbiologists as they include known or putative virulence genes and many antibiotic resistance genes. Thus, the sequence reads obtained for each isolate are used to assemble as much of the genome as possible and these assemblies can be used to identify the accessory genes present in each isolate. The net result is the ability to understand the evolutionary history and spread of, for example, a major multiply antibiotic-resistant strain in unprecedented detail.15
The speed with which genome sequences can be obtained will inevitably increase and, with much longer read lengths, the need to map short sequence reads to a reference genome will be superseded by direct assembly of each genome. As I write this paper, Oxford Nanopore announced the capabilities of their new technology, which directly reads the sequence of nucleotides as a DNA strand passes through a nanopore, with each pore providing sequence reads of 100 kb in a few seconds, although currently with an unacceptably high 4% error rate!
The initial impact of new genome sequencing technology on public health was seen in 2011 during the large outbreak of haemolytic uraemic syndrome in Germany, caused unexpectedly by an E. coli O104 H4 strain, and provided the first example of the appearance and analysis of the genome sequence data while a severe bacterial disease outbreak was still under way.28 In this case it is arguable whether the genome sequences of isolates from the outbreak contributed much as we have so many other tools to characterize novel pathogenic E. coli strains, but the near real-time appearance of genome data would have been invaluable for an outbreak caused by a poorly understood pathogen.
Genomes and the spread of nosocomial pathogens in healthcare settings
Several types of study that previously have been impossible to carry out become possible with the advent of high-throughput whole-genome sequencing. One of these is tracking the spread of methicillin-resistant S. aureus (MRSA) strains (or strains of other nosocomial pathogens) within and between healthcare facilities. This has not been possible as a single MRSA strain typically causes most of the infections acquired within an individual hospital (EMRSA-15 or EMRSA-16 in the UK, for example) and, using standard typing methods, almost all MRSA will look the same, precluding any studies of its spread around the hospital. High-throughput genome sequencing makes such studies possible as the genome sequencing of many isolates of single strains (clones) of pathogens, e.g. S. aureus ST239 (the major MRSA strain in Asian hospitals and elsewhere) and S. pneumoniae ST81 (the globally distributed Spanish multiresistant clone of S. pneumoniae; Spain23F-1), has shown that the genome sequence of each isolate of these strains differs slightly (Figure 5).14,15 Although small studies using whole genome sequences to look at cross-infection in hospitals29 and analyse tuberculosis outbreaks30 have been reported, there are so far no large-scale prospective studies to examine the feasibility, costs and benefits of using whole genome sequences to understand the spread of infections within hospitals or the community.
Figure 5.

Intercontinental spread and hospital transmission of MRSA ST239 isolates. Maximum likelihood phylogenetic tree based on differences (SNPs) in the core genomes of 63 ST239 isolates. The continent from which each isolate was obtained is indicated by the colour of the isolate name: blue, Asia; black, North America; green, South America; red, Europe; and yellow, Australasia. The Thai cluster, representing isolates from a single hospital, is displayed in greater resolution. Two of the isolates in this cluster were from Europe; however, one was recovered from a Thai patient. Even though the isolates are all the same by MLST, among the 63 isolates, variation is found at 4310 nucleotide positions and the sequence of each isolate is different. Reproduced from reference 14 with kind permission from the American Association for the Advancement of Science.
Genomes and sexual networks
Another area where molecular typing has increasingly been making an impact is with bacterial sexually transmitted infections (STIs), particularly gonorrhoea. Many infections with N. gonorrhoeae contribute little to disease transmission and gonorrhoea is largely maintained within communities by the presence of groups of sexually connected individuals who have multiple short-term (and often concurrent) sexual partnerships. The mainstay of the control of gonorrhoea has been contact tracing to identify individuals within these sexual networks, but in major cities such as London or New York, where most gonorrhoea occurs, this is virtually impossible. Several studies have used typing as an adjunct to contact tracing, but more recently it has been shown that molecular typing with a suitably discriminatory method can identify individuals within a community who are within the same sexual network without any contact-tracing data.31 This requires the typing method to be highly discriminatory and the genetic variation being indexed to change rapidly, so that individuals who are not sexually connected will always be infected with different gonococcal genotypes. The identification of a group of individuals infected with gonococci of the same genotype within a city then implies that they are likely to be in the same sexual network.
In the mid-1990s, together with Cathy Ison's laboratory, we developed opa typing32 as a highly discriminatory method for analysing sexual networks, but because it produces complex DNA fragment patterns the comparison of the genotypes of large numbers of isolates is not very straightforward. Consequently, again with Cathy Ison, we developed the DNA sequence-based method NG-MAST (N. gonorrhoeae multi-antigen sequence typing),33 which sequences internal fragments of two gonococcal cell surface protein genes that evolve rapidly (encoding porin and transferrin-binding protein B), due to selection imposed by the human immune system.
This precise method of strain characterization assigns STs based on the alleles at the two loci, and has been used to identify sexual networks in London by typing 2045 isolates recovered from individuals presenting with gonorrhoea to 13 major London STI clinics over a 6 month period. As expected, many isolates (>50%) had unique STs but 13 of the 449 STs were recovered from at least 30 individuals. Strikingly, some of these groups of individuals infected with the same ST were almost entirely composed of men who have sex with men (MSM), whereas others were almost all heterosexual, suggesting that molecular typing (coupled with patient behavioural and demographic data) identified individuals in London within distinct MSM and heterosexual sexual networks.31 A similar study in Amsterdam, but using fingerprinting of the gonococcal porin and Opa genes, has also identified distinct MSM and heterosexual networks.34
Identifying individuals who are infected by the same strain using NG-MAST (or other highly discriminatory typing methods) is not the ideal way to detect individuals in sexual networks, as it provides a static view of the networks within a community, precluding the possibility of showing strains moving from one sexual network to another. A more dynamic view of the spread of gonorrhoea in a local population might be achieved by sequencing the genomes of all gonococcal isolates, constructing a tree using their core genomes, and analysing the tree structure and the clustering patterns of the isolates. Our initial studies with the Sanger Institute have shown that individuals with gonorrhoea who were known sexual contacts do indeed have essentially identical genomes (Figure 6) and that large clusters of individuals with identical, or near identical, gonococcal genome sequences can be identified within a city during a particular time period (N. Bilek, C. J. Bishop, T. R. Connor, S. D. Bentley, B. G. Spratt et al., unpublished data). This approach, applied prospectively, would be a powerful tool for tracking the spread of strains between sexual networks and for rapidly identifying individuals associated with local outbreaks, particularly with strains resistant to third-generation cephalosporins, and implementing public health measures to limit further transmission. However, considerable theoretical work is required to explore the relationship between the structure and dynamics of the underlying sexual networks and the observed tree structure, so that information about the former can be inferred from the latter.
Figure 6.

Genome sequences of gonococci from known sexual contacts. A maximum likelihood tree is shown, based on differences in the core genome of a subset of isolates of a single strain of N. gonorrhoeae (ST12, as defined by NG-MAST) that was circulating in Sheffield in the mid-1990s. All isolates were obtained between April 1995 and December 1997 and included some that were from mutually named sexual contacts, obtained in the clinic within 1 month of each other37 (indicated as those with filled circles of the same colour). N. Bilek, C. J. Bishop, T. R. Connor, S. D. Bentley, B. G. Spratt et al., unpublished data.
Genomes and the clinical microbiology laboratory
At present, the Illumina HiSeq machines generate sequence data far too slowly for the routine needs of clinical microbiology laboratories and produce data that need to be processed by bioinformatic methods that typically would not be available in this setting. The situation will inevitably change because DNA sequencing is currently a highly competitive marketplace, with several competing technologies at various stages of development, only a small number of which are likely to survive to become the machines of choice. At present, none can approach the holy grail of generating extremely large numbers of long sequence reads within an hour, with a very low error rate and simple sample preparation. There is, however, little doubt that this capability will be achieved within the next few years and that this will have major implications for clinical microbiology laboratories.
Ideally, clinical microbiologists need a bench-top machine that has a simple sample preparation procedure, taking DNA from bacteria on a primary isolation plate (or preferably directly using a blood or CSF specimen) and generating useful outputs within a few hours, or preferably minutes. At present, such a machine is not available, but the Illumina MiSeq is likely to provide some of the requirements of clinical microbiology laboratories, and with the addition of integrated software designed specifically for clinical microbiology, could soon become a useful platform for this setting. If, as seems likely, clinical microbiology is seen as a significant market for at least some of the bench-top DNA sequencing platforms that will appear, we could fairly soon see their routine use in larger clinical laboratories, as they have the potential to provide so much information about an unknown presumptive pathogen, including its species, predicted antibiotic resistance profile, virulence gene content, serotype and so on. In addition, once hospitals maintain databases of the genome sequences of isolates of the major nosocomial pathogens that have previously been seen in their hospital, the whole genome sequence can be used to compare the new isolate with those in the database, to rapidly identify likely cross-infection events or emerging outbreaks. Penetration of this technology to cover all hospitals and nursing homes within a health region (and ultimately, nationally) would allow a much better understanding of the transmission of nosocomial pathogens within and between healthcare facilities.
Genome databases and web applications
Just as MLST data are of little use without the online databases, so it is with genome data, and effective use of high-throughput pathogen genome sequencing will require open-access international genome databases for each major pathogen species, as well as the hospital, regional or national databases for public health professionals alluded to above, with analysis tools that allow users without bioinformatic skills to obtain the answers to the kind of questions that clinical microbiologists or infection control teams need to ask. Development of these databases and analysis tools for many thousands of genome sequences (by the end of this year the Wellcome Trust Sanger Institute will have sequenced about 11 000 S. pneumoniae genomes!) is far more challenging than those required for MLST. David Aanensen in my laboratory has developed a prototype web application that is currently being used to database and analyse S. aureus genomes generated by our colleagues at the Wellcome Trust Sanger Institute. This will be used subsequently for other species, to provide genome sequence databases (holding both the variation in the core genomes, the genome assemblies and the accessory gene content of isolates) and analysis tools for individual pathogen species that conceptually are analogous to those developed for MLST.
Although much further development is needed, the application is already being used, particularly a version developed as an online tool to allow collaborators to work together on the analysis of a common set of pathogen genome sequences. A further advantage of this type of collaborator's tool is that a link to the web application, pre-loaded with the set of pathogen genomes, can be included within a publication, allowing anyone to explore the data (Figure 7). Public access to typing data, via a link in a publication to an analogous web application, again developed by David Aanensen, was used recently in a paper on the geographic distribution of the spa types of nearly 3000 S. aureus isolates from patients with invasive disease in 450 hospitals throughout Europe (Figure 8).35
Figure 7.

Collaborator's tool for analysis of sets of bacterial genome sequences. The web application has been loaded with the genome sequences, and genome assemblies, of the 63 isolates of MRSA ST239 shown in Figure 5. The three main panels are fully interactive, so that clicking on an isolate or node on the tree (right panel), which is zoomed in to show the cluster of isolates from the same Thai hospital, automatically shows the map location of the source of the isolate(s) (left panel), and the nucleotide differences in a genome browser (Web Artemis, bottom panel). Phenotypic data (for example, MRSA or MSSA status for S. aureus genomes) and the distribution of any accessory gene (for example, the Panton–Valentine leucocidin genes) can be shown by differential colouring of the terminal nodes on the tree and the flags on the map. The tabs below the map provide additional information on the isolates and further functionalities to help analyse the genome data. D. Aanensen, C. Powell, B. G. Spratt et al., unpublished data.
Figure 8.

Web application to analyse the distribution of spa types of 2890 S. aureus isolates isolated in 450 hospitals in 28 countries across Europe by the EARSS Consortium.35 An individual spa type (t067) has been selected showing its distribution mainly in Spain and Portugal. Red markers are MRSA, green are MSSA. These data, preloaded in the web application, are available at http://www.spatialepidemiology.net/srl-maps.
Final remarks
These are clearly exciting times for academic and clinical microbiology. The arrival of cheap, rapid and simple high-throughput genome sequencing of pathogen isolates will be a transformative technology for clinical microbiology and public health. It will be interesting to see how quickly this technology is taken up by these communities, who have not been notably quick to take on board new developments in molecular biology.
Funding
This work was funded by a Wellcome Trust Principal Research Fellowship (WT089472).
Transparency declarations
None to declare.
Acknowledgements
I thank all those who have worked in my laboratory since the 1970s, my collaborators, and the Wellcome Trust, who have generously funded my work for many years.
References
- 1.Blumberg PM, Strominger JL. Covalent affinity chromatography of penicillin-binding components from bacterial membranes. Methods Enzymol. 1974;34:401–5. doi: 10.1016/s0076-6879(74)34046-3. [DOI] [PubMed] [Google Scholar]
- 2.Spratt BG, Pardee AB. Penicillin-binding proteins and cell shape in E. coli. Nature. 1975;254:516–7. doi: 10.1038/254516a0. doi:10.1093/jac/41.1.49. [DOI] [PubMed] [Google Scholar]
- 3.Spratt BG. Distinct penicillin-binding proteins involved in the division, elongation, and shape of Escherichia coli K12. Proc Natl Acad Sci USA. 1975;72:2999–3003. doi: 10.1073/pnas.72.8.2999. doi:10.1093/jac/dkp350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spratt BG. Escherichia coli resistance to β-lactam antibiotics through a decrease in the affinity of a target for lethality. Nature. 1978;274:713–5. doi: 10.1038/274713a0. doi:10.1128/AEM.00561-11. [DOI] [PubMed] [Google Scholar]
- 5.Hedge PJ, Spratt BG. Resistance to β-lactam antibiotics by re-modelling the active site of an E. coli penicillin-binding protein. Nature. 1985;318:478–80. doi: 10.1038/318478a0. [DOI] [PubMed] [Google Scholar]
- 6.Hakenbeck R, Tarpay M, Tomasz A. Multiple changes of penicillin-binding proteins in penicillin-resistant clinical isolates of Streptococcus pneumoniae. Antimicrob Agents Chemother. 1980;17:364–71. doi: 10.1128/aac.17.3.364. doi:10.1371/journal.pone.0017936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zighelboim S, Tomasz A. Penicillin-binding proteins of multiply antibiotic-resistant South African strains of Streptococcus pneumoniae. Antimicrob Agents Chemother. 1980;17:434–42. doi: 10.1128/aac.17.3.434. doi:10.1093/jac/dkm361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dougherty TJ, Koller AE, Tomasz A. Penicillin-binding proteins of penicillin-susceptible and intrinsically resistant Neisseria gonorrhoeae. Antimicrob Agents Chemother. 1980;18:730–7. doi: 10.1128/aac.18.5.730. doi:10.1093/jac/dkn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown DFJ, Reynolds PE. Intrinsic resistance to β-lactam antibiotics in Staphylococcus aureus. FEBS Lett. 1980;122:275–8. doi: 10.1016/0014-5793(80)80455-8. [DOI] [PubMed] [Google Scholar]
- 10.Spratt BG. Hybrid penicillin-binding proteins in penicillin-resistant gonococci. Nature. 1988;332:173–6. doi: 10.1038/332173a0. [DOI] [PubMed] [Google Scholar]
- 11.Spratt BG, Zhang Q-Y, Hutchison A, et al. Recruitment of a penicillin-binding protein gene from Neisseria flavescens during the emergence of penicillin resistance in Neisseria meningitidis. Proc Natl Acad Sci USA. 1989;86:8988–92. doi: 10.1073/pnas.86.22.8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dowson CG, Hutchison A, Spratt BG. Extensive remodelling of the transpeptidase domain of penicillin-binding protein 2B of a penicillin-resistant South African isolate of Streptococcus pneumoniae. Mol Microbiol. 1989;3:95–102. doi: 10.1111/j.1365-2958.1989.tb00108.x. [DOI] [PubMed] [Google Scholar]
- 13.Maynard Smith J, Smith NH, O'Rourke M, et al. How clonal are bacteria? Proc Natl Acad Sci USA. 1993;90:4384–8. doi: 10.1073/pnas.90.10.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris SR, Feil EJ, Holden MT, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327:469–74. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Croucher NJ, Harris SR, Fraser C, et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;331:430–4. doi: 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selander RK, Musser J, Caugant DA. Population genetics of pathogenic bacteria. Microb Pathog. 1987;3:1–7. doi: 10.1016/0882-4010(87)90032-5. [DOI] [PubMed] [Google Scholar]
- 17.Maiden MCJ, Bygraves JA, Feil E, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci USA. 1998;95:3140–5. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Enright MC, Spratt BG. A multilocus sequence typing scheme for Streptococcus pneumoniae: identification of clones associated with serious invasive disease. Microbiology. 1998;144:3049–60. doi: 10.1099/00221287-144-11-3049. [DOI] [PubMed] [Google Scholar]
- 19.Maiden MCJ, Ibarz-Pavón AB, Unwin R, et al. Impact of meningococcal serogroup C conjugate vaccines on carriage and herd immunity. J Infect Dis. 2008;197:737–43. doi: 10.1086/527401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipsitch M, O'Neill K, Cordy D, et al. Strain characteristics of Streptococcus pneumoniae carriage and invasive disease isolates during a cluster-randomized clinical trial of the 7-valent pneumococcal conjugate vaccine. J Infect Dis. 2007;196:1221–7. doi: 10.1086/521831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Achtman M. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu Rev Microbiol. 2008;62:53–70. doi: 10.1146/annurev.micro.62.081307.162832. [DOI] [PubMed] [Google Scholar]
- 22.Chan M-S, Maiden MCJ, Spratt BG. The development of database-driven web interface software for multilocus sequence typing (MLST) of bacterial pathogens. Bioinformatics. 2001;17:1077–83. doi: 10.1093/bioinformatics/17.11.1077. [DOI] [PubMed] [Google Scholar]
- 23.Feil EJ, Li B, Aanensen DM, et al. eBURST: Inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186:1518–30. doi: 10.1128/JB.186.5.1518-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turner KME, Hanage WP, Fraser C, et al. Assessing the reliability of eBURST using simulated populations with known ancestry. BMC Microbiol. 2007;7:30. doi: 10.1186/1471-2180-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feil EJ, Maiden MCJ, Achtman M, et al. The relative contributions of recombination and mutation to the divergence of clones of Neisseria meningitidis. Mol Biol Evol. 1999;16:1496–502. doi: 10.1093/oxfordjournals.molbev.a026061. [DOI] [PubMed] [Google Scholar]
- 26.Hanage WP, Fraser C, Spratt BG. Sequences, sequence clusters and bacterial species. Phil Trans R Soc Lond B Biol Sci. 2006;361:1917–27. doi: 10.1098/rstb.2006.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bishop CJ, Aanensen DA, Jordan GE, et al. Assigning strains to bacterial species via the internet. BMC Biol. 2009;7:3. doi: 10.1186/1741-7007-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mellmann A, Harmsen D, Cummings CA, et al. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One. 2011;6:e22751. doi: 10.1371/journal.pone.0022751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis T, Loman NJ, Bingle L, et al. High-throughput whole-genome sequencing to dissect the epidemiology of Acinetobacter baumannii isolates from a hospital outbreak. J Hosp Infect. 2010;75:37–41. doi: 10.1016/j.jhin.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 30.Gardy JL, Johnston JC, Ho Sui SJ, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011;364:730–9. doi: 10.1056/NEJMoa1003176. [DOI] [PubMed] [Google Scholar]
- 31.Choudhury B, Risley CL, Ghani AC, et al. Identification of individuals with gonorrhoea within sexual networks: population-based study. Lancet. 2006;368:139–46. doi: 10.1016/S0140-6736(06)69003-X. [DOI] [PubMed] [Google Scholar]
- 32.O'Rourke M, Ison CA, Renton AM, et al. Opa-typing: a high resolution tool for studying the epidemiology of gonorrhoea. Mol Microbiol. 1995;17:865–75. doi: 10.1111/j.1365-2958.1995.mmi_17050865.x. [DOI] [PubMed] [Google Scholar]
- 33.Martin IMC, Ison CA, Aanensen DM, et al. Rapid sequence-based identification of gonococcal transmission clusters in a large metropolitan area. J Infect Dis. 2004;189:1497–505. doi: 10.1086/383047. [DOI] [PubMed] [Google Scholar]
- 34.Kolader ME, Dukers NH, Van der Bij AK, et al. Molecular epidemiology of Neisseria gonorrhoeae in Amsterdam, The Netherlands, shows distinct heterosexual and homosexual networks. J Clin Microbiol. 2006;44:2689–97. doi: 10.1128/JCM.02311-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grundmann H, Aanensen DM, van den Wijngaard CC, et al. Geographic distribution of Staphylococcus aureus causing invasive infections in Europe. PLoS Med. 2010;7:e1000215. doi: 10.1371/journal.pmed.1000215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spratt BG. Temperature-sensitive cell division mutants of Escherichia coli with thermolabile penicillin-binding proteins. J Bacteriol. 1977;131:293–305. doi: 10.1128/jb.131.1.293-305.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bilek N, Martin IM, Bell G, et al. Concordance between Neisseria gonorrhoeae genotypes recovered from known sexual contacts. J Clin Microbiol. 2007;45:3564–7. doi: 10.1128/JCM.01453-07. [DOI] [PMC free article] [PubMed] [Google Scholar]