

Abstract
Objectives
Pyrrolobenzodiazepine (PBD) dimers, tethered through inert propyldioxy or pentyldioxy linkers, possess potent bactericidal activity against a range of Gram-positive bacteria by virtue of their capacity to cross-link duplex DNA in sequence-selective fashion. Here we attempt to improve the antibacterial activity and cytotoxicity profile of PBD-containing conjugates by extension of dimer linkers and replacement of one PBD unit with phenyl-substituted or benzo-fused heterocycles that facilitate non-covalent interactions with duplex DNA.
Methods
DNase I footprinting was used to identify high-affinity DNA binding sites. A staphylococcal gene microarray was used to assess epidemic methicillin-resistant Staphylococcus aureus 16 phenotypes induced by PBD conjugates. Molecular dynamics simulations were employed to investigate the accommodation of compounds within the DNA helix.
Results
Increasing the length of the linker in PBD dimers led to a progressive reduction in antibacterial activity, but not in their cytotoxic capacity. Complex patterns of DNA binding were noted for extended PBD dimers. Modelling of DNA strand cross-linking by PBD dimers indicated distortion of the helix. A majority (26 of 43) of PBD-biaryl conjugates possessed potent antibacterial activity with little or no helical distortion and a more favourable cytotoxicity profile. Bactericidal activity of PBD-biaryl conjugates was determined by inability to excise covalently bound drug molecules from bacterial duplex DNA.
Conclusions
PBD-biaryl conjugates have a superior antibacterial profile compared with PBD dimers such as ELB-21. We have identified six PBD-biaryl conjugates as potential drug development candidates.
Keywords: pyrrolobenzodiazepine dimers, pyrrolobenzodiazepine-biaryl conjugates, DNA adduct formation, MRSA
Introduction
The large majority of antibiotics in current clinical use are derived from bactericidal or bacteriostatic molecules produced as secondary metabolites by microbes, predominantly those belonging to the moulds and actinobacteria.1 Some of the most potent and least toxic compounds from the initial wave of antibacterial drug discovery target cellular structures such as peptidoglycan, unique to bacteria, or selectively interfere with the protein synthesizing machinery of prokaryotes over host cell processes. Such screening identified molecules such as the pyrrolobenzodiazepines (PBDs) tomaymycin, anthramycin and DC-81, which exerted potent antibacterial activity against human pathogens through a capacity to bind to DNA;2–4 some of these compounds have potential as cancer chemotherapeutics, but a high degree of cytotoxicity has rendered them unattractive as antibacterial antibiotics in comparison with other classes of compound. However, the evolution of multidrug-resistant pathogens capable of a rapid and efficient horizontal transmission of genes encoding antibiotic resistance determinants has facilitated the erosion of much of the therapeutic value of front-line antibacterial chemotherapeutic agents in a relatively short time frame.5,6 Increasing multidrug resistance in Gram-negative pathogens such as Pseudomonas aeruginosa and Acinetobacter baumannii has forced the reappraisal of colistin for systemic use; this polymyxin antibiotic, discovered >50years ago and until recently considered too toxic for non-topical use, is now widely used systemically due to the limited therapeutic options available for these infections.7 We have taken our cues from this trend and are evaluating the antibacterial potential of novel chemotherapeutics of the PBD class of DNA-interactive agents.8–10
PBDs exert their biological activity through covalent binding via their N10-C11 imine/carbinolamine moieties to the C2-amino position of a guanine residue within the minor groove of DNA (Figure 1); monomers span three DNA base pairs with a preference for Pu-G-Pu (where Pu = purine and G = guanine; reactive guanine emboldened) sequences and block transcription by the inhibition of RNA polymerase.11 The potency, binding affinity and sequence specificity of PBDs can be enhanced by tethering two PBD units through an inert propyldioxy [-O-(CH2)3-O- diether] or pentyldioxy [-O-(CH2)5-O- diether] linker via their C8/C8′ positions to form dimers that are able to cross-link appropriately separated guanines on opposing DNA strands.12 Such linkers facilitate non-covalent interactions with adjacent adenine residues that determine a preference for A:T-rich sequences. For example, a [-O-(CH2)3-O- diether] linker, such as that incorporated into the structure of SJG-136 (Figure 1), engenders binding predominantly to embedded 5′-Pu-GATC-Py sequences;12,13 extending this linker region to n = 5 enables dimers such as ELB-21 (Figure 1) to span an extra base pair and cross-link sequences such as 5′-Pu-GATTC-Py and 5′-Pu-GAATC-Py.12 Both three- and five-carbon-linked dimers show strong bactericidal activity against a range of Gram-positive pathogens,8,14 but no activity against Gram-negative bacteria due to the barrier function of the outer membrane.8 Of the compounds shown in Figure 1, ELB-21 was significantly more potent than either SJG-136 or DRG-16 and was found to activate resident prophages, up-regulate genes within pathogenicity islands and invoke a RecA-LexA-mediated DNA damage response.10,14 PBD dimers are relatively toxic molecules, as evidenced by comparison of in vitro cytotoxicity against tumour and non-tumour cell lines;15,16 recent evidence has emerged that dimers form sequence-dependent intrastrand and monoalkylated adducts in addition to a variety of interstrand cross-links,14,17 and such multiple binding patterns contribute to their cytotoxicity profiles.
Figure 1.

Mechanism of PBD binding to the N2 of guanine in the DNA minor groove, and structure of the PBD dimers SJG-136, DRG-16 and ELB-21.
The G + C content of DNA from Staphylococcus spp. and Streptococcus spp. is relatively low—30%–38% and 33%–44%, respectively18—consequently, it may be possible to decrease the cytotoxicity of PBDs by restricting non-covalent interactions with A:T-rich regions. We have taken two approaches towards the design of PBD-based conjugates with the capacity to recognize and bind selectively to A:T- and G:C-containing sequences in the DNA of Gram-positive pathogens. In the first instance, we synthesized PBD dimers containing a number of joined heterocyclic building blocks between the PBD units rather than the simple methylene chains utilized in the synthesis of SJG-136, DRG-16 and ELB-21. Thus, the linked heterocyclic units extend the length of the base pairs spanned between the interstrand cross-linked guanines; the heterocyclic building blocks selected are rich in hydrogen bond donating and accepting capability, allowing the potential for the recognition of specific DNA bases spanned by the linker. Secondly, we have designed and synthesized a series of PBD-biaryl conjugates in which poly(N-methylpyrrole) units have been attached to the C8 position of PBD units by a four-carbon linker;19 poly(N-methylpyrrole) units such as 4-(1-methyl-1H-pyrrol-3-yl)benzenamine (MPB) interact in a non-covalent manner with DNA and have a strong preference for G:C tracts. Thus, PBD-MPBs and other PBD-biaryl conjugates are DNA monoalkylating agents with G:C recognition properties and exert cytotoxic effects against human tumour cell lines whilst sparing non-tumour cell lines such as the fibroblast line WI38. We have therefore evaluated the antibacterial properties of members of these DNA mono- and dialkylating libraries against Gram-positive pathogens in relation to their cytotoxicity and, where appropriate, their DNA-binding profiles.
Materials and methods
Bacteria and cell lines
Epidemic methicillin-resistant Staphylococcus aureus (EMRSA) strains EMRSA-15 and EMRSA-16 were isolated from clinical material at the Royal Free Hospital, London;20 MRSA strain BB568 was a gift from Brigitte Berger-Bächi, Institute of Medical Microbiology, University of Zürich, Switzerland. The community-associated MRSA isolate USA300 was purchased from the ATCC as BAA-1556. Vancomycin-intermediate S. aureus (‘VISA’) strain Mu50 is an MRSA clinical isolate with intermediate vancomycin resistance, and was isolated and provided by Keiichi Hiramatsu, Juntendo University, Tokyo. ATCC 29213 is an antibiotic-susceptible S. aureus reference strain. The vancomycin-resistant enterococci (VRE) Enterococcus faecalis VRE1 and Enterococcus faecium VRE10 were isolated at the Royal Free Hospital. Bacteria were grown in Mueller–Hinton (MH) broth (Oxoid) or on MH agar plates at 37°C. The MICs of antibacterial agents were determined by the CLSI broth microplate assay as previously described;9 agents were dissolved in DMSO prior to dilution in broth. At the concentrations used, the solvent had no effect on bacterial growth. Three MIC determinations per strain were performed in duplicate for each compound tested.
For cytotoxicity evaluation, the human lung fibroblast cell line WI38 and ovarian (A2780) and breast (MCF7) human cancer cell lines were employed. The cells were grown at 37°C in a 5% CO2 humidified atmosphere, either in Dulbecco's modified Eagle's medium or modified Eagle's medium (depending on the cell line) supplemented with 10% (v/v) fetal bovine serum (Biosera), 1% (w/v) l-glutamine, 1% (w/v) non-essential amino acids and 0.05% (w/v) hydrocortisone (Invitrogen). Cells were seeded into 96-well plates (total volume 100 μL) and allowed to reach a 30%–40% degree of confluence before the addition of compounds dissolved in sterilized ultrapure water up to a concentration of 100 mg/L. Serial decimal dilutions were employed and 25 μL aliquots added to cells. After 24 h of continuous drug exposure, cells were washed with PBS and placed in 200 μL of drug-free medium for 72 h after the end of drug exposure. The cytotoxicity was determined spectrophotometrically at λ = 570 nm using the MTT colorimetric assay.21 IC50 values were calculated by dose–response analysis using OriginLab 6.0® software (Silverdale Scientific Ltd, Stoke Mandeville, UK).
PBD dimers
Thirteen PBD dimers were examined in this study and structural data for these compounds are shown in Table 1. These include the C2-phenyl (A2) and C2-thiophenyl (A1) derivatives of ELB-21, and symmetric (e.g. SG-2860) and asymmetric (SG-2891) tri-, tetra-, penta- and hexapyrrole dimers containing DNA-interactive heterocyclic units separating the two PBD moieties. ELB-21 was obtained from Spirogen Ltd (London, UK) and was synthesized as described previously.22 As previous studies of the structure–activity relationships of PBD monomers have demonstrated an improvement in activity when C2/C3 endo-unsaturation is combined with an aryl (or planar) substituent at C2,23,24 we functionalized each PBD unit C-ring24 of ELB-21 to yield compounds A1 and A2. The asymmetric C8/C8′-tripyrrole-linked PBD dimer SG-2860, with the capacity to form high-affinity cross-links spanning 11 bp, was synthesized according to Tiberghien et al.25 In this molecule the PBD units are tethered through a pyrrole-imidazole-pyrrole (py-Im-py) linker. SG-2891 is a C11-bisulphite-modified, highly water soluble prodrug of SG-2860 with a decreased rate of DNA adduct formation in comparison with SG-2860 and was modified according to published procedures.26,27 In SG-2910, the DNA cross-linking span has been further extended by including a β-alanine (Ala) residue in the Im-py-Ala-Im-py linker. SG-2242 and the C11-bisulphite-modified derivative SG-2243 were designed to span 13 DNA base pairs by virtue of a py-py-alk-py-py linker (alk; alkyl chain); SG-2907 was prepared utilizing a py-Im-alk-Im-py tether. A further chain extension was introduced through a py-py-Im-alk-Im-py-py linker (SG-2904 and SG-2906). SG-2087 and SG-2088 are 1-methyl-1H-pyrrole-2,4-dicarboxylic acid bis-[(11aS)(7-methoxy-5-oxo-1,2,3,11a-tetrahydro-5H-pyrrolo[2,1-c][1,4]benzodiazepine-8-yl)-amide] and 1-methyl-1H-pyrrole-2,5-dicarboxylic acid bis-[(11aS)(7-methoxy-5-oxo-1,2,3,11a-tetrahydro-5H-pyrrolo[2,1-c][1,4]benzodiazepine-8-yl)-amide], respectively. Further details of the synthesis of these molecules can be found in patent applications WO 2000/012508, WO 2005/085260, WO 2005/085251, WO 2005/085250, WO 2005/085177, WO 2007/039752, WO 2007/039752 and WO 2009/060215.
Table 1.
PBD dimers used in this study
| PBD no. | Code | Chemical structure | Chemical formula | Molecular weight |
|---|---|---|---|---|
| 1 | ELB-21 | 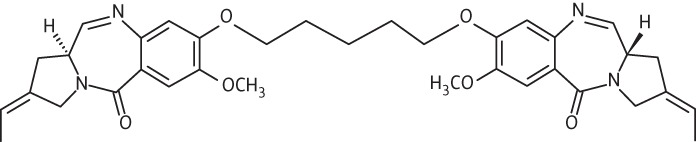 |
C35H40N4O6 | 612.71 |
| 2 | A1 | 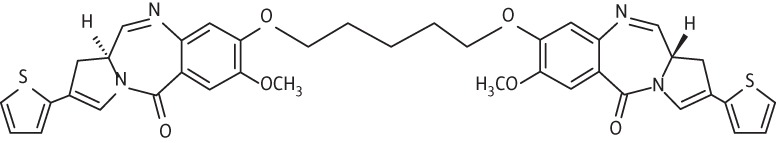 |
C39H36N4O6S2 | 720.21 |
| 3 | A2 | 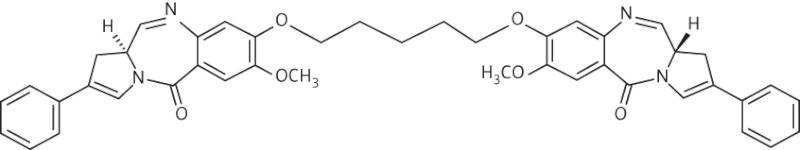 |
C43H40N4O6 | 708.29 |
| 4 | SG-2860 | 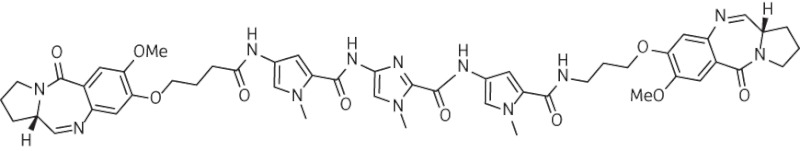 |
C50H56N12O10 | 984.07 |
| 5 | SG-2891 | 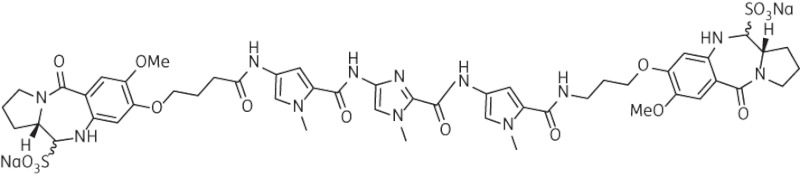 |
C50H58N12O16Na2S2 | 1192 |
| 6 | SG-2910 | 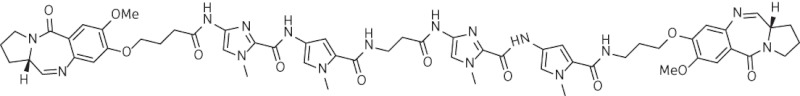 |
C58H66N16O12 | 1179 |
| 7 | SG-2242 | 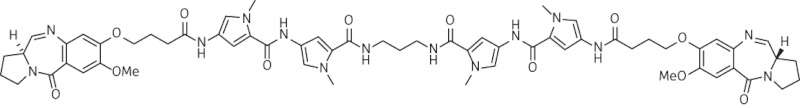 |
C61H70N14O12 | 1191.32 |
| 8 | SG-2243 | 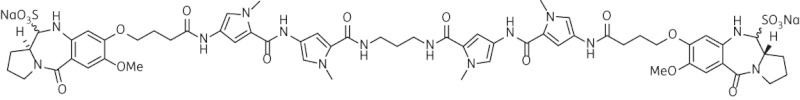 |
C61H72N14O18Na2S2 | 1400 |
| 9 | SG-2907 | 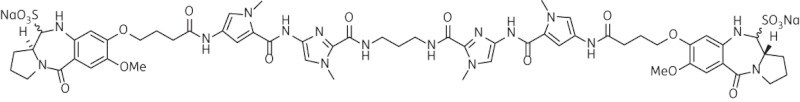 |
C59H70N16O18Na2S2 | 1402 |
| 10 | SG-2904 | 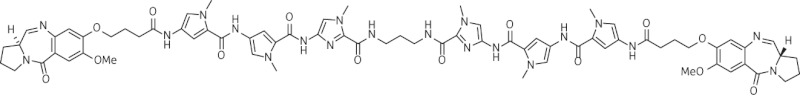 |
C71H80N20O14 | 1436 |
| 11 | SG-2906 | 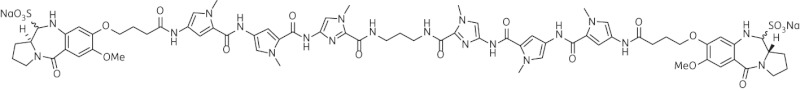 |
C71H82N20O20Na2S2 | 1644 |
| 12 | SG-2087 | 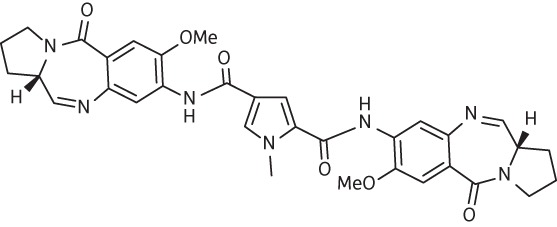 |
C33H34N7O6 | 624 |
| 13 | SG-2088 | 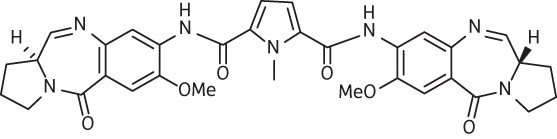 |
C33H34N7O6 | 624 |
PBD-biaryl conjugates
A library of 43 PBD-biaryl conjugates was employed; their structures and key chemical data are shown in Table S1 (available as Supplementary data at JAC Online). The conjugates incorporate members of a set of biaryl building blocks, shown in Table S2 (available as Supplementary data at JAC Online), that are based on phenyl-substituted heterocycles sufficiently long to span two DNA base pairs and facilitate a switch of binding preference from A:T-rich to G:C-rich sequences.
DNase I footprinting
DNase I footprinting with the PBD dimers was performed as previously described28 using the HexA and HexB DNA fragments. The sequences of these fragments are shown in Figure S1 (available as Supplementary data at JAC Online). These synthetic cloned fragments were designed so that between them they contain all 64 symmetrical hexanucleotide sequences. In order to facilitate examination of the binding sites that are located at either end of these sequences, each fragment was cloned in both orientations (HexAfor with HexArev and HexBfor with HexBrev) as previously described.29 These footprinting templates were considered to be especially useful for testing these symmetrical ligands, rather than natural restriction fragments that contain a random mixture of potential binding sites. Radiolabelled DNA fragments were obtained by digesting the parent plasmids with HindIII and SacI (HexA) or EcoR1 and Pst1 (HexB), and labelled at the 3′-end of the EcoR1 or HindIII sites with α-[32P]dATP using reverse transcriptase. The radiolabelled fragments of interest were separated from the remainder of the plasmid DNA on 6% (w/v) polyacrylamide gels. The DNA was eluted and dissolved in 10 mM Tris-HCl, pH 7.5, containing 0.1 mM EDTA to give ∼10 cps/μL, as determined on a hand-held Geiger counter (<10 nM). Ligand–DNA complexes were prepared by mixing 1.5 μL of DNA with 1.5 μL of ligand (diluted in 10 mM Tris-HCl, pH 7.5, containing 10 mM NaCl) to give final ligand concentrations of 3, 1, 0.3 and 0.1 μM. These mixtures were incubated overnight at 20°C before digesting with 2 μL of DNase I (∼0.01 U/mL, diluted in 20 mM NaCl/2 mM MgCl2/2 mM MnCl2). The reaction was stopped after 1 min by adding 4.5 μL of formamide containing 10 mM EDTA and bromophenol blue. Samples were then boiled for 3 min before loading onto an 8% (w/v) denaturing polyacrylamide gel (40 cm) containing 8 M urea. Gels were fixed in 10% (v/v) acetic acid, dried under vacuum and subjected to autoradiography using a phosphorimager screen (Kodak).
Microarray analysis
Total bacterial RNA was purified from each sample, labelled and hybridized to the BμG@S SAv1.1.0 microarray, as described previously.10 This array has been described elsewhere30 and contains PCR products representing all predicted open reading frames from the initial seven S. aureus genome sequencing projects. The array design is available in BμG@Sbase (accession no. A-BUGS-17; http://bugs.sgul.ac.uk/A-BUGS-17) and also ArrayExpress (accession no. A-BUGS-17). Hybridization data were analysed using an Affymetrix 428 scanner and then quantified using BlueFuse for Microarrays 3.5 software (BlueGnome). Data analysis was performed in GeneSpring GX 7.3 (Agilent) using median-normalized Cy5/Cy3 ratio intensities for three biological replicates. Only genes whose expression ratio showed a ≥2-fold difference with Benjamini and Hochberg false discovery rate ≤0.05% in the presence of the drug were regarded as being significantly different from the control. Fully annotated microarray data obtained using two PBD-biaryl conjugates have been deposited in BμG@Sbase (accession no. E-BUGS-118; http://bugs.sgul.ac.uk/E-BUGS-118) and also ArrayExpress (accession no. E-BUGS-118).
Quantitative real-time PCR (qRT-PCR)
qRT-PCR was performed as described previously.10 The data shown are the median of three biological and two technical replicates; each replicate was performed in duplicate. Gene-specific primer pairs were designed for genes of interest to yield amplicons of 100–150 bp and sequences are shown in Table S3 (available as Supplementary data at JAC Online).
Molecular modelling
Molecular modelling was employed in order to investigate the degree to which duplex B-DNA can accommodate ELB-21 along with the degree of distortion imparted to the B-DNA helical structure for a given sequence. Molecular models were constructed for the B-DNA duplexes 5′-TATAGAATCTATA-3′ and 5′-TATAGAAATCTATA-3′ (target sequence underlined) with and without ELB-21, forming an interstrand cross-link by covalent binding to the two available guanine residues on opposite strands. The AMBER31 package was used to build the initial B-DNA structures. ELB-21, having a conformational propensity to follow the curve of the minor groove, was graphically aligned between the two guanine residues using the ‘xleap’ program prior to forming a covalent bond between the C11 of ELB-21 and C2-NH2 of each guanine. Energy minimization was applied to the DNA without ligand using the ‘sander’ program. For the bound complex, initial minimization steps were performed by application of a high positional restraint to the DNA alone, allowing ELB-21 to adjust to the environment. To model the complex, the DNA restraint was gradually reduced to zero in subsequent minimization steps. After minimization, molecular dynamics simulations were performed over 2 ns with no extra restraints applied. For the purpose of illustration, the dynamics frame at 1 ns was saved and rendered in each case. For all modelling, a long-range non-bonded cut-off was applied along with use of the Generalized Born implicit solvent model and monovalent ion screening (0.2 M). Dynamics simulations were viewed using Visual Molecular Dynamics32 and final images rendered with Chimera.33 PBD-biaryl conjugates were modelled in a similar way.
Results
Antibacterial and cytotoxic activity of PBD dimers
We previously examined the antibacterial activity of three PBD dimers (SJG-136, DRG-16 and ELB-21) in which PBD units were separated by simple three- or five-carbon methylene chains.8 All displayed pronounced bactericidal activity against drug-resistant Gram-positive pathogens such as MRSA and VRE clinical isolates, with ELB-21 being the most potent of the three. As these agents are now known to bind to a range of sequences within duplex DNA in addition to those for which the molecules were designed,10,17 we have synthesized and evaluated the range of PBD dimers shown in Table 1 containing PBD units separated by various heterocyclic moieties, in an attempt to extend the length of the base pairs spanned between the inter- and intrastrand cross-linked guanines and to increase PBD dimer interactions through the choice of heterocyclic spacers with substantial hydrogen bond donating and accepting capabilities.
We confirmed that ELB-21 has potent inhibitory activity against a selection of antibiotic-resistant (EMRSA-15, EMRSA-16 and BB568) and -susceptible (ATCC 29213) S. aureus strains and, at low drug concentrations, prevented the growth of the VRE clinical isolate VRE1 (Table 2). C2-thiophenyl substitution (A1) of the PBD C-rings of ELB-21 made no difference to the MIC values obtained for the Gram-positive pathogens, but the C2-phenyl-substituted compound (A2) was less active against drug-resistant and -susceptible strains of S. aureus (Table 2). Pyrrole-linked PBD dimers in the compound 4–9 series (Table 1) were designed to cross-link sites on duplex DNA of increased length over ELB-21. These molecules have the capacity to traverse biomembranes, as evidenced by their cytotoxicity profile against WI38, MCF7 and A2780 cell lines, but displayed reduced antibacterial activity in comparison with ELB-21 (Table 2). SG-2860, designed to span an 11 bp interstrand site, possessed antibacterial activity comparable to A2; the C11-bisulphite derivative SG-2891 yielded practically identical MICs, indicating that slow formation of the active N10-C11 imine did not compromise the antibacterial action. Increasing the length of the putative DNA cross-linking site by increments from 11 bp (SG-2860) to 19 bp (SG-2906) resulted in a reduction or abrogation of the antibacterial potency. Thus, SG-2242 was markedly less active than SG-2860 and SG-2906 was inactive. Antibacterial, but not cytotoxic, activity was lost when two of the pyrroles in the SG-2242 linker were replaced with imidazole moieties to afford SG-2907. Compounds SG-2087 and SG-2088, in which two DC-81 type24 PBD units were linked by a single pyrrole moiety, displayed no antibacterial activity. With the PBD dimers examined, cytotoxic effects against cancer cell lines MCF7 and A2780 were greater than those found using the more slow-growing lung fibroblast line WI38; all compounds showed a degree of selective cytotoxicity (Table 2).
Table 2.
Antibacterial activity and cytotoxicity of PBD dimers
| Compounda | MIC (mg/L)b |
IC50 (mg/L)c |
||||||
|---|---|---|---|---|---|---|---|---|
| EMRSA-15 | EMRSA-16 | BB568 | ATCC 29213 | VRE1 | WI38 | MCF7 | A2780 | |
| ELB-21 | 0.03 | 0.03 | 0.015 | 0.015 | 0.25 | 0.08–0.16 | <0.02 | <0.02 |
| A1 | 0.03 | 0.03 | 0.03 | 0.015 | 0.125 | ND | ND | ND |
| A2 | 0.125 | 0.125 | 0.25 | 0.125 | 0.125 | ND | ND | ND |
| SG-2860 | 0.25 | 0.25 | 0.25 | 0.25 | >16 | 0.08–0.16 | <0.02 | <0.02 |
| SG-2981 | 0.25 | 0.25 | 0.125 | 0.25 | >16 | 0.08–0.16 | <0.02 | <0.02 |
| SG-2910 | 1 | 1 | 0.5 | 2 | 16 | 0.04–0.08 | 0.08–0.16 | <0.02 |
| SG-2242 | 2 | 2 | 2 | 2 | >16 | 0.08–0.16 | 0.08–0.16 | <0.02 |
| SG-2243 | 2 | 2 | 2 | 2 | 8 | 0.08–0.16 | <0.02 | <0.02 |
| SG-2907 | 16 | 16 | 8 | 8 | >16 | 0.16–0.32 | 0.16–0.32 | <0.02 |
| SG-2904 | 16 | 16 | 16 | 16 | >16 | 0.16–0.32 | 0.08–0.16 | <0.02 |
| SG-2906 | 16 | >16 | 16 | 16 | >16 | 0.16–0.32 | 0.32–0.65 | <0.02 |
| SG-2087 | >16 | >16 | >16 | >16 | >16 | >20 | 5–10 | <0.02 |
| SG-2088 | >16 | >16 | >16 | >16 | >16 | 2.5–5 | 0.65–1.25 | <0.02 |
ND, not determined.
aStructures of PBD dimers are shown in Table 1.
bMIC for S. aureus strains EMRSA-15, EMRSA-16, BB568 and ATCC 29213, and the vancomycin-resistant enterococcal isolate VRE1.
cConcentration of PBD dimer required to kill 50% (IC50) of the population of WI38, MCF7 and A2780 cells.
As ELB-21 was the most potent antibacterial PBD dimer examined in this study and we have previously determined10,14 that it invoked a vigorous RecA-LexA-mediated DNA damage response in EMRSA-16 at both sub- and suprainhibitory concentrations, we employed molecular dynamics simulations (Figure 2) to investigate the potential of the compound to produce distortions of the DNA duplex at two drug-binding sites. Helix distortion is known to engender ssDNA-RecA filaments that inactivate the SOS repressor LexA and phage repressors such as Cl, resulting in activation of the DNA damage response and derepression of resident prophages.34,35 In the course of a dynamics simulation, DNA undergoes a degree of conformational variability accompanied by periodic changes in the width of the minor groove. Alignment of ELB-21 resulted in the C-ring (Figure 2b) of the PBD unit oriented towards the 5′ end of the DNA strand to which it was bound and an S-configuration at the C11 position; a schematic of the covalent bond formed between the C11 of the PBD unit and the guanine residue is shown in Figure S2 (available as Supplementary data at JAC Online). In the short sequences simulated, interstrand covalent cross-links mediated by covalent binding of ELB-21 to opposing guanine residues restricted conformational changes and caused greater narrowing of the minor groove by means of van der Waals interactions with the sides of the groove compared with duplex DNA without the drug. With the 5′-TATAGAATCTATA-3′ target sequence, it is apparent that ELB-21 causes little disruption of the overall integrity of the B-DNA helix and of the base pairing (Figure 2a and b). Narrowing of the minor groove can also be seen where ELB-21 is bound. The model for the 5′-TATAGAAATCTATA-3′ duplex substrate is considerably different (Figure 2c and d). The extra base pair between the cross-links determines that ELB-21 is unable to span the extended space between opposing guanines in the natural B-DNA structure. In the course of the dynamics simulations, the extra strain imposed by this restriction results in distortion of the DNA, particularly in the region spanned by the ligand, with base pairs A7-T22, A8-T21 and T9-A20 becoming dislodged from their normal positions with a loss of hydrogen bond pairing. All duplex terminal residues showed transient loss of base pairing during the dynamics simulations. Thus, the simulation indicates that ELB-21 can only be accommodated at the expense of some conformational distortion of the DNA (Figure 2d).
Figure 2.

Molecular models of duplex DNA 5′-TATAGAATCTATA -3′ (a and b) and 5′-TATAGAAATCTATA-3′ (c and d) without ligand (a and c) and with ELB-21 covalently bound to the NH2 of both guanine residues (orange) and forming an interstrand cross-link (b and d). The structures are frames from dynamics simulations after 1 ns. Bases are enumerated beginning at the 5′ end of one strand and continuing from the 5′ end of the opposing strand. The schematics show a ribbon running through the DNA backbone and slab bases are shown in blue, except for guanines, in orange. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Sequence-selective binding of PBD dimers to duplex DNA
The sequence selectivity of the PBD dimers was studied by DNase I footprinting using the HexA and HexB fragments, which were designed to contain all 64 combinations of symmetrical hexanucleotides.29 The reverse fragments (HexArev and HexBrev) contain the same sequence, but in the opposite orientation. Representative footprinting gels for the PBD dimers are shown in Figure 3 and the general location of the footprints is indicated in Figure S1. It is clear that the PBD dimers affect a large number of sites on these fragments and that several of the protected regions must consist of overlapping binding sites. Due to the ability of these ligands to bind covalently to guanine with high affinity, it is not possible to define the precise binding preference of each ligand, though some general features can be determined. In general, the size of the footprints correlated with the length of the linkers separating the two PBD monomeric units, with larger footprints observed with longer linkers. Similarly, the sodium bisulphite salt form of the PBD dimers and their corresponding imine forms (e.g. SG-2860 and SG-2891, and SG-2242 and SG-2243) provided identical footprint patterns in terms of footprint lengths and location. This observation is expected, as the bisulphate forms are considered as the pro-drug of the corresponding imine form of PBD dimers and are expected to hydrolyse to generate the imine form under experimental conditions.36
Figure 3.

DNase I footprints showing the interaction of the PBD dimers with HexA and HexB fragments. The sequences of these fragments are shown in Figure S1. Concentrations of the ligand (μM) are indicated at the top of each gel lane. Tracks labelled ‘con’ show DNase I cleavage in the absence of added ligand, while ‘GA’ tracks are markers specific for purines (G + A). (a) HexAfor and HexArev. (b) HexBfor and HexBrev. The DNA fragments were labelled at their 3′-ends with 32P.
The length of the footprints observed for these compounds ranged from short regions of protection of 3–7 bases to very large footprints spanning ∼38 bp that contain at least seven potential binding sites (Figure S1). This range of sequence protection can be explained by the suggestion that the reactive imine moiety of PBDs is able to covalently attach to the guanine of every X-G-X binding site, and the presence of a large number of such potential binding sites within a long DNA fragment results in the complete inhibition of DNase I cleavage. A detailed analysis of the footprinting sites revealed protection at sequences that correspond to both inter- and intrastrand cross-links (e.g. intrastrand 5′-GTACTAG-3′, 5′-GCTATAG-3′, 5′-GATATATG-3′, 5′-GCTTAAG-3′ and 5′-GCTTATAAG-3′, and interstrand 5′-GTTTAAAC-3′, 5′-GTTAAC-3′, 5′-GTATAC-3′ and 5′-GTATATAC-3′) and monoalkylating sites (e.g. 5′-GCCAAATTTA-3′), which is in agreement with the type of adducts reported previously.17 However, due to the complex and relatively large nature of these footprints, it was not possible to assign specific protection sequences to individual molecules, though it should be noted that although they have many protected regions in common, the detailed patterns are not the same for each ligand.
Antibacterial and cytotoxic activity of PBD-biaryl conjugates
Each of the 43 members of the PBD-biaryl conjugate library was examined for its capacity to inhibit the growth of four MRSA clinical isolates (EMRSA-15, EMRSA-16, BB568 and USA300), one vancomycin-intermediate resistant MRSA (Mu50) and two VRE isolates (VRE1 and VRE10). MIC values obtained for these bacteria are shown in tabular form in Table S4 (available as Supplementary data at JAC Online). With one exception, the MICs obtained for the seven Gram-positive strains of any one compound were comparable; the antibacterial activities of the compounds are therefore categorized in grid form in Figure 4 as effective (bacteria susceptible; ≤4 mg/L), partially effective (8–16 mg/L) or ineffective (≥32 mg/L). Twenty-six compounds produced MIC values of ≤4 mg/L for all test strains. Some were highly potent, e.g. 35 (BztMC-Im-PBD; BztMC = methyl 5-aminobenzothiophene-2-carboxylate) and 36 (MPB-Im-PBD) produced MICs of 0.06 mg/L for all strains tested and sub-mg/L values were obtained with a further 12 compounds. In the large majority of cases, activities against staphylococci and streptococci were comparable. In one case (59; Py-Py-Py-Py-MPB-PBD; Py = 1-methylpyrrol-3-amine), a broad range of intermediate susceptibility values against the various strains was found. Fourteen compounds possessed no detectable antibacterial activity.
Figure 4.

Antibacterial activity (MICs) of PBD-biaryl conjugates as determined using five staphylococcal and two enterococcal clinical isolates. Compounds eliciting MIC values of ≤4 mg/L for all seven Gram-positive bacteria were considered effective. The numbering system for the components of the compound library is shown in Table S1. PBD, pyrrolo(2,1-c)(1,4)benzodiazepine; Py, 1-methylpyrrol-3-amine; PyM, (1,5-dimethylpyrrol-2-yl)methanamine; Im, 1-methylimidazol-4-amine; In, 1-methylindol-5-amine; Bzf, benzofuran-5-amine; Bzt, benzothiophen-5-amine; BztMC, methyl 5-aminobenzothiophene-2-carboxylate; MPB, 4-(1-methyl-1H-pyrrol-3-yl)benzenamine; FB, 4-(3-furyl)benzenamine; TB, 4-(3-thienyl)benzenamine; MTaB, 3-(2-methylthiazol-4-yl)benzenamine; TdB, 4-(thiadiazol-4-yl)benzenamine; MB, 4-morpholinobenzenamine; MPpB, 4-(4-methylpiperazin-1-yl)benzenamine; MMB, 4-(morpholinomethyl)benzenamine; MEB, 4-(2-morpholinoethoxy)benzenamine; MEPM, [4-(2-morpholinoethoxy)phenyl]methanamine; MPA, 6-morpholinopyridin-3-amine.
The PBD-biaryl conjugates have been developed as a series of linear molecules in which combinations of the various heterocyclic building blocks have been incorporated in order to optimize, through their unique curvature, the number of van der Waals contacts within G:C tracts of the DNA minor groove. The sequence of the units incorporated into conjugates influences potency, e.g. utilizing 4-(3-furyl)benzenamine (FB), MPB and PBD in the configuration MPB-FB-PBD produces the highly active (MIC = 0.5 mg/L) molecule 56, but the FB-MPB-PBD arrangement 48 is inactive (≥32 mg/L). Similarly, the activity of conjugates containing Im, MPB and PBD is increased >30-fold when the order of these moieties is changed from Im-MPB-PBD (43) to MPB-Im-PBD (36). This trend was also evident with the Py-containing conjugates Py-MPB-PBD (41; MIC = 1 mg/L) and MPB-Py-PBD (29; 0.125 mg/L). All conjugates containing 1-methylindol-5-amine (In) were inactive, regardless of the presence of other moieties (e.g. compounds 14, 25, 34, 44 and 55). In some instances, the introduction of an MPB unit between another heterocycle and PBD resulted in a loss of antibacterial activity: benzothiophen-5-amine (Bzt)-PBD (15) possessed MICs of 0.25–2 mg/L and Bzt-Py-PBD (27) yielded some of the lowest MICs in this study, but Bzt-MPB-PBD (45) was inactive; FB-PBD is potent (17; 0.125–1 mg/L), but, as noted above, FB-MPB-PBD is inactive. However, substitution of the non-terminal Py heterocycle in Py-Py-PBD (24) to yield Py-MPB-PBD (41) made little difference to the bioactivity of this type of conjugate. In a number of instances, activity was significantly increased by the introduction of units between the heterocycle and the PBD unit, particularly in two-component conjugates. For example, the antibacterial activities of 4-(2-morpholinoethoxy)benzenamine (MEB)-Im-PBD (39; 0.25 mg/L) and MEB-MPB-PBD (51; 2 mg/L) contrasted with the lack of activity of MEB-PBD (23).
Mode of killing of EMRSA-16 by PBD-biaryl conjugates
The effect of the highly-active PBD-biaryl conjugates 28, 29, 36 and 57 on the viability of EMRSA-16 was determined. Mid-logarithmic-phase bacteria were exposed to a range of drug concentrations (0.5× MIC, 1× MIC, 2× MIC and 4× MIC), and the cell density (OD600) and viability (cfu/mL) determined at various times during incubation with agitation (200 rpm) at 37°C. Compounds 29, 36 and 57 exhibited potent bactericidal activity at suprainhibitory concentrations; 28 elicited relatively weak killing activity (2–3 log reduction in viable count) only at the highest concentration used (Figure 5). With all four compounds, there were increases in EMRSA-16 viability over the 24 h incubation period following the reductions that occurred over the initial 8 h (Figure 5). This regrowth was not due to the emergence of resistant phenotypes, as MIC values obtained for bacteria recovered from the 24 h cultures were identical to those for the inoculum.
Figure 5.

Response of EMRSA-16 to subinhibitory and suprainhibitory concentrations of compounds 57 (a and b), 28 (c and d), 29 (e and f) and 36 (g and h). Compounds were added to mid-logarithmic-phase cultures (OD600 ∼ 0.1; 3 h), OD600 measured (a, c, e and g) and viable numbers determined (b, d, f and h): filled circles, no drug; open circles, 0.5× MIC; filled triangles, 1× MIC; open triangles, 2× MIC; and filled squares, 4× MIC. Error bars represent ±1 SD; n = 3.
ELB-21 and other PBD dimers produce similar bactericidal effects with EMRSA-16 and other S. aureus strains,8,10 and appear to act above the MIC threshold by damaging DNA to such an extent that it cannot be repaired by the bacterial RecA-LexA-mediated SOS response system;14 these compounds also induce genes associated with the staphylococcal pathogenicity island SaPI4 and prophage genes, resulting in the release of viable phage from treated cells.10,14 We therefore examined the gene transcriptional response of EMRSA-16 to the mono-adduct-forming PBD-biaryl conjugates 28 and 57 to determine if these compounds elicit similar effects. There was a significant ≥2-fold change in the expression of 174 genes following the exposure of logarithmic-phase bacteria to subinhibitory concentrations (0.5× MIC) of the potent bactericidal conjugate 57; 120 genes were up-regulated and 102 of these were genes associated with resident S. aureus prophages φSa2 and φSa3.37 Lists of up- and down-regulated genes are shown in Tables S5 and S6 (both available as Supplementary data at JAC Online), and the complete data set can be found in ArrayExpress (accession no. E-BUGS-118). Five genes involved in DNA damage repair, including uvrA (encoding excinuclease ABC subunit A), uvrB (excinuclease ABC subunit B), gyrA (DNA gyrase subunit A) and rir2 (ribonucleoside-diphosphate reductase β chain), were up-regulated, as were three genes encoding cell wall turnover proteins. Fifty-four genes showed reduced expression following exposure to 57; a high proportion was involved in membrane transport and bioenergetics, capsular polysaccharide synthesis, protein synthesis and protein modification. Fewer genes were modulated by subinhibitory concentrations of the less overtly bactericidal compound 28, with a total of 91 genes up-regulated ≥2-fold; there was no significant inhibition of gene expression. All genes up-regulated by 28 were also up-regulated by compound 57, but in general to a lesser degree. Changes in the expression of nine genes in logarithmic-phase cultures of EMRSA-16 following exposure to conjugates 28 and 57 were validated using qRT-PCR; genes with varying degrees of modulation of expression were selected (Table 3). Pearson correlation coefficient analysis demonstrated a significant correlation (P ≤ 0.0001, two-tailed) and an R2 value of 0.9537, equivalent to a 95.37% correlation between the two methods of assessment of gene expression.
Table 3.
Comparison of microarray and qRT-PCR data following exposure of EMRSA-16 to PBD-biaryl conjugates 28 and 57 at concentrations of 0.5× MIC
| Gene | Compound | Microarray | qRT-PCRa |
|---|---|---|---|
| grlA | 57 | 1.03 | 0.75 |
| 28 | 1.14 | 1.37 | |
| grlB | 57 | 0.92 | 1.57 |
| 28 | 1.05 | 1.54 | |
| gyrA | 57 | 3.44* | 4.20** |
| 28 | 1.10 | 1.11 | |
| gyrB | 57 | 3.63 | 7.79** |
| 28 | 1.03 | 1.49 | |
| samB | 57 | 17.25* | 128.41** |
| 28 | 5.34* | 28.28** | |
| uvrA | 57 | 2.95* | 4.97** |
| 28 | 2.17* | 2.91** | |
| uvrB | 57 | 3.55* | 7.83** |
| 28 | 2.38* | 3.95** | |
| cap5L | 57 | 0.28* | 0.11** |
| 28 | 0.60 | 0.80 | |
| mnhD | 57 | 0.35* | 0.23** |
| 28 | 0.91 | 0.75 |
amRNA expression levels were normalized to 16S rRNA.
*P < 0.05.
**P < 0.001.
Models of energy-minimized interactions between consensus nucleotide sequences10 and both the most potent bactericidal agent MPB-Im-PBD (36) and the markedly less active Py-Py-PBD (24) indicated that these molecules cause less distortion of the DNA helix compared with members of the PBD dimer series; modelling suggested that the more potent compound 36 may produce little or no distortion of the helix (Figure 6). Calculation of the free energy of binding showed that 36 is favoured over 24 (a difference of 15 kcal/mol).
Figure 6.
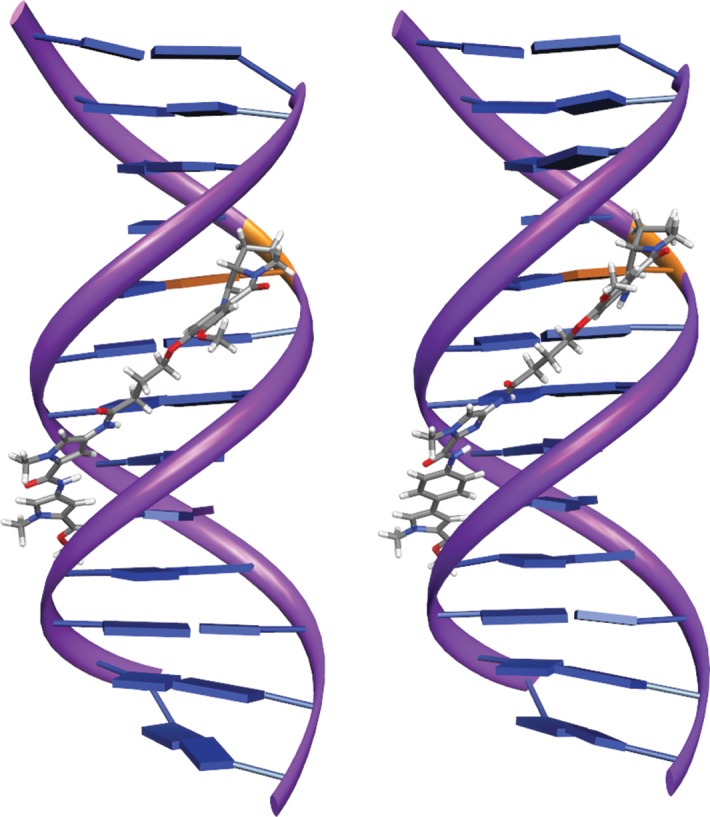
In silico energy-minimized structures of the PBD binding target sequence TATAGAATCTATA and compound 24 Py-Py-PBD (left) and compound 36 MPB-Im-PBD (right). The covalent binding site is shown in orange. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Discussion
The capacity of PBD dimers in which the PBD monomers are separated by short C8/C8′ diether linkages containing odd numbers of methylenes (n = 3 or 5; Figure 1) to form interstrand and intrastrand cross-linked adducts and monoalkylated adducts with duplex DNA by virtue of covalent aminal linkages with guanine C2-NH2 functionalities has been thoroughly characterized.12,17,22 The type and distribution of these adducts is dependent on the length of the linker and the positioning of two reactive guanine bases on the same or opposite strands, and their separation by intervening base pairs.38 The available evidence strongly suggests that both the cytotoxic39,40 and antibacterial activities10,14 of such PBD dimers are dependent on these covalent interactions with DNA. Indeed, interstrand cross-links induced by SJG-136 have been demonstrated in peripheral blood lymphocytes during clinical trials of this agent.41 ELB-21 elicits potent antibacterial activity that is restricted to Gram-positive species, many of current medical importance. We identified and validated a large number of interstrand and intrastrand ELB-21 binding sequences within the genome of S. aureus (∼40 000) that probably account for the MICs of 0.015–0.03 mg/L for the staphylococcal strains shown in Table 2. This compound and other structurally related agents (Figure 1) display significant cytotoxicity against human cell lines,15,16 although this has not precluded the promotion of SJG-136 to human Phase II evaluation in ovarian cancer and leukaemia with an excellent safety profile.42 We sought to restrict the cytotoxicity of antibacterial PBD dimers through targeting of extended DNA binding sites, which appear less frequently within the staphylococcal and human genomes, by increasing the length of the linker region; we also examined the antibacterial activity of ELB-21 analogues through modification of the PBD units.
Increasing the molecular distance between the two PBD units in the PBD dimer series shown in Table 1 led to a progressive reduction in antibacterial activity against the five Gram-positive strains examined, even though, generally, the compounds retained their cytotoxic capacity (Table 2). Dimers in the SG-2860–2906 numerical series were significantly less potent against all strains tested compared with ELB-21 and showed strong if complex patterns of binding to duplex DNA, as evidenced by DNase I footprinting (Figures 3 and S1), indicating that the relatively weak antibacterial activity was most likely due to a lack of capacity to traverse the cell wall and/or cytoplasmic membrane of the staphylococcal and enterococcal targets. Two compounds, SG-2087 and SG-2088, possessed no discernible antibacterial activity and produced no significant DNA footprints at the highest drug concentrations used, indicating that the presence of PBD units within PBD dimers is not a prerequisite for antibacterial activity through DNA cross-linking. Thus, ELB-21 remains the most potent PBD dimer with respect to activity against Gram-positive pathogens and no improvement in its potency could be elicited by functionalizing the C-ring of the PBD units in the manner described earlier (Table 2). The in vitro cytotoxicity and bioactivity of SJG-136 and ELB-21 are similar. SJG-136 is progressing through the clinic for cancer indications and has been found to be relatively well tolerated in human subjects42 in comparison with other cytotoxic drugs; ELB-21 may have the potential, therefore, for clinical use as a ‘drug of last resort’ against intractable, life-threatening infections caused by MRSA, VRE and other dangerous Gram-positive pathogens. However, we have also focused in this study on a new generation of PBD-based DNA-interactive molecules, PBD-biaryl conjugates. Representatives of this class are remarkably well tolerated in mice at high concentrations (D. E. Thurston, unpublished observations) and we therefore expected them to yield a more favourable toxicity profile than PBD dimers such as ELB-21 when evaluated as antibacterial agents.
A high proportion (26 from 43) of compounds from the library detailed in Figure 4 and Table S1 yielded MICs of ≤4 mg/L, and many of these were effective at <1 mg/L (Table S4). Six compounds were particularly potent against the MRSA/VRE panel and their structures are shown in Figure 7. It is our intention to select one or two of these agents for further profiling and for preclinical and, potentially, clinical evaluation. Interestingly, as these compounds possessed between two and nine orders of magnitude less cytotoxicity against non-proliferating as compared with proliferating human cell lines, we exposed 24 h TUPLF/AB zebrafish (Danio rerio) embryos to these six conjugates, monitoring viability and morphology over a 5 day period; we found no evidence of toxicity when concentrations up to 20-fold MIC were employed (J. B. Moreira and P. W. Taylor, unpublished observations). The PBD-biaryl conjugates were designed to preferentially bind to G:C-rich sequences. We have shown by fluorescence resonance energy transfer-based thermal denaturation that they stabilize G:C-rich sequences up to 13-fold compared with A:T-rich sequences and by ion-pair HPLC-MS that the MPB units in compounds such as 29, 36 and 57 (Figure 7) prefer G:C base pairs close to the PBD covalent attachment site. DNase I footprinting demonstrated strong binding in G:C-rich regions that varied consistently in relation to the relative position of the building block within the molecule (K. M. Rahman, K. R. Fox and D. E. Thurston, unpublished observations). Molecular dynamics simulations with MPB-containing PBD-biaryl conjugates indicate that these molecules are more easily accommodated within the minor groove than PBD dimers such as ELB-21 and cause less distortion to the DNA duplex (compare Figure 6 with Figure 2). It is important, however, that the outcomes of these simulations be verified by experimental observation.
Figure 7.

Structures of PBD-biaryl conjugates with potent activity (∼0.06 mg/L) against a panel of Gram-positive pathogens. The structures for BztMC-Im-PBD (35), BztMC-Py-PBD (27), Bzf-Py-PBD (26), Py-Py-MPB-PBD (57), MPB-Py-PBD (29) and MPB-Im-PBD (36) have been extracted from Table S1.
Some clear relationships emerged from comparison of the PBD-biaryl structures and their antibacterial activity: (i) the potency is highly influenced by the positions of the incorporated units; (ii) all In-containing conjugates are inactive; (iii) a single MPB unit is essential for enhanced activity (e.g. PBD-Im-Im had only weak activity of 4–16 mg/L and a PBD-MPB-MPB conjugate was inactive against both MRSA and VRE, but PBD-Py-MPB and PBD-Im-MPB were highly active with MICs of ∼0.06 mg/L); (iv) a five-membered heterocycle between the MPB and PBD units provides optimum activity; (v) switching the position of the MPB unit (e.g. PBD-MPB-Im in place of PBD-Im-MPB) reduces activity by 16–32-fold; (vi) introduction of a second five-membered heterocycle after MPB increases potency by 10–50-fold (PBD-MPB-Im-Py compared with PBD-MPB-Py); and (vii) more than two heterocycles after the MPB reduces potency. These preliminary data underline the potential of the strongly bioactive PBD-biaryl conjugates as prototypical agents for the treatment of systemic Gram-positive infections.
The limited acute toxicity data currently available for PBD-biaryl compounds (D. E. Thurston, unpublished observations) indicate that their therapeutic index is likely to be superior to that for PBD dimers and our future efforts to develop PBD conjugates as antibacterial chemotherapeutics for multidrug-resistant Gram-positive pathogens will therefore focus on these agents. Molecular dynamics simulations suggest that PBD-biaryl compound 36 (MPB-Im-PBD) causes virtually no distortion of the DNA duplex following binding to the target guanine within the minor groove, even though this conjugate possesses potent antibacterial activity. If this a general feature of bioactive PBD-biaryl conjugates, it is likely to impact advantageously on the safety profile. Conformational distortion is known to engender activation of the bacterial DNA damage response;34,35 transcriptomic analysis of gene expression following exposure of EMRSA-16 to 28 and 57 strongly suggests that, similar to ELB-21,10,14 the bactericidal effects are due to the failure of the target bacteria to excise and repair regions of DNA to which conjugates are covalently attached. Less pronounced induction of the RecA-LexA response by PBD-biaryl conjugates in comparison with ELB-21 probably reflects a lack of helix distortion following covalent attachment of the drug. Similarly to ELB-21,10,14 PBD-biaryl conjugates also engendered activation of prophage genes within the EMRSA-16 genome.
Funding
This study was supported by Wellcome Trust project grant 078669/Z/05/Z and by BSAC project grant GA840.
Transparency declarations
P. W. H. and S. J. G. are employees of Spirogen Ltd, and D. E. T. is a consultant to the company. P. W. H. and D. E. T. own stock in Spirogen Ltd. All other authors: none to declare.
Supplementary data
Tables S1 to S6 and Figures S1 and S2 are available as Supplementary data at JAC Online (http://jac.oxfordjournals.org/).
Acknowledgements
We acknowledge BμG@S (the Bacterial Microarray Group at St George's, University of London) for supply of the microarray and the Wellcome Trust for funding the multicollaborative microbial pathogen microarray facility under its Functional Genomics Resources Initiative.
References
- 1.Greenwood D. Antimicrobial Drugs – Chronicle of a Twentieth Century Medical Triumph. Oxford: Oxford University Press; 2008. [Google Scholar]
- 2.Leimgruber W, Stefanovic V, Schenker F, et al. Isolation and characterization of anthramycin, a new antitumor antibiotic. J Amer Chem Soc. 1965;87:5791–3. doi: 10.1021/ja00952a050. doi:10.1021/ja00952a050. [DOI] [PubMed] [Google Scholar]
- 3.Arima K, Kosaka M, Tamura G, et al. Studies on tomaymycin, a new antibiotic. I. Isolation and properties of tomaymycin. J Antibiot (Tokyo) 1972;25:437–44. doi: 10.7164/antibiotics.25.437. doi:10.7164/antibiotics.25.437. [DOI] [PubMed] [Google Scholar]
- 4.Brazhnikova MG, Konstantinova NV, Mesentsev AS. Sibiromycin: isolation and characterization. J Antibiot (Tokyo) 1972;25:668–73. doi: 10.7164/antibiotics.25.668. doi:10.7164/antibiotics.25.668. [DOI] [PubMed] [Google Scholar]
- 5.Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med. 2004;10(Suppl 12):S122–9. doi: 10.1038/nm1145. doi:10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 6.Davies J, Davies G. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010;74:417–33. doi: 10.1128/MMBR.00016-10. doi:10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, Nation RL, Turnidge JD, et al. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect Dis. 2006;6:589–601. doi: 10.1016/S1473-3099(06)70580-1. doi:10.1016/S1473-3099(06)70580-1. [DOI] [PubMed] [Google Scholar]
- 8.Hadjivassileva T, Thurston DE, Taylor PW. Pyrrolobenzodiazepine dimers: novel sequence-selective, DNA-interactive, cross-linking agents with activity against Gram-positive bacteria. J Antimicrob Chemother. 2005;56:513–8. doi: 10.1093/jac/dki256. doi:10.1093/jac/dki256. [DOI] [PubMed] [Google Scholar]
- 9.Hadjivassileva T, Stapleton PD, Thurston DE, et al. Interactions of pyrrolobenzodiazepine dimers and duplex DNA from methicillin-resistant Staphylococcus aureus. Int J Antimicrob Agents. 2007;290:672–8. doi: 10.1016/j.ijantimicag.2007.01.005. doi:10.1016/j.ijantimicag.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Doyle M, Feuerbaum E-A, Fox KR, et al. Response of Staphylococcus aureus to subinhibitory concentrations of a sequence-selective, DNA minor groove cross-linking pyrrolobenzodiazepine dimer. J Antimicrob Chemother. 2009;64:949–59. doi: 10.1093/jac/dkp325. doi:10.1093/jac/dkp325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puvvada MS, Forrow SA, Hartley JA, et al. Inhibition of bacteriophage T7 RNA polymerase in vitro transcription by DNA-binding pyrrolo[2,1-c][1,4]-benzodiazepines. Biochemistry. 1997;36:2478–84. doi: 10.1021/bi952490r. doi:10.1021/bi952490r. [DOI] [PubMed] [Google Scholar]
- 12.Smellie M, Bose DS, Thompson AS, et al. Sequence-selective recognition of duplex DNA through covalent interstrand cross-linking: kinetic and molecular modeling studies with pyrrolobenzodiazepine dimers. Biochemistry. 2003;42:8232–9. doi: 10.1021/bi034313t. doi:10.1021/bi034313t. [DOI] [PubMed] [Google Scholar]
- 13.Martin C, Ellis T, McGurk CJ, et al. Sequence-selective interaction of the minor-groove interstrand cross-linking agent SJG-136 with naked and cellular DNA: footprinting and enzyme inhibition studies. Biochemistry. 2005;44:4135–47. doi: 10.1021/bi0479813. doi:10.1021/bi0479813. [DOI] [PubMed] [Google Scholar]
- 14.Rosado H, Rahman K, Feuerbaum E-A, et al. The minor groove-binding agent ELB-21 forms multiple interstrand and intrastrand covalent cross-links with duplex DNA and displays potent bactericidal activity against methicillin-resistant Staphylococcus aureus. J Antimicrob Chemother. 2011;66:985–96. doi: 10.1093/jac/dkr044. doi:10.1093/jac/dkr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pepper CJ, Hambly RM, Fegan CD, et al. The novel sequence-specific DNA cross-linking agent SJG-136 (NSC 694501) has potent and selective in vitro cytotoxicity in human B-cell chronic lymphocytic leukemia cells with evidence of a p53-independent mechanism of cell kill. Cancer Res. 2004;64:6750–5. doi: 10.1158/0008-5472.CAN-04-1713. doi:10.1158/0008-5472.CAN-04-1713. [DOI] [PubMed] [Google Scholar]
- 16.Arnould S, Spanswick VJ, Macpherson JS, et al. Time-dependent cytotoxicity induced by SJG-136 (NSC 694501): influence of the rate of interstrand cross-link formation on DNA damage signaling. Mol Cancer Ther. 2006;5:1602–9. doi: 10.1158/1535-7163.MCT-06-0018. doi:10.1158/1535-7163.MCT-06-0018. [DOI] [PubMed] [Google Scholar]
- 17.Rahman KM, Thompson AS, James CH, et al. The pyrrolobenzodiazepine dimer SJG-136 forms sequence-dependent intrastrand DNA cross-links and monoalkylated adducts in addition to interstrand cross-links. J Amer Chem Soc. 2009;131:13756–66. doi: 10.1021/ja902986x. doi:10.1021/ja902986x. [DOI] [PubMed] [Google Scholar]
- 18.Haywood-Farmer E, Otto SP. The evolution of genomic base composition in bacteria. Evolution. 2003;57:1783–92. doi: 10.1111/j.0014-3820.2003.tb00586.x. [DOI] [PubMed] [Google Scholar]
- 19.Wells G, Martin CRH, Howard PW, et al. Design, synthesis, and biophysical and biological evaluation of a series of pyrrolobenzodiazepine-poly(N-methylpyrrole) conjugates. J Med Chem. 2006;49:5442–61. doi: 10.1021/jm051199z. doi:10.1021/jm051199z. [DOI] [PubMed] [Google Scholar]
- 20.Stapleton PD, Shah S, Anderson JC, et al. Modulation of β-lactam resistance in Staphylococcus aureus by catechins and gallates. Int J Antimicrob Agents. 2004;23:462–7. doi: 10.1016/j.ijantimicag.2003.09.027. doi:10.1016/j.ijantimicag.2003.09.027. [DOI] [PubMed] [Google Scholar]
- 21.Skehan P, Storeng R, Scudiero DA, et al. New colorimetric cytotoxicity assay for anticancer drug screening. J Natl Cancer Inst. 1990;82:1107–12. doi: 10.1093/jnci/82.13.1107. doi:10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 22.Gregson SJ, Howard PW, Gullick DR, et al. Linker length modulates DNA cross-linking reactivity and cytotoxic potency of C8/C8′ ether-linked C2-exo-unsaturated pyrrolo[2,1-c][1,4]benzodiazepine (PBD) dimers. J Med Chem. 2004;47:1161–74. doi: 10.1021/jm030897l. doi:10.1021/jm030897l. [DOI] [PubMed] [Google Scholar]
- 23.Thurston DE, Bose DS, Howard PW, et al. Effect of A-ring modifications on the DNA-binding behavior and cytotoxicity of pyrrolo[2,1-c][1,4]benzodiazepines. J Med Chem. 1999;42:1951–64. doi: 10.1021/jm981117p. doi:10.1021/jm981117p. [DOI] [PubMed] [Google Scholar]
- 24.Antonow D, Thurston DE. Synthesis of DNA-interactive pyrrolo[2,1-c][1,4]benzodiazepines (PBDs) Chem Rev. 2011;111:2815–64. doi: 10.1021/cr100120f. doi:10.1021/cr100120f. [DOI] [PubMed] [Google Scholar]
- 25.Tiberghien AC, Evans DA, Kiakos K, et al. An asymmetric C8/C8′-tripyrrole-linked sequence-selective pyrrolo[2,1-c][1,4]benzodiazepine (PBD) dimer DNA interstrand cross-linking agent spanning 11 DNA base pairs. Bioorg Med Chem Lett. 2008;18:2073–7. doi: 10.1016/j.bmcl.2008.01.096. doi:10.1016/j.bmcl.2008.01.096. [DOI] [PubMed] [Google Scholar]
- 26.Howard PW, Chen Z, Gregson SJ, et al. Synthesis of a novel C2/C2′-aryl-substituted pyrrolo[2,1-c][1,4]benzodiazepine dimer prodrug with improved water solubility and reduced reaction rate. Bioorg Med Chem Lett. 2009;19:6463–6. doi: 10.1016/j.bmcl.2009.09.012. doi:10.1016/j.bmcl.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 27.Antonow D, Kaliszczak M, Kang GD, et al. Structure–activity relationships of monomeric C2-aryl pyrrolo[2,1-c][1,4]benzodiazepine (PBD) antitumor agents. J Med Chem. 2010;53:2927–41. doi: 10.1021/jm901722v. doi:10.1021/jm901722v. [DOI] [PubMed] [Google Scholar]
- 28.Hampshire AJ, Rusling DA, Broughton-Head VJ, et al. Footprinting: a method for determining the sequence selectivity, affinity and kinetics of DNA-binding ligands. Methods. 2007;42:128–40. doi: 10.1016/j.ymeth.2007.01.002. doi:10.1016/j.ymeth.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Hampshire AJ, Fox KR. Preferred binding sites for the bifunctional intercalator TANDEM determined using DNA fragments that contain every symmetrical hexanucleotide sequence. Anal Biochem. 2008;374:298–303. doi: 10.1016/j.ab.2007.10.008. doi:10.1016/j.ab.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 30.Witney AA, Marsden GL, Holden MT, et al. Design, validation, and application of a seven-strain Staphylococcus aureus PCR product microarray for comparative genomics. Appl Environ Microbiol. 2005;71:7504–14. doi: 10.1128/AEM.71.11.7504-7514.2005. doi:10.1128/AEM.71.11.7504-7514.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Case DA, Darden TA, Cheatham TE, III, et al. AMBER 9. San Francisco: University of California; 2006. [Google Scholar]
- 32.Humphrey W, Dalke A, Schulten K. VMD – visual molecular dynamics. J Molec Graph. 1996;14:33–8. doi: 10.1016/0263-7855(96)00018-5. doi:10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 33.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. doi:10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 34.Lebbink JHG, Georgijevic D, Natrajan G, et al. Dual role of MutS glutamate 38 in mismatch discrimination and in the authorization of repair. EMBO J. 2006;25:409–19. doi: 10.1038/sj.emboj.7600936. doi:10.1038/sj.emboj.7600936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nohmi T. Environmental stress and lesion-bypass DNA polymerases. Ann Rev Microbiol. 2006;60:231–53. doi: 10.1146/annurev.micro.60.080805.142238. doi:10.1146/annurev.micro.60.080805.142238. [DOI] [PubMed] [Google Scholar]
- 36.Hartley JA, Hamaguchi A, Coffils M, et al. SG2285, a novel C2-aryl-substituted pyrrolobenzodiazepine dimer prodrug that cross-links DNA and exerts highly potent antitumor activity. Cancer Res. 2010;70:6849–58. doi: 10.1158/0008-5472.CAN-10-0790. doi:10.1158/0008-5472.CAN-10-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baba T, Takeuchi F, Kuroda M, et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002;359:1819–27. doi: 10.1016/s0140-6736(02)08713-5. doi:10.1016/S0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
- 38.Rahman KM, James CH, Thurston DE. Effect of base sequence on the DNA cross-linking properties of pyrrolobenzodiazepine (PBD) dimers. Nucl Acids Res. 2011;39:5800–12. doi: 10.1093/nar/gkr122. doi:10.1093/nar/gkr122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartley JA, Spanswick VJ, Brooks N, et al. SJG-136 (NSC 694501), a novel rationally designed DNA minor groove interstrand cross-linking agent with potent and broad spectrum antitumour activity: part 1: cellular pharmacology, in vitro and initial in vivo antitumour activity. Cancer Res. 2004;64:6693–9. doi: 10.1158/0008-5472.CAN-03-2941. doi:10.1158/0008-5472.CAN-03-2941. [DOI] [PubMed] [Google Scholar]
- 40.Alley MC, Hollingshead MG, Pacula-Cox CM, et al. SJG-136 (NSC 694501), a novel rationally designed DNA minor groove interstrand cross-linking agent with potent and broad spectrum antitumour activity: part 2: efficacy evaluations. Cancer Res. 2004;64:6700–6. doi: 10.1158/0008-5472.CAN-03-2942. doi:10.1158/0008-5472.CAN-03-2942. [DOI] [PubMed] [Google Scholar]
- 41.Redon CE, Nakamura AJ, Zhang YW, et al. Histone yH2AX and poly(ADP-ribose) as clinical pharmacodynamic biomarkers. Clin Cancer Res. 2010;16:4532–42. doi: 10.1158/1078-0432.CCR-10-0523. doi:10.1158/1078-0432.CCR-10-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janjigian Y, Lee W, Kris M, et al. A phase I trial of SJG-136 (NSC#694501) in advanced solid tumors. Cancer Chemother Pharmacol. 2010;65:833–8. doi: 10.1007/s00280-009-1088-4. doi:10.1007/s00280-009-1088-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.