

Abstract
Glutamate excitotoxicity, a major component of many neurodegenerative disorders, is characterized by excessive calcium influx selectively through NMDARs. However, there is a substantial uncertainty concerning why other known routes of significant calcium entry, in particular, VGCCs, are not similarly toxic. Here, we report that in the majority of neurons in rat hippocampal and cortical cultures, maximal L-type VGCC activation induces much lower calcium loading than toxic NMDAR activation. Consequently, few depolarization-activated neurons exhibit calcium deregulation and cell death. Activation of alternative routes of calcium entry induced neuronal death in proportion to the degree of calcium loading. In a small subset of neurons, depolarization evoked stronger calcium elevations, approaching those induced by toxic NMDA. These neurons were characterized by elevated expression of VGCCs and enhanced voltage-gated calcium currents, mitochondrial dysfunction and cell death. Preventing VGCC-dependent mitochondrial calcium loading resulted in stronger cytoplasmic calcium elevations, whereas inhibiting mitochondrial calcium clearance accelerated mitochondrial depolarization. Both observations further implicate mitochondrial dysfunction in VGCC-mediated cell death. Results indicate that neuronal vulnerability tracks the extent of calcium loading but does not appear to depend explicitly on the route of calcium entry.
Introduction
Disruption of Ca2+ homeostasis is a critical event in neuronal degeneration following ischemic or traumatic brain injury (Greve and Zink, 2009; Szydlowska and Tymianski, 2010), as well as in several neurological disorders (Bezprozvanny, 2009). Under many pathological conditions, excessive glutamate release induces excitotoxicity, a condition that is characterized by massive Ca2+ influx mainly through NMDARs. Two major hypotheses that consider excessive intracellular Ca2+ as a trigger for neuronal death, namely, “calcium loading” (Hartley et al., 1993) and “source specificity” (Tymianski et al., 1993), have been debated for well over a decade. The first, inferred from the correlation between toxicity and the magnitude of the Ca2+ load, is in line with current models of excitotoxic cell death in which Ca2+ overload-induced mitochondrial dysfunction also plays a central role (for review, see Nicholls, 2009; Pivovarova and Andrews, 2010). On the other hand, there is considerable evidence for the idea that excitotoxic death is “source specific” because it is selectively linked to Ca2+ entry through NMDARs, most likely by a specialized mechanism that depends on activation of synaptic neuronal nitric oxide synthase (nNOS) (Sattler et al., 1999; Aarts et al., 2002; Soriano et al., 2008).
From the viewpoint of the Ca2+ loading hypothesis, the prominent role of NMDARs in excitotoxic injury might reflect the fact that most other potential routes of Ca2+ entry generally restrict the amount of Ca2+ that can be transported (Hyrc et al., 1997; Stout and Reynolds, 1999). This view can explain why Ca2+ loading through AMPARs, for example, becomes strong and toxic only when their channels are rendered Ca2+ permeable (Carriedo et al., 2000), as well as why in hippocampal neurons vulnerability increases with age in parallel with VGCC expression and Ca2+ currents (Thibault et al., 2001; Brewer et al., 2007; Stanika et al., 2009). However, this straightforward hypothesis is not obviously consistent with certain results taken to support route specificity, for example, data showing that equivalent amounts of Ca2+ are highly toxic when loaded via NMDARs but not when loaded via VGCCs (Sattler et al., 1998; Aarts et al., 2002; Tymianski, 2009).
In the present study we provide evidence that loading a given amount of Ca2+ induces proportionate mitochondrial Ca2+ accumulation and dysfunction regardless of the route of Ca2+ entry, and that this correlates with cell vulnerability. This principle explains the generally limited impact of VGCCs, since results show that in the majority of hippocampal neurons the Ca2+ load attainable via this route is much lower than that via NMDARs, and generally insufficient to induce mitochondrial dysfunction, nNOS activation, and cell death. However, in a small subset of neurons—one that becomes larger as cells age—VGCC activation does lead to Ca2+ loading approaching that induced by toxic NMDA, and also results in mitochondria-mediated cell death. These results support the proposition that differences in Ca2+ loading can adequately account for the privileged toxicity of NMDARs compared with VGCCs or other physiologically plausible pathways of Ca2+ entry.
Materials and Methods
Cell culture.
Primary cultures of rat hippocampal and cortical neurons were prepared as previously described (Pivovarova et al., 2004). In brief, suspensions of a mixed population of brain cells prepared by papain dissociation of hippocampi or cortex from ∼20 d embryonic Sprague Dawley rats of either sex were plated onto previously prepared layers of glia in MEM supplemented with 5% horse serum, 1% fetal bovine serum, 2 mm Glutamax (Life Technologies), 136 μm uridine, 54 μm 2-deoxy-5-fluoro-uridine, and the growth factor cocktail N3. Cells were maintained in a 37°C incubator with 10% CO2. Half of the culture medium was replaced twice weekly. All procedures, including animal euthanasia, were performed in accordance with an NINDS Animal Care and Use Committee Protocol. Experiments were performed on cultures 14–28 DIV.
Stimulation protocols, solutions, and viability assays.
For experiments, cultures were transferred to a HEPES-buffered saline solution (HBSS) containing (in mm): 137 NaCl, 10 HEPES, 2.0 CaCl2, 1.0 MgSO4, 5.4 KCl, 0.3 Na2HPO4, 0.22 KH2PO4, 10 glucose, and 26 sucrose (320 mOsM, pH 7.4). Maximal VGCC activation was performed according to published protocols (Sattler et al., 1998; Hardingham et al., 2002) by depolarizing the plasma membrane with a Ca2+-containing (10 mm) high K+ (90 mm) solution (prepared by substituting Na+ with K+) in HBSS in the presence of 10 μm CNQX (an AMPAR blocker) and 20 μm MK-801 (an NMDAR blocker), along with the L-type Ca2+-channel activators (S)-(−)-Bay K 8644 (1 μm) or FPL 64176 (5 μm). Excitotoxic activation of NMDARs was performed for 30 min in Mg2+-free HBSS containing 10 μm glycine and 100 μm NMDA in the presence of 10 μm nimodipine (an L-type VGCC blocker) and 10 μm CNQX.
A modification of the protocol of Kiedrowski et al. (2004) was used to load Ca2+ via reverse mode Na+/Ca2+ exchange (NCX). Neurons were first incubated with 200 μm glutamate and 1 mm ouabain in Ca2+-free (2 mm EGTA) HBSS for 20 min to Na+ load cells, followed by brief exposure to 20 μm MK-801 (to block NMDARs) before reintroduction of Ca2+ (10 mm in HBSS in the presence of MK-801) to activate NCX. A “chemical LTD” protocol that degrades PSDs, thereby unlinking NMDARs from nNOS and preventing synaptic nNOS activation (Colledge et al., 2003; Di Biase et al., 2008), consisted of a brief exposure (5 min) to 100 μm NMDA.
For viability assays, cultures after stimulation were washed in HBSS and returned to the incubator in growth medium for 20–24 h. Cell death was assayed by propidium iodide (3.3 μg/ml) staining. Immunostaining for NeuN, a neuron-specific protein, was used to assay the total number of neurons. Cell counting was performed using ImageJ software (http://rsb.info.nih.gov/ij/index.html).
Nimodipine, CNQX, MK-801, (S)-(−)-Bay K 8644, FPL 64176, ionomycin, and CGP 37157 were purchased from Tocris Bioscience and NeuN from Millipore Bioscience Research Reagents. All other reagents were from Sigma-Aldrich.
Fluorescence microscopy.
Intracellular cytosolic free Ca2+ concentrations were estimated using the low-affinity Ca2+ probe fura-2FF (TEFLabs). For dye loading, cultures were incubated in 4 μm fura-2FF AM in HBSS for 30 min at 37°C and subsequently imaged using a Zeiss Axiovert 200 light microscope equipped with a 40× objective. Images were recorded by means of a CoolSNAP HQ2 CCD camera (Photometrics) controlled by EasyRatioPro software (Photon Technology International). All experiments were performed at 25°C. For quantitative imaging, fura-2FF was excited alternately at 340 and 380 nm and fluorescence emission was collected at 510 nm. Ca2+ concentrations were quantified from the ratio of 340/380 fluorescence intensities using established methods (Grynkiewicz et al., 1985). During imaging, cultures were continuously perfused with HBSS containing added drugs as needed. Images were collected at 5–30 s intervals during the initial part of the experiment and at 60 s intervals during the final part. Due to the inherent uncertainties of calibration procedures, absolute concentrations should be considered as estimates.
Mitochondrial membrane potentials (MMPs) were visualized with the membrane-permeant cationic dye rhodamine-123 (Rh123) (Invitrogen; excitation 514 nm, emission 530 nm). Cultures were incubated for 15 min at 37°C in 25 μm Rh123 in HBSS and washed an additional 15 min before imaging. Rh123 is accumulated and quenched in polarized mitochondria; upon depolarization, the dye is released from mitochondria leading to increased cytosolic fluorescence in proportion to the degree of depolarization. Intracellular nitric oxide (NO) production was assayed using DAF-FM diacetate (4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate, Invitrogen; excitation 488 nm, emission 505 nm) (Marks et al., 2005). Cultures were incubated with 10 μm DAF-FM in HBSS for 30 min at 37°C and washed for 15 min. Imaging of Rh123 and DAF-FM was performed at room temperature using a Zeiss LSM 510 confocal microscope equipped with a 40× objective.
Immunocytochemistry.
Cells were fixed for 10 min at −20°C with methanol, then washed three times with PBS and permeabilized with 0.5% Triton X-100 in blocking solution (2% BSA, 2% NGS in PBS) for 1 h. Immunostaining was performed using a rabbit primary antibody against the α1C subunit of the L-type (Cav1.2) calcium channel (1:200, Sigma-Aldrich). Cultures were incubated with primary antibodies at room temperature for 1 h and after washing were stained with secondary goat anti-rabbit IgG FITC-conjugated antibodies (1:500, Millipore Bioscience Research Reagents) at room temperature for 2 h.
Electrophysiology.
Cells were voltage-clamped in whole-cell configuration. Calcium channel activity was recorded by 2–4 MΩ micropipettes manufactured from borosilicate glass capillary tubes with micropipette puller P-87 (Sutter Instruments). Cells were depolarized from a holding potential of −70 mV to +10 mV with 20 mV steps, and recordings were filtered at 5 kHz, digitized at 10 kHz using an Axopatch 200B amplifier, and integrated with a Digidata 1440A data acquisition system (Molecular Devices). The composition of the external solution was as follows (in mm): 25 tetraethylammonium chloride, 5 4-aminopyridine, 2 MgCl2, 3 KCl, 5 CaCl2, 20 HEPES, 100 NaCl, 0.001 TTX, at pH 7.4. Pipette solution was (in mm): 56 CsCl, 47 Cs2SO4, 20 HEPES, 1 MgCl2, 4 ATP, 0.3 GTP, 0.5 EGTA, 0.2 CaCl2, pH 7.2.
Electron microscopy and electron probe x-ray microanalysis.
For electron microscopy and electron probe x-ray microanalysis (EPMA), cultures were rapidly frozen, cryosectioned, imaged, and analyzed in an analytical electron microscope as previously described (Pivovarova et al., 2004). X-ray spectra were recorded and processed by established procedures (Pivovarova et al., 1999), providing total concentrations of cytoplasmic and mitochondrial Ca, Na, Mg, P, Cl, and K in units of mmol/kg dry weight.
Statistics.
One-way ANOVA followed by post hoc Dunnett's multiple-comparisons test was used for normally distributed data. For non-normally distributed data the nonparametric Kruskal–Wallis rank ANOVA with post hoc Dunn's test was used. Data are given as mean ± SEM. Analysis was performed using InStat software (GraphPad Software). In general, experiments were performed on at least three cultures prepared from different animals.
Results
Neuronal vulnerability parallels cytosolic Ca2+ elevations
We first compared the elevation of cytoplasmic free Ca2+ induced by activation of VGCCs and NMDARs in cultured rat hippocampal and cortical neurons using the low-affinity Ca2+ probe fura-2FF. VGCCs were activated by strong depolarization according to published protocols that used 90 mm K+ together with the VGCC activator Bay K 8644 in 10 mm Ca2+-containing saline (Sattler et al., 1998) to maximize Ca2+ influx. The majority of 14 DIV cells exhibited a typical Ca2+ spike, followed by decay to a steady-state level slightly higher than basal concentrations (Fig. 1A); a small subset of cells (whose significance is discussed below) maintained much higher Ca2+ levels, >5 μm (Fig. 1A). The average Ca2+ concentration after high K+ exposure was ∼4 μm (Fig. 1A,C). Replacement of Bay K 8644 with FPL 64176 (Hardingham et al., 2002) produced similar results (data not shown). Depolarization-induced Ca2+ elevations were blocked by the L-type VGCC blocker nimodipine (Fig. 1A) but were unaffected by the N/P-type Ca2+ channel blocker ω-conotoxin MVIIC (data not shown), indicating that Ca2+ influx was mainly mediated by L-type VGCCs.
Figure 1.

Strong VGCC activation induces lower Ca2+ elevations and less death than NMDAR activation. A, Time course of 90 mm K+-induced free Ca2+ elevations (fura-2FF) in individual 14 DIV hippocampal neurons (gray traces) and their average (red; n = 46). Incubation medium was 90 mm KCl, 1 μm Bay K 8644, 20 μm MK-801, 10 μm CNQX in 10 mm Ca2+-containing HBSS. Blue traces, Average Ca2+ elevations induced by 100 μm (dark blue; n = 49) and 10 μm (light blue; n = 47) NMDA. Orange trace, Average Ca2+ response induced by high K+ in the presence of 10 μm nimodipine (n = 35). B, Representative Ca2+ traces from experiments in which Ca2+ entry was induced by 100 μm NMDA (as in A), 10 μm ionomycin (Iono, n = 30), or reverse mode NCX (n = 40; see Materials and Methods for protocol). C, Left, Maximal sustained concentration of free Ca2+ in individual neurons 30 min after the activation of Ca2+ uptake as in A and B). Right, Neuronal death rate 24 h after the same stimuli. No significant cell death was observed in controls without stimulation. D, Concentrations of total calcium in the cytoplasm and mitochondria of neurons immediately after 30 min of stimulation as described in A and B. Data are mean ± SEM, as measured by EPMA. E, Time courses of Rh123 relative fluorescence (F/F0) show no significant changes of MMP in 14 DIV hippocampal neurons induced by 90 K+ depolarization (n = 23) and typical strong MMP depolarization induced by NMDA (100 μm; n = 26). F, Left, Representative traces of NO production (DAF-FM fluorescence; F/F0) induced by 100 μm NMDA (n = 19) alone or in the presence 100 μm l-NAME (n = 14), or by 90 mm K+ depolarization (n = 16). Right, Neuronal death rate 24 h after NMDA stimulation is reduced by pharmacological NOS inhibition (l-NAME) or by disrupting the NMDAR/nNOS linkage using a chemical LTD protocol (see Materials and Methods).
In contrast to VGCC activation, exposure to excitotoxic NMDA induced strong, biphasic cytosolic Ca2+ increases in essentially all neurons, the second phase representing complete Ca2+ deregulation with average Ca2+ concentrations >10 μm (Fig. 1A,C). Typical traces were similar to previous records from ourselves and others (Abramov and Duchen, 2008; Li et al., 2009; Stanika et al., 2009). VGCC and NMDAR activation in 14 DIV cortical neurons produced similar results (data not shown). Importantly, the size and distribution of the Ca2+ elevations induced by these two diverse stimuli (Fig. 1C, left) correlate well with cell viability (Fig. 1C, right). Thus, VGCC activation resulted in the death of only ∼20% of neurons in 14 DIV cultures, whereas NMDAR activation led to massive delayed neuronal death, ∼70% (Fig. 1C, right).
Several additional approaches were used to further test the idea that the size of the Ca2+ load is a dominant factor governing neuronal vulnerability. First, NMDA concentrations were incrementally adjusted downward, progressively reducing Ca2+ elevations and death rates until, at 10 μm NMDA, they were essentially equal to the lower Ca2+ elevations (∼4 μm) and death rates (∼20%) induced by maximal 90 mm K+ depolarization (Fig. 1C). Conversely, exposure to the Ca2+ ionophore ionomycin (10 μm), which leads to strong Ca2+ accumulation by an entirely different route, resulted in cytosolic Ca2+ levels even higher than toxic NMDA, in parallel with a proportionately higher death rate (Fig. 1B,C); in contrast, ionomycin exposure in Ca2+-free medium was entirely nontoxic (Fig. 1C). Finally, an intermediate regime of Ca2+ loading was achieved by exploiting mechanisms that depend on reversal of the plasma membrane NCX. Cells were Na+ loaded by global glutamate receptor activation in a Ca2+-free medium, followed by MK-801 block of activated NMDARs and reintroduction of extracellular Ca2+ to activate NCX in reverse mode (Kiedrowski et al., 2004). This protocol leads, as expected, to intermediate levels of Ca2+ elevation and corresponding cell death (Fig. 1B,C).
Cytosolic and mitochondrial total calcium loading
Although small free cytosolic Ca2+ elevations imply limited calcium uptake, and vice versa, it is important to evaluate total calcium loading directly because the calcium loading hypothesis holds that cell death is triggered by excessive amounts of total intracellular calcium (Hartley et al., 1993). Thus, we used electron probe x-ray microanalysis (EPMA) to show that cytosolic total calcium concentrations closely follow the same trend as free Ca2+ elevations and death rates, namely, that toxic NMDA and ionomycin strongly load cells with calcium (37 ± 3 and 91 ± 4 mmol/kg dry weight, respectively), while even maximal VGCC induces minimal calcium accumulation in the majority of cells (3.8 ± 1.2 mmol/kg) (Fig. 1D, left). Significantly, mitochondrial calcium loading after NMDA, ionomycin, or 90 mm K+ exposure—938 ± 110, 652 ± 83 and 1.4 ± 0.6 mmol/kg, respectively (Fig. 1D, right)—tracked cytosolic changes. As would be predicted by such large differences in mitochondrial calcium accumulation, strong VGCC activation, unlike toxic NMDAR activation, did not induce detectable mitochondrial depolarization (Fig. 1E). To summarize, all measures—free Ca2+, total calcium, mitochondrial calcium, mitochondrial depolarization—support the proposition that neuronal death rates closely parallel cell and mitochondrial calcium loading. In the specific case of VGCCs, even excessively strong activation is not toxic because calcium loading in the large majority of neurons is low and insufficient to trigger mitochondrial dysfunction and cell death.
The role of nitric oxide
Although we consider the evidence presented so far as strong support for the calcium loading hypothesis of excitotoxic injury, it leaves unanswered questions about how calcium loading relates to the well documented source specificity hypothesis. Calcium activation of synaptic nNOS, a PSD-95-linked component of the postsynaptic density (PSD), is thought to underlie the selective toxicity of Ca2+ entry through NMDARs (Forder and Tymianski, 2009). Although the focus of this hypothesis is fundamentally different from the calcium loading hypothesis, the two are not necessarily incompatible—a view supported by the role of nNOS activation in our cells. As shown in Figure 1F, toxic NMDA significantly increased NO levels (as reported by the NO-specific probe DAF; Marks et al., 2005), while NOS inhibition with l-NAME prevented NMDA-induced NO production and reduced cell death by ∼25%. Moreover, synaptic nNOS appears to be the major contributor to NO production since disrupting PSDs using a chemical LTD strategy (see Materials and Methods for protocol) (Colledge et al., 2003; Di Biase et al., 2008) similarly reduced NMDA toxicity (Fig. 1F). As might be expected because depolarization induces minimal Ca2+ loading, VGCC activation did not induce significant NO production. These observations are compatible with both source specificity and calcium loading.
Calcium-dependent injury in older neurons
The developmental program of hippocampal neurons in culture provides additional opportunities to dissect out VGCC-dependent contributions to neuronal injury. To be specific, VGCC-activated Ca2+ influx increases during aging (Thibault and Landfield, 1996; Thibault et al., 2001), as well as in older cells in culture (Stanika et al., 2009). This leads to the expectation that any VGCC-dependent component of neuronal vulnerability should be enhanced as a result of increased Ca2+ elevations in older cells. We confirmed that under our culture conditions older hippocampal neurons (28 DIV) express significantly more functional L-type VGCCs than do 14 DIV cells (Fig. 2A), and that this difference is reflected in considerably larger voltage-activated Ca2+ currents (Fig. 2B). Also in contrast to 14 DIV cells, VGCC activation of older cells resulted in stronger Ca2+ elevations in a larger fraction of neurons, some of these cells exhibiting Ca2+ levels approaching those induced by NMDA (Fig. 2C,D; compare Fig. 1A,C). The larger fraction of 28 DIV cells with elevated Ca2+ was paralleled by a larger death rate, ∼30% (Fig. 2D, right), which is consistent with the correlation seen in 14 DIV cells. Both depolarization-induced Ca2+ elevations (data not shown) and neuronal death (25 ± 2% vs 6 ± 1%, p < 0.001) were prevented by nimodipine.
Figure 2.

VGCC expression and activity are increased in older neurons. A, Confocal images of hippocampal neurons—fixed at 14 DIV and 28 DIV and stained with anti-Cav1.2 antibodies—reveal brighter somatic Cav1.2 fluorescence in older neurons. Bar graph, Quantitation confirms higher relative Cav1.2 fluorescence in 28 DIV cells (n = 24) than in 14 DIV cells (n = 46, p < 0.01). B, Voltage-activated, whole-cell normalized Ca2+ currents (top) increase in 28 DIV neurons compared with 14 DIV neurons. A significant number of 28 DIV neurons exhibit elevated current maxima (bottom). Depolarization was from −70 to +50 mV in 20 mV steps. C, Time course of free Ca2+ elevations after 90 mm K+ depolarization (as in Fig. 1) in individual 28 DIV hippocampal neurons (gray) and their average (red; n = 29) compared with representative Ca2+ trace induced by 100 μm NMDA in cells of the same age (n = 26). D, Maximal sustained concentration of free Ca2+ in individual neurons after 90 mm K+ depolarization in younger (14 DIV) and older (28 DIV) cells and their corresponding death rate.
VGCC-mediated calcium loading and mitochondrial dysfunction
The general correlation between calcium loading and cell death (cf. Fig. 1C) is reminiscent of the well established link between glutamate-induced, calcium overload-dependent mitochondrial dysfunction and excitotoxic death (Nicholls, 2009). This raises the question as to whether VGCC-mediated injury is mechanistically analogous. Twenty-eight DIV cultures enriched in vulnerable cells were used to test whether Ca2+entering via VGCCs can accumulate in and damage mitochondria. In a first approach, these cells were pretreated before VGCC activation with the protonophore FCCP (carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone), which depolarizes mitochondria and thus prevents mitochondrial calcium accumulation (Stout et al., 1998). This pretreatment strongly enhanced depolarization-induced cytosolic Ca2+ elevations so that they became quite large in a subset of cells (Fig. 3A). This response indicates that much of the Ca2+ entering through VGCCs is sequestered in mitochondria.
Figure 3.
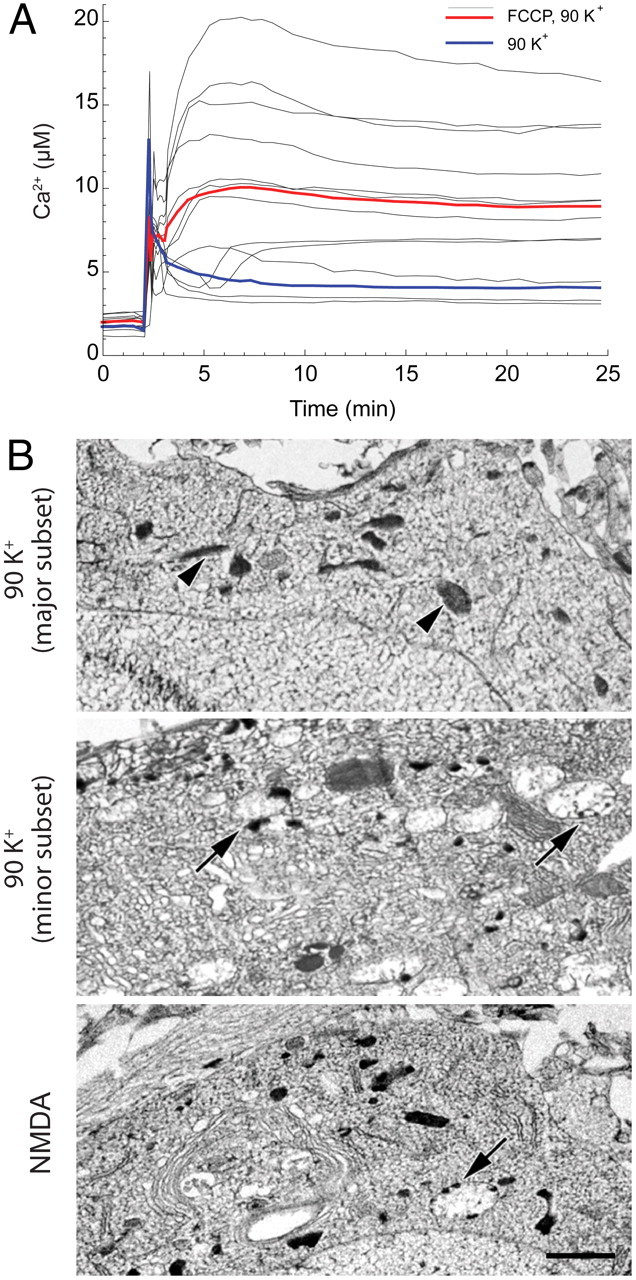
VGCC-mediated calcium overload and mitochondrial structural damage. A, In neurons pretreated with FCCP (1 μm) to eliminate mitochondrial calcium accumulation, 90 mm K+ depolarization (as described in Fig. 1) leads to rapid cytosolic Ca2+ deregulation (∼10 μm) in 28 DIV neurons. Bold traces are the averages from representative coverslips for 90 mm K+ depolarization with (n = 12) or without (n = 14) FCCP. B, Electron micrographs of freeze-dried cryosections prepared from 28 DIV cultured hippocampal neurons rapidly frozen immediately after 30 min exposure to 90 mm K+ (as in Fig. 1) or 100 μm NMDA. Top, Cell body with normal mitochondrial structure (arrowheads). Image is representative of the dominant population of depolarized neurons. Middle, Cell body with numerous swollen mitochondria (arrows), almost always containing calcium phosphate inclusions along the inner aspect of the mitochondrial membrane, illustrates characteristic structural damage in the minor subset of cells. Bottom, Typical cell body after NMDA exposure exhibits swollen mitochondria with inclusions (arrows) similar to those seen in the minor subset of depolarized neurons (compare with middle panel). Scale bar (for all panels), 1 μm.
Electron microscopy provided further evidence that VGCC-mediated Ca2+ loading can damage mitochondria. As discussed previously (Pivovarova and Andrews, 2010), NMDA-mediated mitochondrial Ca2+ overload is generally accompanied by dramatic structural changes, including massive mitochondrial swelling and the formation of calcium-rich inclusions within mitochondrial matrices (see, for example, Fig. 3B, bottom). These changes in mitochondrial structure have proven to be a diagnostically useful predictor of delayed cell death (Pivovarova et al., 2008). In contrast to cells exposed to NMDA, the majority of strongly depolarized neurons maintained normal mitochondrial morphology (Fig. 3B, top), consistent with limited mitochondrial calcium accumulation. However, a fraction of cells similar in size to the previously mentioned minor subset with elevated cytosolic Ca2+ contained a large number of swollen, damaged, and inclusion-containing mitochondria (Fig. 3B, middle). Thus, structural observations suggest that, even though depolarization-induced mitochondrial calcium loads do not in general exceed the mitochondrion's loading capacity, Ca2+ entry through VGCCs is capable of triggering mitochondrial damage in those cases where entry is unusually strong.
The evident difference in cell-to-cell structural responses to depolarization allowed cells to be partitioned into two objectively defined subsets: (1) a dominant subset displaying normal subcellular ultrastructure (Fig. 3B, top); and (2) a second, minor subset containing abundant swollen, structurally damaged mitochondria (Fig. 3B, middle). As a measure of the large structural differences between members of these subsets, the average cross-sectional area of mitochondria in the dominant population was 0.164 ± 0.007 (SD) μm2, compared with 0.611 ± 0.053 μm2 (p < 0.001) in the minor population.
An additional, complementary approach examined the effect of VGCC-dependent calcium loading on the MMP. It is well established (Schinder et al., 1996; Vergun et al., 1999) that toxic NMDAR activation of hippocampal neurons leads to strong MMP depolarization. In contrast, we found that VGCC activation had almost no effect on the MMP in the majority of 14 DIV neurons (Fig. 1E), but did induce a delayed increase of Rh123 fluorescence in a subset (∼30%) of 28 DIV neurons (Fig. 4A) whose size matches the death rate under these conditions (Fig. 2D). When mitochondrial overload was accelerated by blocking mitochondrial Ca2+ release with CGP 37157, an inhibitor of the mitochondrial Na+/Ca2+ exchanger, the delayed onset of depolarization was significantly shortened (Fig. 4B).
Figure 4.
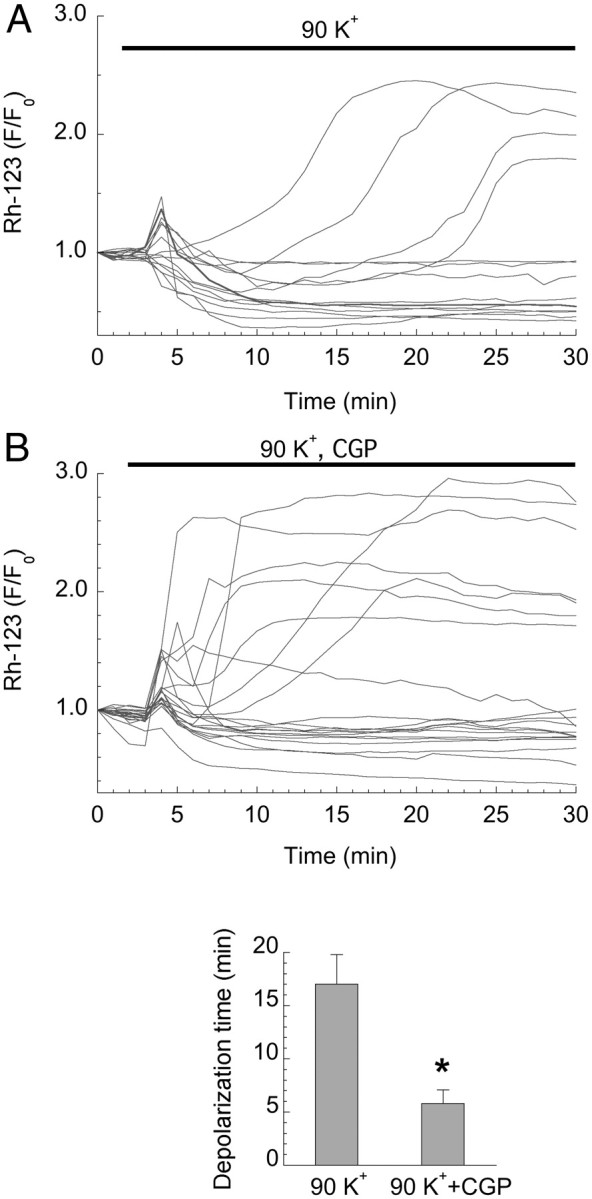
VGCC-mediated calcium overload and mitochondrial dysfunction. A, Time courses of Rh123 fluorescence (F/F0) in 28 DIV neurons reveal MMP depolarization in ∼30% of cells after 90 mm K+ depolarization (as described in Fig. 1). Depolarization is typically delayed by 10–20 min. B, Enhancing mitochondrial calcium loading by blocking the mitochondrial Na+/Ca2+ exchanger with CGP 37157 (10 μm) significantly accelerates mitochondrial depolarization (bar graph, p < 0.01), but does not change the fraction of depolarized cells, ∼35%, which is consistent with the minor subset exhibiting structural damage. Traces from two replicate experiments.
As a final and direct test of whether mitochondria can accumulate damaging amounts of calcium that enter through VGCCs, EPMA was used to measure and compare NMDA- and depolarization-induced changes in total calcium loading in 28 DIV cells. Strong depolarization led to only a slight increase in cytoplasmic total calcium in the dominant population of cells (Table 1, Row 1 vs Row 2); this increase was much lower than the total cytoplasmic calcium elevations induced by excitotoxic NMDA (Table 1, Row 4), but reminiscent of the generally small depolarization-induced increases in free cytosolic Ca2+. Consistent with the variability of the free Ca2+ response to depolarization (Fig. 1A), neurons of the minor subset characterized by severely damaged mitochondria, and only these neurons, exhibited strongly elevated cytoplasmic total calcium (Table 1, Row 3).
Table 1.
Effect of depolarization on elemental concentrations in cytoplasm and mitochondria of cultured 28 DIV hippocampal neurons
| Row # | n | (mmol/kg dry weight) |
||||||
|---|---|---|---|---|---|---|---|---|
| Na | Mg | Cl | P | K | Ca | |||
| Cytoplasm | ||||||||
| 1 | Control | 45 | 23 ± 4 | 39 ± 2 | 63 ± 5 | 483 ± 15 | 511 ± 16 | 2.2 ± 1.2 |
| 90 K+ | ||||||||
| 2 | Major subseta | 46 | 13 ± 3 | 25 ± 2* | 201 ± 16* | 335 ± 18* | 574 ± 24 | 3.8 ± 1.2 |
| 3 | Minor subsetb | 35 | 42 ± 5* | 16 ± 2* | 239 ± 16* | 236 ± 16* | 469 ± 25 | 16.0 ± 3.0* |
| 4 | NMDA | 47 | 644 ± 73* | 25 ± 4* | 484 ± 55* | 333 ± 28* | 67 ± 12* | 41.0 ± 3.0* |
| Mitochondria | ||||||||
| 5 | Control | 46 | 9 ± 3 | 29 ± 2 | 23 ± 2 | 328 ± 9 | 326 ± 12 | 0.1 ± 0.8c |
| 90 K+ | ||||||||
| 6 | Major subseta | 49 | 6 ± 2 | 28 ± 3 | 81 ± 8* | 304 ± 11 | 313 ± 17 | 1.4 ± 0.6 |
| 7 | Minor subsetb | 35 | 8 ± 2 | 53 ± 5* | 85 ± 12* | 643 ± 48* | 290 ± 21 | 650 ± 95* |
| 8 | NMDA | 39 | 268 ± 24* | 43 ± 5* | 229 ± 21* | 869 ± 58* | 38 ± 4* | 1057 ± 99* |
Cultures (28 DIV) were frozen immediately after 30 min exposure to high K+ (90 mm KCl, 1 μm Bay K-8644 in 10 mm Ca2+ HBSS) in the presence of 20 μm MK-801, 10 μm CNQX, or to 100 μm NMDA in Mg-free HBSS in the presence of 10 μm nimodipine. Data are given as mean ± SEM in mmol/kg dry weight. The number of neurons analyzed was 10–15 for each condition. n is the number of analyses.
aCells characterized by normal subcellular structure.
bCells characterized by abundant structurally damaged mitochondria.
cBelow detection limit.
*Different from control (p < 0.05) for cytoplasm by one-way ANOVA with post hoc Dunnett's multiple-comparisons test, and for mitochondria by nonparametric Kruskal–Wallis rank ANOVA with post hoc Dunn's test for unbalanced data.
Mitochondria in the dominant subset of cells accumulated little calcium following depolarization (Table 1, Row 5 vs Row 6), which is consistent with the small elevations in cytoplasmic calcium. Thus, the calcium content of the structurally normal cells of this population clustered around relatively low concentrations (Fig. 5A, open circles). However, in the minor subset with elevated cytoplasmic calcium, the concentration of mitochondrial calcium was strongly increased (Fig. 5A, filled circles), approaching that induced by NMDA (Fig. 5B; Table 1, Row 7 vs Row 8). The punctate, heterogeneous distribution of calcium within these mitochondria resembled that typically observed after strong mitochondrial calcium loading (compare Fig. 3B, middle vs bottom) (Pivovarova et al., 2004; Kristian et al., 2007). Importantly, there was a linear correlation between cytoplasmic and mitochondrial calcium concentrations in neurons of the minor subset (Fig. 5A, filled circles). In summary, structural, functional, and analytical data support the conclusion that VGCC-mediated Ca2+ entry is fully capable of inducing mitochondrial dysfunction, but generally does not do so because the mitochondrial calcium load, limited by the relatively low Ca2+ permeability of L-type VGCCs, is too small.
Figure 5.

VGCC-mediated calcium loading and mitochondrial calcium accumulation. EPMA reveals that 90 mm K+ exposure (as described in Fig. 1) induces calcium accumulation in the cytoplasm and mitochondria of the minor depolarization-responsive subset (see also Table 1, Rows 3 and 7) of 28 DIV neurons. A, Cell-by-cell correlation plot of mean concentrations of total calcium in cytoplasm versus mitochondria of individual cells from the minor subset characterized by damaged mitochondria (filled circles) and the dominant subset characterized by normal structure (open circles). Each data point represents the average of 3–5 measurements in the cytoplasm or 3–5 mitochondria from an individual neuron. Cytoplasmic and mitochondrial calcium concentration elevations in the minor subset are well correlated (r2 = 0.76). B, Calcium concentration in individual mitochondria of the minor subset are significantly elevated compared with control, with concentrations in most members of this group comparable to that induced by 100 μm NMDA. Each data point represents a single measurement in an individual mitochondrion. The number of data points at each condition is given in parentheses. C, D, Intracellular concentrations of Na and K were not affected by 90 mm K+ depolarization, including in cells with elevated calcium, but were reversed after NMDA exposure (p < 0.001). EPMA data are means ± SEM.
A role for sodium in VGCC-mediated injury?
In addition to Ca2+ overload, there is clear evidence that neuronal vulnerability to NMDA is influenced by changes in the cytosolic concentrations of other ions, Na+ being particularly important. Elevated cytosolic Na+ increases Na+/K+-ATPase pump activity, thereby creating an additional ATP demand. In some circumstances this will lead to energy deficiency, which in turn will exacerbate mitochondrial dysfunction and enhance neuronal death (Nicholls, 2008). Thus, elevated Na+ might represent an additive factor that raises vulnerability to NMDAR activation. However, EPMA measurements in our cells show that in contrast to the effects of NMDA, which reverses the normal intracellular sodium/potassium distribution, strong depolarization induced at most minor changes in total sodium or potassium concentrations in both compartments of both subsets (Table 1), and in particular in the minor subset that exhibits high concentrations of calcium and damaged mitochondria (Fig. 5C,D). This observation is consistent with the relative cation permeabilities of NMDARs, which conduct Na+ and K+ as well as Ca2+, compared with VGCCs, which are highly Ca2+ selective. It further suggests that ATP depletion is likely to have limited impact on the depolarization-dependent component of neuronal injury, at least during the acute phase.
Discussion
In this study we have demonstrated that in the majority of cultured hippocampal or cortical neurons, even maximal VGCC activation (90 mm K+ plus Bay K 8644) induces limited calcium loading and minimal NO production. This outcome is comparable to that evoked by sublethal (10 μm) NMDA exposure but much milder than the effects of toxic NMDA. Consequently, VGCC-mediated Ca2+ entry is minimally toxic. Moreover, alternative independent routes of Ca2+ entry such as reverse mode NCX, which produces intermediate levels of Ca2+ loading, or ionophore (ionomycin) exposure, which produces very high levels of Ca2+ loading, also induced death rates corresponding to the degree of calcium loading. The results recapitulate the now generally observed linkage between calcium loading and toxicity, thereby providing new insight into the longstanding puzzle as to why Ca2+ influx via VGCCs is so much less toxic than influx via NMDARs. In addition, we find that depolarization-induced calcium loading is highly variable from one neuron to the next, and thus there exists, at least in culture, a small but significant number of cells that exhibit strong calcium loading and attendant toxicity. In these cells, injury appears to follow the canonical excitotoxicity pathway involving mitochondrial calcium overload, structural damage, and subsequent dysfunction.
In view of the results just summarized, it is timely to reconsider the relationship between generalized Ca2+ loading and the known, selective toxicity of NMDAR-mediated Ca2+ entry, in particular emphasizing that present data offer a logical reconciliation of the source specificity and Ca2+ loading hypotheses. Specifically, our results indicate that, previous reports aside (Sattler et al., 1998), it is not physically possible, except in rare cases, to transport enough Ca2+ through VGCCs to Ca2+ load mitochondria to anywhere near toxic levels. In contrast, NMDARs are exceptionally toxic at least partly because they allow a much larger Ca2+ flux than VGCCs or, for that matter, any other plausible route of Ca2+ entry. However this view does not preclude additional, synergistic toxicity due to selective activation of specific signal transduction pathways. Perhaps the best example of such synergy might be the death mechanism—widely accepted and confirmed in our cells—that depends on nNOS/PSD coupling (Forder and Tymianski, 2009). It remains to be seen whether other death pathways can be explained within this framework. Examples include NMDAR-dependent toxicity selectively through extrasynaptic receptors (Stanika et al., 2009; Hardingham and Bading, 2010) or in cells lacking a PSD-like signaling complex (Soriano et al., 2008).
Because our approach facilitated single cell analysis, we were able to detect the small subset of vulnerable neurons that responded to depolarization with strong Ca2+ accumulation. Cells of this subset accumulated Ca2+—mainly through L-type VGCCs, since Ca2+ elevations were largely blocked by nimodipine—to concentrations that were two- to threefold higher than in the rest of the cells. This subset of neurons increased in size in older (28 DIV) cultures, more of which also exhibited higher L-type VGCC expression and larger Ca2+ currents in response to depolarization. The latter finding is consistent with earlier reports that have documented increased age-dependent VGCC-mediated Ca2+ currents in parallel with increased VGCC expression in CA1 hippocampal neurons (Thibault and Landfield, 1996; Thibault et al., 2001).
The age-dependent increase in the fraction of depolarized cells showing high Ca2+ elevations and evident structural damage to mitochondria provided an opportunity to validate the proposition that when VGCC-mediated Ca2+ loading is excessive it will be toxic mainly via the mitochondrial dysfunction pathway. The logic is that the structural damage typical of NMDA-induced excitotoxic injury should be a distinguishing characteristic of those neurons that show large VGCC-dependent Ca2+ elevations, and that these two correlated characteristics should be more frequent in older cultures. Indeed, in the minor subset of 28 DIV depolarized cells characterized by swollen, structurally damaged mitochondria, the level of total mitochondrial calcium was many hundred times higher than in the nonresponsive population. Moreover, mitochondria were readily depolarized in a fraction of cells similar in size to the depolarization-induced death rate, viz., ∼30%. These observations imply a pivotal role for mitochondrial calcium overload, a role that was demonstrated by inhibiting mitochondrial calcium uptake (with FCCP), which enhances cytosolic Ca2+ levels and by inhibiting mitochondrial Ca2+ release (with CGP 37157), which amplifies mitochondrial calcium overload and thus accelerates dysfunction. Our new results complement previous reports from ourselves and others demonstrating a significant, synergistic contribution of VGCC-mediated Ca2+ deregulation to NMDA-dependent excitotoxicity in older neurons (Brewer et al., 2007; Stanika et al., 2009), and are consistent with the emerging view that NMDARs and VGCCs are organized at postsynaptic membranes as independent signaling complexes (Di Biase et al., 2008). Increases in VGCC-mediated Ca2+ transients have also been implicated in Ca2+ dysregulation during brain aging and in age-related dementia (Thibault et al., 1998, 2007). The age dependence of channel expression raises, not for the first time, a cautionary note concerning the importance of accounting for the influence of the developmental program on any given laboratory's culture models of neurodegenerative conditions.
Beyond issues of brain aging, present findings are important because they strongly suggest that Ca2+ entering through routes other than NMDARs may play a role in calcium-dependent neurodegeneration. Without minimizing the primacy of glutamate receptors in the majority of brain injuries, it seems plausible that contributions from other pathways will have significant implications for devising therapies for a variety of neurodegenerative conditions in which mitochondrial Ca2+ homeostasis is compromised. In Parkinson's disease for example, dopaminergic neurons of the substantia nigra are selective vulnerable because pacemaking through L-type VGCCs marginalizes Ca2+ homeostasis and promotes mitochondria-derived oxidative stress (Guzman et al., 2010), while in neurons deficient in the Parkinson's-associated mitochondrial gene PINK-1, VGCC activation is sufficient to induce mitochondrial dysfunction and neurodegeneration (Gandhi et al., 2009). As another clinically important example, Ca2+-permeable AMPA receptors—whether endogenously expressed (Carriedo et al., 2000) or acquired by ischemia-exposed CNS neurons (Kwak and Weiss, 2006)—promote injury by mechanisms similar to NMDA exposure.
Most of the experiments described here were performed on primary hippocampal neurons in culture. Because much of the data pointing to route-specific injury mechanisms has been obtained on cortical preparations (Sattler et al., 1998, 1999), we repeated some of the most critical Ca2+ imaging experiments on cortical neurons. As the results were essentially similar for both cortical and hippocampal neurons, we find no evidence for tissue-specific differences in the calcium overload response.
Finally, by way of technical commentary, our Ca2+ imaging results, obtained using low-affinity Ca2+ probes, are generally consistent with several earlier studies that used similar Ca2+ indicators in neuronal preparations (Carriedo et al., 1998; Stout and Reynolds, 1999). Additional insight comes from direct EPMA measurements of total intracellular calcium. Beyond mere validation, EPMA measurements of total calcium in specific compartments—individual mitochondria being a prime example—when combined with fluorescence assays of free Ca2+, are especially informative for processes like calcium-dependent toxicity where knowing the total amount of calcium sequestered and its location is every bit as critical as free Ca2+ concentration changes. In this context, the EPMA approach (Fernandez-Segura and Warley, 2008) has significant advantages over other methods for assaying total calcium (Pivovarova and Andrews, 2010). Among these are the imaging capabilities of the electron microscope, which provide resolution well beyond the single-organelle level—and therefore certainty regarding the cell type and subcellular compartment analyzed while not being limited to ensemble averages—together with well documented, reliable quantitation routines and sub-millimolar sensitivity.
Footnotes
This research was supported by the Intramural Research Program of the NIH, NINDS. We are grateful to Christine A. Winters and Dr. Paul E. Gallant for excellent technical assistance.
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- Abramov AY, Duchen MR. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim Biophys Acta. 2008;1777:953–964. doi: 10.1016/j.bbabio.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med. 2009;15:89–100. doi: 10.1016/j.molmed.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer LD, Thibault O, Staton J, Thibault V, Rogers JT, Garcia-Ramos G, Kraner S, Landfield PW, Porter NM. Increased vulnerability of hippocampal neurons with age in culture: temporal association with increases in NMDA receptor current, NR2A subunit expression and recruitment of L-type calcium channels. Brain Res. 2007;1151:20–31. doi: 10.1016/j.brainres.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Carriedo SG, Yin HZ, Sensi SL, Weiss JH. Rapid Ca2+ entry through Ca2+-permeable AMPA/Kainate channels triggers marked intracellular Ca2+ rises and consequent oxygen radical production. J Neurosci. 1998;18:7727–7738. doi: 10.1523/JNEUROSCI.18-19-07727.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriedo SG, Sensi SL, Yin HZ, Weiss JH. AMPA exposures induce mitochondrial Ca2+ overload and ROS generation in spinal motor neurons in vitro. J Neurosci. 2000;20:240–250. doi: 10.1523/JNEUROSCI.20-01-00240.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Biase V, Obermair GJ, Szabo Z, Altier C, Sanguesa J, Bourinet E, Flucher BE. Stable membrane expression of postsynaptic CaV1.2 calcium channel clusters is independent of interactions with AKAP79/150 and PDZ proteins. J Neurosci. 2008;28:13845–13855. doi: 10.1523/JNEUROSCI.3213-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Segura E, Warley A. Electron probe X-ray microanalysis for the study of cell physiology. Methods Cell Biol. 2008;88:19–43. doi: 10.1016/S0091-679X(08)00402-0. [DOI] [PubMed] [Google Scholar]
- Forder JP, Tymianski M. Postsynaptic mechanisms of excitotoxicity: Involvement of postsynaptic density proteins, radicals, and oxidant molecules. Neuroscience. 2009;158:293–300. doi: 10.1016/j.neuroscience.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, Duchen MR, Abramov AY. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greve MW, Zink BJ. Pathophysiology of traumatic brain injury. Mt Sinai J Med. 2009;76:97–104. doi: 10.1002/msj.20104. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyrc K, Handran SD, Rothman SM, Goldberg MP. Ionized intracellular calcium concentration predicts excitotoxic neuronal death: observations with low-affinity fluorescent calcium indicators. J Neurosci. 1997;17:6669–6677. doi: 10.1523/JNEUROSCI.17-17-06669.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiedrowski L, Czyz A, Baranauskas G, Li XF, Lytton J. Differential contribution of plasmalemmal Na/Ca exchange isoforms to sodium-dependent calcium influx and NMDA excitotoxicity in depolarized neurons. J Neurochem. 2004;90:117–128. doi: 10.1111/j.1471-4159.2004.02462.x. [DOI] [PubMed] [Google Scholar]
- Kristian T, Pivovarova NB, Fiskum G, Andrews SB. Calcium-induced precipitate formation in brain mitochondria: composition, calcium capacity, and retention. J Neurochem. 2007;102:1346–1356. doi: 10.1111/j.1471-4159.2007.04626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak S, Weiss JH. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr Opin Neurobiol. 2006;16:281–287. doi: 10.1016/j.conb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Li V, Brustovetsky T, Brustovetsky N. Role of cyclophilin D-dependent mitochondrial permeability transition in glutamate-induced calcium deregulation and excitotoxic neuronal death. Exp Neurol. 2009;218:171–182. doi: 10.1016/j.expneurol.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks JD, Boriboun C, Wang J. Mitochondrial nitric oxide mediates decreased vulnerability of hippocampal neurons from immature animals to NMDA. J Neurosci. 2005;25:6561–6575. doi: 10.1523/JNEUROSCI.1450-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. Oxidative stress and energy crises in neuronal dysfunction. Ann N Y Acad Sci. 2008;1147:53–60. doi: 10.1196/annals.1427.002. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim Biophys Acta. 2009;1787:1416–1424. doi: 10.1016/j.bbabio.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Andrews SB. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010;277:3622–3636. doi: 10.1111/j.1742-4658.2010.07754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Hongpaisan J, Andrews SB, Friel DD. Depolarization-induced mitochondrial Ca accumulation in sympathetic neurons: spatial and temporal characteristics. J Neurosci. 1999;19:6372–6384. doi: 10.1523/JNEUROSCI.19-15-06372.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Nguyen HV, Winters CA, Brantner CA, Smith CL, Andrews SB. Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J Neurosci. 2004;24:5611–5622. doi: 10.1523/JNEUROSCI.0531-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Stanika RI, Watts CA, Brantner CA, Smith CL, Andrews SB. Reduced calcium-dependent mitochondrial damage underlies the reduced vulnerability of excitotoxicity-tolerant hippocampal neurons. J Neurochem. 2008;104:1686–1699. doi: 10.1111/j.1471-4159.2007.05080.x. [DOI] [PubMed] [Google Scholar]
- Sattler R, Charlton MP, Hafner M, Tymianski M. Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J Neurochem. 1998;71:2349–2364. doi: 10.1046/j.1471-4159.1998.71062349.x. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Martel MA, Papadia S, Vaslin A, Baxter P, Rickman C, Forder J, Tymianski M, Duncan R, Aarts M, Clarke P, Wyllie DJ, Hardingham GE. Specific targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ ligand. J Neurosci. 2008;28:10696–10710. doi: 10.1523/JNEUROSCI.1207-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, Andrews SB. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci U S A. 2009;106:9854–9859. doi: 10.1073/pnas.0903546106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout AK, Reynolds IJ. High-affinity calcium indicators underestimate increases in intracellular calcium concentrations associated with excitotoxic glutamate stimulations. Neuroscience. 1999;89:91–100. doi: 10.1016/s0306-4522(98)00441-2. [DOI] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272:1017–1020. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- Thibault O, Porter NM, Chen KC, Blalock EM, Kaminker PG, Clodfelter GV, Brewer LD, Landfield PW. Calcium dysregulation in neuronal aging and Alzheimer's disease: history and new directions. Cell Calcium. 1998;24:417–433. doi: 10.1016/s0143-4160(98)90064-1. [DOI] [PubMed] [Google Scholar]
- Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21:9744–9756. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Gant JC, Landfield PW. Expansion of the calcium hypothesis of brain aging and Alzheimer's disease: minding the store. Aging Cell. 2007;6:307–317. doi: 10.1111/j.1474-9726.2007.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M. International Stroke Conference 2009. Translation of high impact targets into clinical trials. February 17–20, 2009, San Diego, CA. IDrugs. 2009;12:217–219. [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergun O, Keelan J, Khodorov BI, Duchen MR. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J Physiol. 1999;519:451–466. doi: 10.1111/j.1469-7793.1999.0451m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]