

Abstract
Corynebacterium diphtheriae is one of the most prominent human pathogens and the causative agent of the communicable disease diphtheria. The genomes of 12 strains isolated from patients with classical diphtheria, endocarditis, and pneumonia were completely sequenced and annotated. Including the genome of C. diphtheriae NCTC 13129, we herewith present a comprehensive comparative analysis of 13 strains and the first characterization of the pangenome of the species C. diphtheriae. Comparative genomics showed extensive synteny and revealed a core genome consisting of 1,632 conserved genes. The pangenome currently comprises 4,786 protein-coding regions and increases at an average of 65 unique genes per newly sequenced strain. Analysis of prophages carrying the diphtheria toxin gene tox revealed that the toxoid vaccine producer C. diphtheriae Park-Williams no. 8 has been lysogenized by two copies of the ωtox+ phage, whereas C. diphtheriae 31A harbors a hitherto-unknown tox+ corynephage. DNA binding sites of the tox-controlling regulator DtxR were detected by genome-wide motif searches. Comparative content analysis showed that the DtxR regulons exhibit marked differences due to gene gain, gene loss, partial gene deletion, and DtxR binding site depletion. Most predicted pathogenicity islands of C. diphtheriae revealed characteristics of horizontal gene transfer. The majority of these islands encode subunits of adhesive pili, which can play important roles in adhesion of C. diphtheriae to different host tissues. All sequenced isolates contain at least two pilus gene clusters. It appears that variation in the distributed genome is a common strategy of C. diphtheriae to establish differences in host-pathogen interactions.
INTRODUCTION
Corynebacterium diphtheriae is an important human pathogen of the genus Corynebacterium and the causal agent of the communicable disease diphtheria (85). Classical diphtheria is an upper respiratory tract illness initially characterized by a sore throat, low-grade fever, and an adherent membrane (called a pseudomembrane) on the tonsils, pharynx, and/or nasal cavity (24). The most prominent virulence factor of toxigenic C. diphtheriae strains is a potent A-B exotoxin named diphtheria toxin. It inhibits protein biosynthesis by ADP-ribosylation of elongation factor EF-2 and kills susceptible host cells (28). As diphtheria toxin is encoded by corynephages, the toxigenicity of C. diphtheriae strains is dependent on their lysogenization by a tox+ corynephage (40). Although the tox gene is part of the phage genome, the regulation of diphtheria toxin expression is under bacterial control, as the corresponding iron-sensing regulator DtxR is encoded by a gene on the C. diphtheriae chromosome. Therefore, transcription of the tox gene is directly linked to bacterial iron homeostasis; i.e., low iron concentrations induce the expression of diphtheria toxin (73).
C. diphtheriae was first visualized in stained specimens from pseudomembranes by the bacteriologist Edwin Klebs in 1883 (35), and in 1884, C. diphtheriae was isolated by Friedrich Loeffler and shown to be the cause of diphtheria (43). In 1890, Emil von Behring isolated the first diphtheria antitoxin from blood samples of an infected horse (84). A few years later, William H. Park and Anna W. Williams isolated a C. diphtheriae strain that produced an unusually large amount of diphtheria toxin, later named the Park-Williams no. 8 (PW8) strain (56). Since 1923, a diphtheria toxoid vaccine has been produced from diphtheria toxin treated with formalin to inactivate the toxicity and to maintain the immunogenicity of the protein (34). C. diphtheriae PW8 and derivatives thereof are widely used for the initial production of diphtheria toxin by submerged fermentation, because of their ability to secrete large amounts of toxin into the culture supernatant (31, 52). C. diphtheriae PW8 is lysogenized by corynephage ωtox+, which moderately differs in its restriction map from the common βtox+ phage (60). The ωtox+ phage integrated into two nontandem attachment sites within the chromosome of C. diphtheriae PW8, suggesting that the enhanced toxin synthesis is due to a gene dosage effect of the tox gene (59). Another prominent C. diphtheriae strain is C7(β)tox+, which was introduced into laboratory research on diphtheria in 1953. This strain is based on the originally avirulent C. diphtheriae culture no. 770 (17) and was lysogenized experimentally with corynephage βtox+ in a study on phage-host relationships of C. diphtheriae strains (3). Since then, C. diphtheriae C7(β)tox+ has been used worldwide as a reference strain for genetic research on iron metabolism and the regulation of tox gene expression. An initial genome comparison of C. diphtheriae C7(−) and C. diphtheriae PW8 based on genomic hybridization showed remarkable differences in the distribution of predicted pathogenicity islands (PAIs) and provided the first insights into the diversity of clinical isolates (31).
Up to now, diphtheria has been very effectively controlled in developed countries by an efficient immunization program (83). However, the disease has made a dramatic return in recent years, in particular within Eastern Europe. The largest outbreak since the advent of mass immunization started within Russia and the newly independent states of the former Soviet Union in the 1990s (12). In 2003, the genome of a clinical isolate related to this outbreak (C. diphtheriae NCTC 13129) was sequenced at the Sanger Institute to identify candidate virulence factors besides the toxin itself, like iron transport systems and fimbrial proteins (7).
Although C. diphtheriae is of great medical importance and (genetic) research has been performed for more than a century, very little is currently known about the molecular basis of pathogenicity and factors contributing to virulence of nontoxigenic C. diphtheriae isolates. In the present study, we extended the genetic knowledge of the species C. diphtheriae by performing comparative analysis of the complete genome sequences of isolates from patients with classical diphtheria, endocarditis, or pneumonia. In the following sections, we present the results of this sequencing project and the comparison of 13 genome sequences, including those of the widely used strains C. diphtheriae PW8 and C. diphtheriae C7(β)tox+. These data open the way to describe the genome of C. diphtheriae at the species level by pangenomics. The microbial pangenome is defined as the full complement of genes in a bacterial species and comprises the “core genome,” containing genes present in all isolates of a species, and the “dispensable genome,” containing genes present only in a subset of genomes (48). This global view on the gene content of C. diphtheriae provides a more accurate account of features associated with the lifestyle and virulence of a human pathogen (26). Moreover, this pangenomics project provides a novel perspective on the evolution of the human-pathogenic species C. diphtheriae.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All C. diphtheriae strains sequenced in this study are listed in Table 1. The isolates were provided by the strain collection of the University of Rio de Janeiro State (Rio de Janeiro, Brazil). C. diphtheriae PW8 and C7(β)tox+ were obtained from the strain collection of the Food and Drug Administration (Bethesda, MD). All C. diphtheriae strains were routinely grown at 37°C in brain heart infusion (BHI) broth or on Columbia agar supplemented with 5% sheep blood.
Table 1.
Overview of sequenced C. diphtheriae strains and general features of the genome sequences
| Strain | Origin of strain | Genome size (bp) | No. of genes | No. of singletons | No. of transposases | Types of CRISPRsa | GenBank accession no. | Reference(s) |
|---|---|---|---|---|---|---|---|---|
| NCTC 13129 | Isolated from a diphtheria patient in the United Kingdom, 1997; tox+ | 2,488,635 | 2,368 | 124 | 36 | I (7); II (26) | BX248353 | 6, 8 |
| C7(β)tox+ | Derivative of the avirulent isolate C7, 1954; widely used laboratory strain; tox+ | 2,499,189 | 2,350 | 126 | 69 | I (6) | CP003210 | 3, 15 |
| PW8 | Isolated from a diphtheria patient in New York, 1896; widely used toxoid vaccine producer; tox+ | 2,530,683 | 2,361 | 101 | 79 | III (15) | CP003216 | 56 |
| CDC-E8392 | Isolated from a diphtheria patient; originally from the Centers for Disease Control and Prevention; tox+ | 2,433,326 | 2,270 | 52 | 66 | III (12) | CP003211 | 58 |
| 31A | Isolated from a diphtheria patient (vaccinated adult) in Rio de Janeiro, 1978; tox+ | 2,535,346 | 2,402 | 104 | 93 | I (28) | CP003206 | 47 |
| 241 | Isolated from a diphtheria patient in Rio de Janeiro, 1981; toxnegative | 2,426,551 | 2,260 | 6 | 87 | I (15); II (4) | CP003207 | 47 |
| VA01 | Isolated from a diphtheria patient in Rio de Janeiro, 1999; toxnegative | 2,395,441 | 2,196 | 27 | 66 | I (7) | CP003217 | 80 |
| HC01 | Isolated from a blood sample from a patient with fatal endocarditis in Rio de Janeiro, 1993 | 2,427,149 | 2,260 | 7 | 82 | I (15); II (4) | CP003212 | 47 |
| HC02 | Isolated from a blood sample from a patient with endocarditis in Rio de Janeiro, 1999 | 2,468,612 | 2,244 | 69 | 77 | I (5) | CP003213 | 58 |
| HC03 | Isolated from a blood sample from a patient with endocarditis in Rio de Janeiro, 2000 | 2,478,364 | 2,268 | 35 | 83 | III (42) | CP003214 | 58 |
| HC04 | Isolated from a blood sample from a patient with fatal endocarditis in Rio de Janeiro, 2003 | 2,484,332 | 2,280 | 13 | 79 | III (15) | CP003215 | 58 |
| INCA 402 | Isolated from a bronchial wash specimen from a cancer patient with pneumonia in Rio de Janeiro, 2000 | 2,449,071 | 2,235 | 44 | 65 | III (17) | CP003208 | 80 |
| BH8 | Isolated from an inpatient in Rio de Janeiro; antibiotic-resistant strain | 2,485,519 | 2,375 | 85 | 97 | I (1) | CP003209 | This study |
The number of repeats is given in parentheses.
Preparation of chromosomal DNA for genome sequencing.
The purification of chromosomal DNA from C. diphtheriae was performed as described previously (80). Briefly, 50-ml aliquots of bacterial cultures grown for 48 to 72 h in BHI broth were centrifuged at 4°C and 2,000 × g for 20 min. The resulting cell pellets were resuspended in 0.6 ml of Tris-NaCl buffer (10 mM Tris [pH 7.0], 10 mM EDTA, 300 mM NaCl) and transferred to VK01 Precellys lysing tubes. The bacterial cells were lysed by means of a Precellys 24-Dual tissue homogenizer using two cycles of 6,500 rpm for 15 s with an interval of 30 s. The chromosomal DNA was subsequently purified by extraction with phenol-chloroform-isoamyl alcohol (25:24:1) and precipitated with ethanol. For the isolation of chromosomal DNA from C. diphtheriae PW8 and C7(β)tox+, both strains were grown overnight in 3 ml of BHI medium at 37°C, with shaking. Cells were pelleted by centrifugation at 5,000 × g for 5 min, and the supernatant was discarded. The cell pellet was resuspended in phosphate-buffered saline (PBS) containing 10 mg/ml lysozyme and incubated at 37°C for 30 min. Following the incubation, the cells were pelleted as described above and the supernatant was discarded. The cell pellet was resuspended in 25 μl of PBS, and chromosomal DNA was isolated using the MasterPure DNA purification kit (Epicentre Technologies, Madison, WI). DNA concentrations were determined with a Tecan Infinite 200 microplate reader.
Genome sequencing of the selected C. diphtheriae strains.
Purified genomic DNA from C. diphtheriae was sequenced with the Genome Sequencer FLX Instrument and titanium chemistry (Roche Applied Science) using a quarter of a PicoTiter Plate per strain. Single-stranded template DNA libraries were established by using 5 μg of genomic DNA. The preparation of DNA libraries was carried out according to standard protocols from Roche Applied Science. DNA concentrations of the DNA libraries were measured with the Agilent RNA 6000 Nano kit. The resulting genomic DNA sequences were assembled with the Newbler Assembler software (version 2.5.3) using default parameters.
For subsequent gap closure, in silico predictions of the contig order were computed by the related reference contig arrangement tool r2cat (30) using the default parameters of the integrated q-gram filter and the C. diphtheriae NCTC 13129 genome sequence as a reference. Based on a sliding-window approach that determines the position of a contig on the reference genome, all matching regions were displayed in an interactive synteny plot, wherein the contigs were oriented automatically according to their matches (30). The remaining gaps in the genome sequences were closed by PCR strategies using genomic template DNAs and Phusion hot-start high-fidelity DNA polymerase (Finnzymes). The PCR assays were performed according to standard protocols from Finnzymes using 1 M betain for efficient denaturation of DNA secondary structures. All contigs and additional DNA sequences were uploaded into the Consed program (20) to finish the genome sequences of the selected C. diphtheriae strains.
Annotation and bioinformatics analysis of the complete genome sequences.
Initial automated annotations of the assembled genome sequences of the C. diphtheriae strains were performed with the GenDB 2.2 system (49). It combines different gene prediction strategies that were executed by means of REGANOR (42), GLIMMER 2.1 (11), and the CRITICA program suite (2) in conjunction with postprocessing by the RBSfinder tool (69). Functional characterization of the predicted proteins was performed by automated searches in public databases, including Swiss-Prot, TrEMBL, Pfam, TIGRFAM, KEGG, COG, CDD, and Interpro (49). Metabolic pathways were annotated by means of in silico reconstructions of metabolic networks with the software CARMEN using metabolic pathway information from the KEGG database and manually curated SBML templates (64). The predicted C. diphtheriae proteins were mapped onto the SBML templates using bidirectional best BLASTP hits and the scoring matrix BLOSUM62 with an E-value cutoff of 1 × 10−10. The comparative annotation tool provided by the software EDGAR was used for manual data curation and a consistent annotation of the sequenced C. diphtheriae genomes (4). Clustered regularly interspaced short palindromic repeats (CRISPRs) and cas genes were detected with the CRISPRFinder tool (23). Secreted proteins were detected with SignalP 4.0 using default settings for Gram-positive bacteria (57). Genomic islands and candidate pathogenicity islands were identified with the pathogenicity island prediction software PIPS (68). For this purpose, PIPS performs a combined analysis based on the presence of the following features: (i) deviation from the mean G+C content, (ii) deviation from the codon usage using Colombo-SIGI-HMM, (iii) prediction of flanking tRNAs based on tRNAscan-SE, (iv) prediction of transposases using HMMER3 and Pfam models, (v) prediction of virulence factors based on mVirDB, and (vi) the absence of the putative islands in nonpathogenic bacteria of related species using Artemis and ACT (68). The genome sequence of the nonpathogenic species Corynebacterium glutamicum ATCC 13032 was used as a reference (33).
Comparative genomics and pangenomics.
All comparative analyses of the sequenced C. diphtheriae genomes were performed with EDGAR software (4). Comparative analysis at the protein level was based on an all-against-all comparison of the predicted proteomes. The algorithm used was BLASTP with the standard scoring matrix BLOSUM62 and an initial E-value cutoff of 1 × 10−5. All BLAST hits were normalized in relation to the best score possible, i.e., the score of a hit of the query gene against itself (4). By dividing the scores of further hits by this best score, a similarity to the best in percent is obtained, the so-called score ratio value (SRV) that reflects the quality of the hit (41). Two genes were considered orthologous when exhibiting a bidirectional best BLAST hit with single SRVs exceeding the precalculated corynebacterial cutoff of 76 (4). The core genome was calculated, based on this information, as the set of genes that are orthologous in all sequenced C. diphtheriae strains. A phylogenetic tree was constructed by aligning all orthologous genes of C. diphtheriae and masking nonmatching parts of the alignments. The remaining data were concatenated and used for the calculation of a distance matrix, which provides the input for the neighbor-joining method as implemented in the PHYLIP package (13). All genes of the reference genome C. diphtheriae C7(β)tox+ were selected as a basis for calculating the pangenome of C. diphtheriae. These genes were compared to those of a second C. diphtheriae genome, and all nonorthologous genes were added to the pangenome. By iteratively repeating this process for all remaining genomes, the gene composition of the final pangenome was deduced (4). The development of the pangenome in dependence on the number of sequenced genomes was calculated by Heaps' law as proposed previously (76). The relevant parameters κ and γ, as well as the confidence intervals, were estimated using the nonlinear least-squares curve fitting with the statistical computing language R.
Whole-genome alignments of the C. diphtheriae sequences were calculated using the software Mauve with default parameters (10). Mauve facilitates the accurate detection of rearrangement breakpoints when genomes have unequal gene contents based on an alignment objective score, called a sum-of-pairs breakpoint score. For comparative analysis of the gene composition of DtxR regulons, DNA motif discovery was performed with a hidden Markov model using previously detected DtxR binding sites (6) as input for the HMMER software suite (14). ClustalW2 (39) was used to align the protein sequences of pilus shaft proteins and their related sortases. Phylogenetic trees of pilin-specific sortases, shaft proteins, tip pilins, and base pilins were generated with the neighbor-joining algorithm using MEGA 4.0 software (72).
Nucleotide sequence accession numbers.
The annotated genome sequences of the C. diphtheriae strains have been deposited in the GenBank database with the following accession numbers: C7(β)tox+, CP003210; PW8, CP003216; CDC-E8392, CP003211; 31A, CP003206; 241, CP003207; VA01, CP003217; HC01, CP003212; HC02, CP003213; HC03, CP003214; HC04, CP003215; INCA 402, CP003208; and BH8, CP003209.
RESULTS
General features of the sequenced C. diphtheriae genomes.
In this study, 12 C. diphtheriae genomes, including the widely used laboratory strain C7(β)tox+ and the prominent toxoid vaccine producer PW8, were sequenced by pyrosequencing with the 454 GS FLX system (Table 1). As infections with C. diphtheriae can cause different diseases in humans (29, 50), the strains selected for genome sequencing were originally isolated from patients with classical diphtheria, blood cultures of patients with endocarditis, and a rare case of pneumonia in a cancer patient (Table 1). The sequencing depth of the C. diphtheriae genomes ranged from coverages of 29-fold to 55-fold, while the average number of assembled contigs ranged from 33 to 73 (data not shown). After gap closure, the genomic sequences were assembled to circular chromosomes 2.395 Mb (C. diphtheriae VA01) to 2.535 Mb (C. diphtheriae 31A) in size (Table 1). The average G+C content of each genome is in the range of 53%, which is consistent with the G+C content detected previously in the genome of C. diphtheriae NCTC 13129 (7). To ensure consistent annotations of all C. diphtheriae genomes, the “comparative annotation function module” of the EDGAR software was used for functional assignments of protein-coding genes (4). Manual annotation of the 12 C. diphtheriae chromosomes and concurrent reannotation of the NCTC 13129 genome (9) revealed a median number of 2,294 protein-coding genes for each strain, with the lowest number, 2,196 genes, annotated in the genome of C. diphtheriae VA01 and the highest number, 2,402 genes, in the genome of C. diphtheriae 31A (Table 1). For the laboratory strain C. diphtheriae C7(β)tox+, annotation of the 2,499,189-bp chromosome revealed 2,350 protein-coding genes, while the chromosome of the toxoid vaccine producer C. diphtheriae PW8 has a size of 2,530,683 bp, with 2,361 predicted protein-coding genes (Table 1).
Phylogenetic tree of the sequenced C. diphtheriae strains.
To evaluate the relationship of the 13 hitherto-sequenced C. diphtheriae strains, a dendrogram based on allelic differences in the following standard set of housekeeping loci (32) was calculated: ATP synthase α chain (atpA), DNA polymerase III α subunit (dnaE), chaperone protein DnaK (dnaK), elongation factor G (fusA), 2-isopropylmalate synthase (leuA), 2-oxoglutarate dehydrogenase E1 and E2 components (sucA), and DNA-directed RNA polymerase β subunit (rpoB). Allelic numbers were assigned by performing BLASTN similarity searches against corresponding genes collected in the multilocus sequence typing (MLST) database mlstdbNet (32) and used to deduce a phylogenetic tree with the PHYLIP package (Fig. 1A). C. diphtheriae INCA 402, isolated from a pneumonia patient and assigned to the biotype belfanti (82), constituted a distinct subline within the phylogenetic tree, separating it from the other sequenced strains originally isolated from cases of diphtheria and endocarditis. The latter strains show a high degree of genetic diversity, even if they were isolated in the same hospital from patients with similar clinical symptoms, like strains HC01, HC02, HC03, and HC04 (Fig. 1A). The most closely related pair of strains comprises the tox-negative isolates C. diphtheriae 241 (diphtheria) and C. diphtheriae HC01 (endocarditis), which show identical MLST profiles (Fig. 1A).
Fig 1.

Phylogenetic trees of hitherto-sequenced C. diphtheriae strains based on allelic profiles of housekeeping genes (A) and variations in the deduced core genome (B). Allelic profiles of the housekeeping genes were determined according to references deposited in the C. diphtheriae MLST database mlstdbNet. The dendrogram was calculated with the PHYLIP package using the unweighted-pair group method with arithmetic mean (UPGMA). The core genome of C. diphtheriae was deduced with EDGAR software and includes 1,632 genes. The dendrogram was calculated with the EDGAR system using the neighbor-joining method.
Another method to determine the phylogenetic relationship of corynebacterial strains is spoligotyping, which is based on arrays of so-called clustered regularly interspaced short palindromic repeats (CRISPRs) (51). These arrays are composed of direct repeats that are separated by nonrepetitive, similar-sized spacers. Together with their associated cas genes, CRISPR arrays can confer resistance to phages by RNA interference-like mechanisms (74). Targets for spoligotyping are the spacer regions between the direct repeats, as variations in the number or nucleotide sequence of spacers may provide patterns for the differentiation between subtypes of bacterial isolates (22). Three different types of CRISPR arrays were detected in the genomes of the sequenced C. diphtheriae strains (Table 1). CRISPR type I is composed of three cas genes (cas1 to cas3), and the number of associated spacers ranged from 1 (C. diphtheriae BH8) to 28 (C. diphtheriae 31A). The type I CRISPR array was detected in the genomes of eight strains (Table 1). C. diphtheriae strains NCTC 13129, 241, and HC01 harbor the additional CRISPR type II, which contains eight cas genes (cas4 to cas11). The number of repeats in these arrays ranged from 4 (C. diphtheriae 241 and HC01) to 26 (C. diphtheriae NCTC 13129). A perfect match of both CRISPR types was detected in the closely related strains C. diphtheriae 241 and HC01, as these genomes encode exactly the same CRISPR arrays. CRISPR type III is present in five C. diphtheriae genomes (Table 1), with various numbers of repeats ranging from 12 (C. diphtheriae CDC-E8392) to 42 (C. diphtheriae HC03). The type III CRISPR array is flanked by eight cas genes (cas12 to cas19). The comparison of the identified spacer sequences revealed that only 48 out of the 219 spacers are shared by two or three C. diphtheriae strains (data not shown), supporting the view that CRISPR arrays provide a solid basis to discriminate effectively between different C. diphtheriae isolates (51).
Pairwise comparison of the sequenced C. diphtheriae strains.
To obtain more detailed insights into the genetic diversity of the sequenced C. diphtheriae strains, the software EDGAR was used to calculate the so-called “similarities” (genes shared by two strains), “differences” (genes not shared by a strain pair), and “pair-uniques” (genes only present in two selected strains) for all possible pairs of strains (Fig. 2). The mean number of similarities between two C. diphtheriae strains comprises 1,903 ± 54 orthologous genes, while the mean number of differences comprises 644 genes (with a high standard deviation, ±134), indicating the large variability of the gene content in the selected C. diphtheriae strains. The highest number of orthologous genes (i.e., 2,201) and the smallest number of differences (i.e., 64) were calculated for the strain pair C. diphtheriae 241 and C. diphtheriae HC01 (Fig. 2), supporting their close relationship already observed in the phylogenetic tree based on allelic profiles (Fig. 1A). The highest number of differences (838) was detected in the strain pair C. diphtheriae C7βtox+ and C. diphtheriae HC02, although both belong to the same subline of the phylogenetic tree (Fig. 1A). The lowest number of similarities, with only 1,831 shared genes, was calculated for the strain pair C. diphtheriae PW8 and C. diphtheriae 241 (Fig. 2), both isolated from patients with classical diphtheria but in different countries and about 100 years apart (Table 1). The number of pair-uniques was strikingly low, with a mean number of five genes shared by only two C. diphtheriae strains. The detected sets of pair-uniques mainly comprise genes for transposases or their inactivated derivatives and genes encoding uncharacterized transporters (data not shown). The highest number of pair-uniques (101) was detected in the closely related strains C. diphtheriae 241 and C. diphtheriae HC01 (Fig. 2). On the other hand, no pair-uniques were detected in 15 pairs of C. diphtheriae strains, indicating that the current collection of sequenced genomes largely reflects the gene content of the species C. diphtheriae.
Fig 2.

Pairwise comparison of the gene contents of hitherto-sequenced C. diphtheriae strains. Similarities denote the number of genes shared by a particular pair of strains, differences display the number of genes not shared within a pair of strains, and pair-uniques correspond to orthologous genes shared only by a distinct strain pair. All calculations were carried out with the software tool EDGAR. The highest and lowest values of each category are listed and specifically marked.
For further comparative genomics studies, the annotated protein-coding sequences were grouped into three categories: “core genes” (conserved in all sequenced C. diphtheriae strains), “singletons” (genes present in a single strain only), and “distributed genes” (shared by a subset of two or more strains). The bioinformatics analysis of the respective categories provided knowledge of the core genome, strain-specific functions eventually associated with pathogenicity, and the pangenome of the species C. diphtheriae.
Deduced core genome of the species C. diphtheriae.
The number of core genes of C. diphtheriae was determined with the software EDGAR using bidirectional best BLASTP hits with the precalculated score ratio value of 76 as a cutoff. Based on a series of calculations using all genomes individually as a reference, the core genome of the hitherto-sequenced C. diphtheriae strains comprises 1,632 genes that are therefore highly conserved in this species. A phylogenetic tree of the C. diphtheriae strains was constructed using a concatenated multiple alignment of the detected core genes and a distance matrix, which was calculated with the Kimura algorithm (Fig. 1B). This genome-based approach revealed a different phylogenetic relationship of the 13 C. diphtheriae strains compared with the MLST profile (Fig. 1A). The use of a multitude of core genes for genome comparison apparently provided a greater taxonomic resolution of the clinical isolates and showed a close relationship between the widely used strains C. diphtheriae PW8 and C. diphtheriae C7(β)tox+ (Fig. 1B).
To deduce the development of the core genome in dependence on the number of sequenced C. diphtheriae strains, the median number of core genes in each genome was calculated based on the permutation of all possible genome comparisons (Fig. 3). These data revealed that the number of core genes is approaching a curve with 422.803 × e(−x/4.258) + 1,611.762, with x being the number of sequenced C. diphtheriae genomes and e being Euler's number. Hence, the number of core genes present in the species C. diphtheriae will comprise about 1,611 protein-coding genes when adding further genome sequences to the current data set. This value revealed a distinct genetic backbone of the species C. diphtheriae, which includes approximately 70% of the gene repertoire of the sequenced strains. In other words, about 30% of the gene content of C. diphtheriae strains is variable to some extent and therefore belongs to the dispensable portion of the genomes. The number of core genes of C. diphtheriae is notably high compared to the previously published corynebacterial backbone of 835 genes calculated with genomic data from the pathogenic and nonpathogenic species C. diphtheriae, Corynebacterium jeikeium, Corynebacterium efficiens, and C. glutamicum (86). However, this difference can be explained by the very close relationship of the C. diphtheriae isolates belonging to the same corynebacterial species. The core genome of C. diphtheriae comprises genes for components of the central carbon and energy metabolism, biosynthesis routes for amino acids, cofactors, purines, and pyrimidines, the basic machineries involved in cell wall formation, DNA replication, DNA repair, transcription, and protein biosynthesis, as well as conserved transcriptional regulatory systems. A remarkably high number of 493 core genes encode proteins with only poorly characterized functions (data not shown).
Fig 3.

Development of the number of core genes and singletons as a function of the number of sequenced C. diphtheriae strains. The respective numbers were calculated for two strains and then iteratively for an increasing number of sequenced genomes, added one by one. The deduced equations denote the exponential decay model based on the median number of core genes and singletons, when increasing numbers of genomes were compared.
The highly conserved genomic backbone of the species C. diphtheriae was visualized by a whole-genome alignment of the 13 sequenced strains with the software Mauve (Fig. 4). Less conserved areas of the genomes are mostly located around the origin of replication (oriC), indicated by the multitude of short conserved blocks interrupted by nonconserved segments (Fig. 4). These genomic areas often include hot spots of insertion sequences and remnants of these elements. Bacterial insertion sequences and their encoded transposases facilitate the formation of DNA inversions, deletions, and replicon fusions, thereby promoting genomic rearrangements (66). A total number of 979 transposase genes were identified in the 13 C. diphtheriae genomes (Table 1), representing about 3.3% of the protein-coding regions of an individual strain and probably playing a significant role in genome evolution. The number of transposase genes varied from 65 in C. diphtheriae INCA 402 to 97 in the antibiotic-resistant strain C. diphtheriae BH8 (Table 1). A nucleotide sequence comparison among the transposase genes revealed a large diversity of mobile elements present in C. diphtheriae, as the remarkable number of 413 different insertion sequences was found in the genome data. Most elements are characterized by a low copy number and are present in only a few strains. On the other hand, the ISCod5 element represented by DIP0843 and orthologous genes is distributed in all sequenced C. diphtheriae strains, albeit with different copy numbers.
Fig 4.
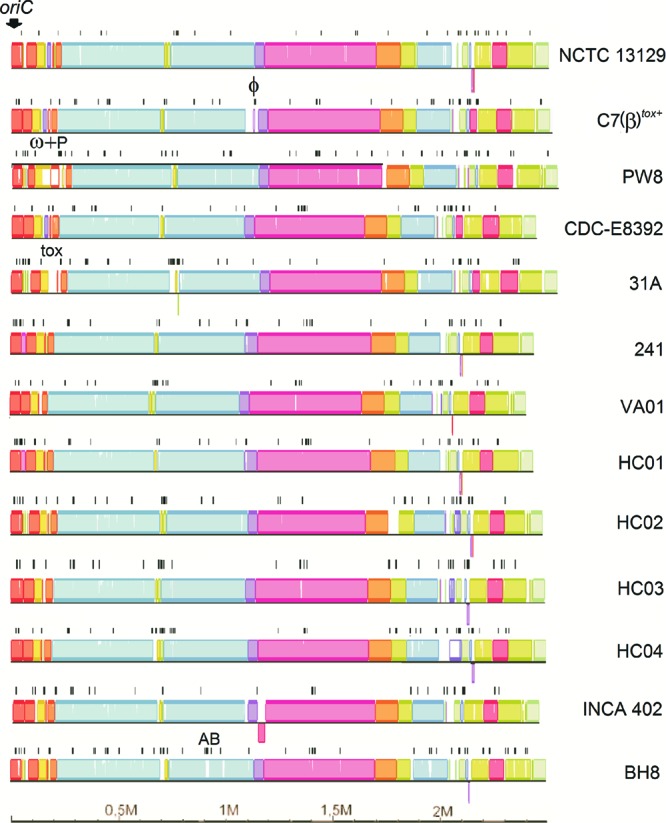
Whole-genome alignment of the 13 sequenced C. diphtheriae strains. The nucleotide sequence alignment was calculated with the software Mauve using the genome of C. diphtheriae C7(β)tox+ as a reference. Each genome is presented in linear view, and homologous DNA segments are shown as colored blocks. The position of the origin of replication (oriC) is indicated. Identified transposase genes are marked by black lines. DNA segments mentioned in the text are labeled as follows: ϕ, additional prophage in C7(β)tox+; ω+P, ω prophages and adjacent pilus gene cluster in PW8; tox, tox+ prophage in 31A; AB, antibiotic resistance gene region in BH8.
To analyze the correlation between the presence of insertion sequences and less conserved areas of the C. diphtheriae genomes, the positions of transposase genes were plotted onto the genome sequences of the 13 C. diphtheriae strains (Fig. 4). A preferred integration of insertion sequences around oriC and between conserved blocks of core genes was observed in all C. diphtheriae genomes. This preference might be due to a gene dosage effect of genes around oriC during DNA replication. Previous studies showed that the copy numbers of origin-proximal genes in rapidly growing cells were 2-fold to 4-fold enhanced compared to those of genes flanking the terminus region (67). Mobile elements are also important components facilitating the spread of antibiotic resistance genes by horizontal gene transfer (63). The genome of the antibiotic-resistant isolate C. diphtheriae BH8 contains a gene region comprising the known resistance determinants cmx, sul1, and tet(W), probably conferring resistances to chloramphenicol, sulfonamides, and tetracyclines in corynebacteria (65). This gene region is also flanked by insertion sequences, suggesting that it has been acquired by C. diphtheriae BH8 via transpositional integration into the chromosome (Fig. 4).
Singletons of the sequenced C. diphtheriae strains.
The bioinformatics detection of singletons revealed the average number of 61 ± 43 strain-specific genes per sequenced C. diphtheriae isolate (Table 1). To characterize the development of the number of singletons in dependence on the number of sequenced C. diphtheriae genomes, the median number of strain-specific genes was determined using the permutation of all possible genome comparisons (Fig. 3). The respective calculation indicates that the number of singletons is approaching a curve with 474.448 × e(−x/2.897) + 65.198, with x being the number of sequenced C. diphtheriae genomes and e being Euler's number. Hence, the median number of singletons estimated to occur in additionally sequenced C. diphtheriae isolates comprises about 65 protein-coding genes. The lowest number of singletons was found in the genome of C. diphtheriae 241, with only 6 out of the 2,260 annotated genes denoted as strain specific (Table 1). However, the number of singletons in C. diphtheriae C7(β)tox+ is approximately twice the calculated mean of 65 singletons per additionally sequenced strain, as the highest number of strain-specific genes (126) was detected in this genome. About 61% of these genes were classified as transposases (14 genes) or prophage-related coding regions (63 genes). The respective corynephage integrated into an attachment site within the tRNALeu gene at about 1.08 Mb on the genomic map of C. diphtheriae C7(β)tox+ (Fig. 4). The presence of the 57-kb prophage region in the chromosome of C. diphtheriae C7(β)tox+ was evident from the whole-genome alignment, as it revealed no similarities to the other genomes at the nucleotide level. In principle, the detected prophage singletons are clustered in the C. diphtheriae genomes and indicate that different phages or remnants thereof are present in the genomes of the selected strains.
In the case of C. diphtheriae PW8, the number of singletons was also above average and calculated as 101 (Table 1). The majority of these genes encode subunits of putative adhesive pili, which play an important role in adhesion of C. diphtheriae to host tissues (46). The respective gene cluster is characterized by numerous mobile elements leading to gene disruptions and deletions in this genomic region (CDPW8_0225 to CDPW8_0252). It is located in the immediate vicinity of the duplicated ω prophage harboring the tox genes for diphtheria toxin (CDPW8_0179 and CDPW8_0220) and displayed no similarities to any of the other genomes (Fig. 4). This result indicates that singletons may contribute to important strain-specific features which are relevant for the pathogenicity of C. diphtheriae.
Deduced pangenome of the species C. diphtheriae.
The pangenome of a bacterial species includes core genes (conserved in all strains), singletons (strain-specific genes), and distributed genes (48). The latter coding regions are shared by at least two strains and are thus part of the variable gene content of C. diphtheriae, in addition to the 793 genes assigned as singletons (Table 1). To calculate the full complement of protein-coding regions in the sequenced C. diphtheriae strains, the number of distributed genes was finally determined as 2,361. Accordingly, the sum total of genes included in the pangenome of the sequenced C. diphtheriae strains comprises 4,786 genes. This number is about three times the size of the deduced core genome. The further trend of the pangenome size was calculated based on Heaps' law: n = κ × Nγ, with N being the number of sequenced genomes (77). According to the number of genes annotated in the sequenced C. diphtheriae genomes, κ was determined as 2,130.67 and γ as 0.306. Hence, the number of genes added to the pangenome with additionally sequenced C. diphtheriae strains will increase with an α of 0.69 per newly sequenced genome (Fig. 5). This value is consistent with the continually increasing number of singletons per sequenced strain (Fig. 3) and indicates an open pangenome for the species C. diphtheriae (77). A pangenome is considered open when each newly sequenced bacterial strain can be expected to reveal some genes unique within the species, regardless of the number of already-analyzed genomes (25).
Fig 5.

Heaps' law plot representing the development of the pangenome of C. diphtheriae. The total number of genes found according to the pangenome analysis is shown for increasing numbers of sequenced C. diphtheriae genomes. Medians of the distributions are shown by squares; permutations are indicated.
Comparison of the sequenced tox+ corynephages.
Prophages harboring the tox gene for diphtheria toxin were identified in C. diphtheriae strains C7(β)tox+, CDC-E8392, PW8, and 31A, in addition to the tox+ prophage known from C. diphtheriae NCTC 13129 (Fig. 6). Two copies of corynephage ωtox+ were detected in the genome of C. diphtheriae PW8, as deduced previously from restriction endonuclease maps of phage DNA (60). The two ωtox+ phages are almost identical, as they show only five nucleotide mismatches in their 36-kb genomes. The nontandem copies of the prophage are separated by a 2-kb gene region coding for a putative membrane protein (CDPW8_0180) that is flanked by two copies of a tRNAArg gene in all C. diphtheriae genomes. The tRNAArg gene located downstream of CDPW8_0180 and its orthologous counterparts usually provide the attachment site attB for corynephages ω and β (61). In the case of C. diphtheriae PW8, the ωtox+ phage integrated into both tRNAArg genes present on either side of CDPW8_0180, explaining the nontandem arrangement of the prophage genomes. Nucleotide sequence alignments of the identified tox+ prophages revealed that the ωtox+ phage of C. diphtheriae PW8 is homologous to corynephage βtox+ that has been integrated into the genome of the previously avirulent strain C. diphtheriae C7 (Fig. 6). This observation confirms an early study demonstrating by restriction mapping that the two phages differ in only three genomic regions (60). Likewise, the prophage detected in C. diphtheriae CDC-E8392 is highly similar to corynephage βtox+, whereas the previously reported prophage of C. diphtheriae NCTC 13129 shows greater divergence from the nucleotide sequence and gene content of the βtox+ phage (Fig. 6).
Fig 6.

Genome alignment of tox+ prophages identified in the sequenced C. diphtheriae strains. The nucleotide sequence alignment was calculated with the software Mauve. The height of the plot denotes the similarities of the aligned DNA sequences. The tox gene is located at the right-hand end of the prophage genome. The proposed modular structure of the corynephage present in C. diphtheriae NCTC 13129 is indicated by annotated brackets.
A remarkably different tox+ corynephage was detected in the genome of C. diphtheriae 31A (Fig. 6). The highest similarity at the nucleotide level to β-like phages is observed at the right-hand end of the prophage genome. This region harbors the diphtheria toxin gene (Fig. 6) and is specified by a decreased G+C content of 42.54%. It is interesting that all tox genes detected in this study showed a perfect nucleotide sequence identity, with the exception of a single nucleotide exchange in the respective gene of C. diphtheriae CDC-E8392. On the other hand, several proteins encoded by genes in the prophage region of C. diphtheriae 31A are homologous to those identified in prophage CULC22IV, which is present in the genome of the closely related tox-negative strain C. ulcerans BR-AD22. The respective prophage has a size of 41 kb, comprises 53 genes, and has been integrated into a tRNAThr gene of Corynebacterium ulcerans (79). It has been proposed previously that the diphtheria toxin gene was acquired by corynephage β due to the terminal location of tox in the genome of the prophage and the significantly decreased G+C content of this gene region (7). The detection of an identical tox gene in the prophage of C. diphtheriae 31A indicates that the acquisition of the diphtheria toxin gene occurred independently in two different corynephages or that gene shuffling occurred frequently in corynephages.
Variations in gene composition of the iron-dependent DtxR regulons.
The diphtheria toxin repressor DtxR of C. diphtheriae is known as an iron-dependent regulator that controls the transcription of the diphtheria toxin gene tox and a complex gene-regulatory network involved in iron homeostasis (6). In the case of a low iron concentration, DtxR is inactivated and transcription of the tox gene is induced (15). As iron is an essential cofactor for proteins involved in important cellular functions, such as respiration and DNA biosynthesis, iron limitation is a common strategy by the mammalian host to suppress bacterial growth (1). Therefore, pathogenic bacteria have to compete for iron in the host to establish an infection, a mechanism that is coupled with the expression of diphtheria toxin in tox+ strains of C. diphtheriae (54). To perform a genome-wide detection of DtxR binding sites, the functional annotations of the C. diphtheriae genome sequences were combined with bioinformatics motif searches based on hidden Markov models (6). This approach resulted in the prediction of 36 different DtxR binding sites in the genome sequences of the 13 C. diphtheriae strains, of which 26 binding sites were detected in front of highly conserved genes or gene clusters (Fig. 7). These highly conserved parts of the DtxR regulon comprise the diphtheria toxin gene in tox+ strains and two iron-dependent ABC-type transport systems: the previously described frgCBAD genes (38) and the newly identified iutABCDE gene cluster. Likewise, genes involved in hemin utilization (hmuTUV), the corresponding hemin binding protein genes (htaABC), and the hmuO gene, which encodes heme oxygenase, are highly conserved (Fig. 7). In addition to the htaA and htaB genes, several strains feature the chtA and chtB genes that code for homologs of HtaA and HtaB and also comprise DtxR binding sites in their promoter regions. Likewise, some C. diphtheriae strains harbor the DtxR-regulated gene clusters irp1ABC and iusABCDE, both encoding ABC-type iron uptake systems (Fig. 7).
Fig 7.

Comparison of predicted DtxR regulons encoded in the sequenced C. diphtheriae genomes. Genes and gene clusters specified by the presence of DtxR binding sites are listed with their proposed physiological functions. The presence of DtxR binding sites is represented by gray boxes. White boxes denote gene clusters and corresponding DtxR binding sites missing in the respective genomes. Specifically marked are the duplication of the tox gene in PW8 (2 × tox), the deletion in the sidBA gene region (ΔsidBA), and the integration of an insertion sequence into the regulatory region of the nitrate reductase gene cluster (IS). Genes assigned to the same cluster are linked with hyphens. The position of the DtxR binding site is marked by double slashes if it is located between two divergently oriented gene clusters. The asterisks label gene clusters with experimental information on DtxR regulation.
Moreover, pathogenic bacteria can synthesize and secret high-affinity iron chelators for efficient iron acquisition, termed siderophores (8). Up to now, the sidBA genes and the ciuEFG gene cluster were assigned functions in siderophore biosynthesis of C. diphtheriae (37, 38). The ciuEFG gene cluster and the associated siderophore-dependent iron uptake system genes ciuABCD are highly conserved parts of the DtxR regulon in all sequenced C. diphtheriae strains, while the sidBA genes were completely lost in the genomes of C. diphtheriae 241, HC01, and BH8 (Fig. 7). The sidBA gene region also comprises the cdtQ and cdtP genes, encoding a permease and an ATPase, respectively. The DtxR binding site of this cluster is located between the sidB gene and the ddpABCD gene cluster, probably coding for an iron-dependent ABC transport system. Interestingly, the genes cdtQP and ddpABCD and a remnant of the sidB gene including the associated DtxR binding site were found in the genomes of C. diphtheriae C7(β)tox+ and INCA 402 (Fig. 7). These strains contain an additional gene cluster for siderophore biosynthesis and secretion, which is located downstream of the gene coding for the iron-regulated protein 2 (IRP2). This cluster comprises nine genes, of which the largest coding regions, irp2C and irp2F, yield amino acid sequence similarities with nonribosomal peptide synthetases from Burkholderia cenocepacia PC184 (AAKX00000000) and Ralstonia solanacearum IPO1609 (NW_002196568). A putative iron-dependent siderophore uptake system is encoded by the associated irp2JKLMN genes. C. diphtheriae strains containing all three gene clusters for siderophore biosynthesis and transport are CDC-E8392 and 31A, while the other isolates lack either one or two of the described gene clusters (Fig. 7). Furthermore, the iron-regulated nitrate reductase genes narKGHJI are conserved in all sequenced C. diphtheriae genomes. The assigned DtxR binding site is depleted by the integration of an insertion sequence located upstream of the gene cluster in the genomes of C7(β)tox+, PW8, VA01, INCA 402, and BH8 (Fig. 7). Accordingly, the reconstruction and comparison of DtxR regulons participating in iron homeostasis of C. diphtheriae led to the detection of variations in gene composition due to gene gain, gene loss, partial gene deletion, and DtxR binding site depletion.
Distribution and gene content of genomic islands.
Further factors besides the tox gene are apparently important for the virulence of C. diphtheriae, as invasive infections caused by nontoxigenic strains have been increasingly reported over the past years (19). Additional virulence factors can be encoded in genomic islands (GEIs) of the C. diphtheriae genome, which often show characteristics of horizontal gene transfer (7). The recently developed software PIPS was used for the detection of GEIs in the sequenced C. diphtheriae strains (68). In total, 57 GEIs were identified in the 13 C. diphtheriae genomes (see Table S1 in the supplemental material). Comparative content analysis of the detected GEIs revealed that some islands are strain specific, whereas others are completely or partially conserved in more than one strain. Eight GEIs can be regarded as highly conserved in all C. diphtheriae genomes (Fig. 8). According to the predicted gene content, the GEIs can be classified as pathogenicity islands, resistance islands, phage islands, or metabolic islands. Many GEIs (19 out of 57) assigned as phage islands encode typical phage products, and the respective regions of the C. diphtheriae genomes can be regarded as remnants of prophages. GEIs encoding proteins involved in specific metabolic pathways were assigned as metabolic islands of the C. diphtheriae genome. GEI48, for instance, carries genes involved in the degradation of 3-hydroxyphenylpropionic acid to succinate, pyruvate, and acetaldehyde, whereas other metabolic islands encode enzymes involved in polysaccharide degradation (GEI04) or components of sugar transport systems (GEI41). The majority of the resistance determinants of C. diphtheriae are encoded by genes located within GEI32, GEI38, and GEI54. These genomic islands encode proteins for heavy metal ion resistance, such as cadmium, copper, mercury, and arsenic resistance, and the clustered antibiotic resistance determinants in the genome of C. diphtheriae BH8.
Fig 8.

Genomic islands detected in the sequenced C. diphtheriae genomes and comparison of the predicted gene contents. The genomic islands were identified with the software PIPS, and the deduced similarities are shown as percentages.
The detected GEIs also include the previously identified pathogenicity islands (PAIs) of C. diphtheriae NCTC 13129 (Fig. 8). These PAIs comprise the βtox+ prophage region (GEI01), the spaDEF (GEI02) and spaABC (GEI10) gene clusters encoding adhesive pili, and the cdtQP-sidBA-ddpABCD gene cluster for siderophore biosynthesis and transport (GEI12) (7). Moreover, the software PIPS detected additional PAIs in the genome of C. diphtheriae NCTC 13129, including the irp6ABC genes for a siderophore-dependent iron uptake system (GEI15) and the second siderophore biosynthesis and transport gene cluster, ciuABCDEFG (GEI19). The search for pathogenicity islands in the genomes of C. diphtheriae led to the detection of 10 additional islands not present in strain NCTC 13129. These PAIs include the third siderophore biosynthesis and transport gene cluster, i.e., irp2 (GEI26), additional iron transport systems (GEI34, GEI 36, GEI37, and GEI42), a collagen adhesion protein gene (GEI27), and additional gene clusters for adhesive pili (GEI30, GEI47, GEI49, and GEI52). Therefore, the extended search for PAIs in the genomes of the sequenced C. diphtheriae strains revealed new gene clusters with characteristics of horizontal gene transfer, which are probably involved in iron acquisition and the formation of adhesive pili.
Conservation and heterogeneity of pilus gene clusters in C. diphtheriae clinical isolates.
Adhesive pili play pivotal roles in bacterial colonization, pathogenesis, and biofilm development (45). Pilus assembly in corynebacteria occurs by a two-step mechanism, whereby pilins are polymerized and then covalently anchored to cell wall peptidoglycan (62). In C. diphtheriae, a pilin-specific sortase catalyzes the polymerization of the pilus, consisting of the shaft protein, tip pilin, and base pilin (45). By amino acid sequence homology and BLASTP searches using the pilin motif and cell wall sorting signal as queries, we identified at least two pilus gene clusters in each of the sequenced genomes from 12 C. diphtheriae clinical isolates, with C. diphtheriae HC04 harboring four pilus gene clusters (Fig. 9). To designate each pilin according to known pilins of the reference strain C. diphtheriae NCTC 13129, ClustalW2 was employed to align the protein sequences for pilus shaft proteins and their cognate sortases. Their phylogenetic trees were then reconstructed with the neighbor-joining algorithm using MEGA 4.0 (72); similarly, the phylogenetic trees of minor pilins were produced. Built on these analyses, we assigned each pilus gene cluster according to the pilus shaft protein, i.e., SpaA, SpaD, or SpaH type (Fig. 9). Although SpaA pilus gene clusters were found in 9 out of 12 strains, with C. diphtheriae HC04 harboring two SpaA loci, namely, SpaA1 and SpaA2, SpaA pilins are mostly conserved (Fig. 10). Interestingly, SpaD pilins are the most divergent group among the three types of shaft proteins (Fig. 10) and found in all 12 strains (Fig. 9). Only a few C. diphtheriae strains harbor the SpaH-type gene clusters, and they appear to form two clades in the phylogenetic tree, i.e., SpaH and SpaH′ type (Fig. 9 and 10).
Fig 9.

Overview of pilus gene clusters found in the sequenced C. diphtheriae strains in relation to the reference strain C. diphtheriae NCTC 13129. Homologous pilin genes are indicated by color; sortase genes are shown in dark gray. Genes encoding hypothetical proteins in the pilus gene cluster of C. diphtheriae PW8 are shown in light gray; mobile elements are labeled in yellow. Genes similar to SpaD and SpaH types are denoted with primes. Asterisks and hatched arrows indicate fragmented genes.
Fig 10.

Analysis of pilus shaft proteins (A) and the corresponding sortases (B). ClustalW2 was used to align the protein sequences for major pilin subunits and the predicted sortases of 13 C. diphtheriae strains. Their phylogenetic trees were reconstructed with the neighbor-joining algorithm using MEGA 4.0 software. Locus tags are color-coded to indicate the pilus types of the reference strain C. diphtheriae NCTC 13129. Proteins similar to SpaH, SrtD, and SrtE are denoted with primes.
Significantly, cognate pilin-specific sortases display the same trend, with sequences of pilin-specific sortase SrtA for SpaA highly conserved (Fig. 9 and 10). As expected, pilin-specific sortases for SpaD pili (SrtB and SrtC) are the most divergent enzymes, followed by the cognate sortase enzymes for the SpaH pili (SrtD and SrtE). Intriguingly, while all tip pilin SpaC homologs are greatly conserved, the tip pilins for the other pilus types are varied (Fig. 11). The majority of SpaF homologs form a clade that is closer to SpaC homologs, but they are much different from two clades of SpaG pilins, one of which is extremely conserved (Fig. 11). A second SpaF-like tip protein is encoded as part of the SpaD-type clusters in C. diphtheriae VA01 and 31A. With the exception of C. diphtheriae BH8, the base SpaB pilins are mostly conserved, and the same is true for SpaI pilins, which are present in only a few isolates (Fig. 11). As anticipated, the base pilins for the SpaD-type pili, i.e., SpaE pilins, are also highly divergent (Fig. 11). Of note, SpaE pilin of the vaccine strain C. diphtheriae PW8 is closer to SpaI homologs. It is also noteworthy that C. diphtheriae PW8 contains a degenerated SpaD gene cluster with multiple intact and disrupted genes encoding SpaD, SpaE, and SpaF pilins and sortases SrtB and SrtE (Fig. 9), in addition to a SpaA locus with a disrupted spaC gene (31). Sequences of mobile DNA elements are also detected in the SpaD locus of C. diphtheriae PW8, suggesting horizontal gene transfer for gene duplication.
Fig 11.

Phylogenetic trees of the tip (A) and base (B) pilus proteins. ClustalW2 was used to align the protein sequences for tip and base pilins of 13 C. diphtheriae strains. Phylogenetic trees were reconstructed with the neighbor-joining algorithm using MEGA 4.0 software. Locus tags are color-coded to indicate the pilus types of the reference strain C. diphtheriae NCTC 13129. Proteins similar to SpaG and SpaI are denoted with primes.
DISCUSSION
The concept of the pangenome was introduced into genomic research by Tettelin and coworkers in 2005 and defined as the full complement of genes in a bacterial species consisting of the core genome and the dispensable genome (75). The core genome contains all genes present in a collection of analyzed strains of a bacterial species, defines the major phenotypic traits of an organism, and is essential for basic cellular functions, such as growth and reproduction or maintenance and survival (53). The variability of genome sizes of different strains is caused by the dispensable genome, which significantly contributes to the diversity of a bacterial species and provides pathways or functions which can confer selective advantages involved in strain-specific niche adaptations (16, 48). The dispensable genome is mainly based on the gene pool available for inclusion into the bacterial genome by mechanisms of horizontal gene transfer (48). Moreover, genome reduction by gene loss, genome rearrangements, and expansion of functional capabilities through gene duplication are forces that have shaped the microbial genome during its evolution (16). In general, the bacterial pangenome can be classified as closed or open (77). A pangenome is considered to be closed if the number of new genes added per newly sequenced genome converges to zero. Therefore, a closed pangenome indicates a static genomic content that is no longer expendable by genome sequencing. It is thus possible to acquire the full gene pool of such a bacterial species by adding a sufficient number of sequenced genomes. A recent systematic study on the pangenomes of 34 bacterial species demonstrated that one-third of the considered microorganisms have a closed pangenome, including the two actinobacteria Bifidobacterium animalis and Mycobacterium tuberculosis (5, 25). A pangenome is classified as open if new genes were recognized with newly sequenced genomes of a bacterial species (77). An open pangenome is associated with dynamic gene content and was previously calculated for the actinobacterium Bifidobacterium longum (5). The present study compares the gene contents of 13 C. diphtheriae genomes, including the widely studied strains C. diphtheriae PW8 and C. diphtheriae C7(β)tox+. Calculating the development of the corynebacterial gene content according to Heaps' law, the pangenome of C. diphtheriae was classified as open. The mean number of newly detected genes per genome is particularly low when considering the spectrum of countries, different diseases, and the points in time the sequenced C. diphtheriae strains were isolated. This result suggests that the calculated pangenome with its present gene content largely reflects the genetic diversity of the species C. diphtheriae. Differences in the gene contents of C. diphtheriae strains were mainly found in genomic islands located around the origin of replication and showing several characteristics of horizontal gene transfer (68). The specific location of genomic islands could be related to structural constraints of the bacterial chromosome or the accessibility of different chromosomal regions to foreign DNA elements such as insertion sequences. The detected genomic islands are embedded in a highly stable genomic backbone accounting for 70% of the gene content of C. diphtheriae. This profound stability of the core genome is characteristic of corynebacterial species lacking recombination enzymes generally involved in genomic rearrangements (81). The absence of these enzymes in C. diphtheriae prevents a rapid evolution of the core genome by recombination and suggests that intraspecific differences are probably related not to a large extent to variations in the genome's architecture but to differences in the distinct gene repertoires of different strains. The detected genomic islands account significantly for the genetic diversity of C. diphtheriae, as the dispensable genome constitutes about 30% of the gene content of each strain. A comprehensive comparative work on the open pangenome of E. coli estimated a core genome of 1,472 conserved gene families and a pangenome of 13,296 gene families comprising the full complement of genes (44). Some of these variable genes tend to be colocalized on genomic islands of the E. coli chromosome. In contrast to C. diphtheriae, the variable gene content of this enterobacterial species makes up more than 90% of the pangenome (44).
A marked difference between the C. diphtheriae strains is the presence of tox+ prophages that were identified in five genome sequences. Analysis of the prophage regions revealed that the ωtox+ phage of C. diphtheriae PW8 is homologous to the common βtox+ phage but present twice as a nontandem repeat. These genome data confirm a previous report, suggesting a second integration of the tox+ corynephage and thus a gene dosage effect as a cause for the high-level synthesis of diphtheria toxin in C. diphtheriae PW8 (59). Moreover, the genomic sequences revealed the presence of a new tox+ corynephage that was identified in the genome of C. diphtheriae 31A. The genome sequence of the prophage shows similarities to the βtox+ corynephage and the cryptic prophage ϕCULC22IV from C. ulcerans BR-AD22 (79). Extensive similarity to the βtox+ phage was observed in the tox gene region, whereas the components of the basic phage machinery are more similar to ϕCULC22IV. This result indicates that the genome architecture of tox+ corynephages is more diverse and that additional phages contribute to the spread of the tox gene in the human population. Diversity of the gene repertoire was also detected in regulons contributing to iron homeostasis in C. diphtheriae, including genes for iron transport systems, iron-dependent proteins, and the diphtheria toxin. These variations are based on differences in gene composition due not only to gene gain or gene loss but also to the depletion of DNA binding sites for the responsible regulator DtxR. As iron plays a vital role in bacterial infections due to its restricted availability in the host and functions as major environmental signal, proteins involved in iron acquisition are recognized as essential virulence factors during the infection of a mammalian host (21). Therefore, variations in the regulatory network of DtxR might lead to differences in iron supply of the bacterial cell, thereby influencing the expression of the tox gene and the virulence of the C. diphtheriae strains.
A remarkable outcome of this study is the detection of a great variety of pilus gene clusters encoding adhesive pili in C. diphtheriae. The pilin gene clusters of the tox+ isolate C. diphtheriae NCTC 13129 were intensely studied in recent years (18, 71, 78). It was demonstrated that the assembly process depends on pilin-specific sortases as well as the housekeeping sortase and that the SpaA-type pilus is necessary for the specific adherence of C. diphtheriae to human pharyngeal epithelial cells (46, 70). The protein components of the pilus, i.e., shaft protein, tip pilin, and base pilin, showed a great diversity in their amino acid sequences, and most of their encoding genes were assigned as singletons during the pangenome analysis. This result implies that important variations exist on the cell surface of toxigenic and nontoxigenic C. diphtheriae strains that are relevant for the initial step of an infection. Different degrees of attachment of C. diphtheriae strains to HEp-2 cell monolayers were reported previously (27). Differences in adhesion of C. diphtheriae C7(−) and C. diphtheriae PW8 to Detroit 562 cells were also reported, and PW8 showed a reduced level of adherence compared with C7(−) (29). Mutations in the base pilin SpaB and the tip pilin SpaC of the SpaA-type pilus were shown to reduce the adhesive activity of C. diphtheriae (46). In C. diphtheriae PW8, the spaC gene is characterized by a frameshift mutation (31), and almost all genes of the complex SpaD pilus gene cluster of PW8 are apparently inactivated by transpositional integration of insertion sequences. However, factors other than pili can contribute to cell adhesion of C. diphtheriae, including members of the resuscitation-promoting factor-interacting protein family (DIP1281) and the cell wall-associated hydrolase family (DIP1621). Both enzymes are encoded by the core genome and probably contribute indirectly to adhesion of C. diphtheriae, as they are involved in organizing the corynebacterial cell surface (36, 55). The high degree of diversity of pilus gene clusters in C. diphtheriae shows that the process of adhesion could be more diverse than initially anticipated. Comparative analysis of adhesion properties of the sequenced C. diphtheriae strains are now necessary to experimentally detect and describe differences in adhesion to mammalian host cells. The enormous collection of variable pilus gene clusters detected in this pangenome project may reveal novel and more detailed insights into adhesion properties of C. diphtheriae.
Supplementary Material
ACKNOWLEDGMENTS
E.T. acknowledges the receipt of a scholarship of the CLIB Graduate Cluster Industrial Biotechnology. S.D.C.S. was supported by a CAPES-DAAD scholarship. J.B. and S.J. acknowledge funding by the German Federal Ministry of Education and Research “GenoMik-Transfer” project.
We thank Burkhard Linke (CeBiTec) for bioinformatics support. The valuable contribution of the CeBiTec sequencing team to this pangenome project is gratefully acknowledged.
Footnotes
Published ahead of print 13 April 2012
Supplemental material for this article can be found at http://jb.asm.org/.
REFERENCES
- 1. Andrews SC, Robinson AK, Rodriguez-Quinones F. 2003. Bacterial iron homeostasis. FEMS Microbiol. Rev. 27:215–237 [DOI] [PubMed] [Google Scholar]
- 2. Badger JH, Olsen GJ. 1999. CRITICA: coding region identification tool invoking comparative analysis. Mol. Biol. Evol. 16:512–524 [DOI] [PubMed] [Google Scholar]
- 3. Barksdale WL, Pappenheimer AM., Jr 1954. Phage-host relationships in nontoxigenic and toxigenic diphtheria bacilli. J. Bacteriol. 67:220–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blom J, et al. 2009. EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics 10:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bottacini F, et al. 2010. Comparative genomics of the genus Bifidobacterium. Microbiology 156:3243–3254 [DOI] [PubMed] [Google Scholar]
- 6. Brune I, et al. 2006. The DtxR protein acting as dual transcriptional regulator directs a global regulatory network involved in iron metabolism of Corynebacterium glutamicum. BMC Genomics 7:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cerdeño-Táarraga AM, et al. 2003. The complete genome sequence and analysis of Corynebacterium diphtheriae NCTC13129. Nucleic Acids Res. 31:6516–6523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crosa JH, Walsh CT. 2002. Genetics and assembly line enzymology of siderophore biosynthesis in bacteria. Microbiol. Mol. Biol. Rev. 66:223–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. D'Afonseca V, et al. 2012. Reannotation of the Corynebacterium diphtheriae NCTC13129 genome as a new approach to studying gene targets connected to virulence and pathogenicity in diphtheria. Open Access Bioinformatics 4:1–13 [Google Scholar]
- 10. Darling AE, Mau B, Perna NT. 2010. Progressive Mauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147 doi:10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27:4636–4641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dittmann S, et al. 2000. Successful control of epidemic diphtheria in the states of the Former Union of Soviet Socialist Republics: lessons learned. J. Infect. Dis. 181:S10–S22 [DOI] [PubMed] [Google Scholar]
- 13. Felsenstein J. 1989. PHYLIP—Phylogeny Inference Package (version 3.2). Cladistics 5:164–166 [Google Scholar]
- 14. Finn RD, et al. 2010. The Pfam protein families database. Nucleic Acids Res. 38:D211–D222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fourel G, Phalipon A, Kaczorek M. 1989. Evidence for direct regulation of diphtheria toxin gene transcription by an Fe2+-dependent DNA-binding repressor, DtoxR, in Corynebacterium diphtheriae. Infect. Immun. 57:3221–3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fraser-Liggett CM. 2005. Insights on biology and evolution from microbial genome sequencing. Genome Res. 15:1603–1610 [DOI] [PubMed] [Google Scholar]
- 17. Freeman VJ. 1951. Studies on the virulence of bacteriophage-infected strains of Corynebacterium diphtheriae. J. Bacteriol. 61:675–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gaspar AH, Ton-That H. 2006. Assembly of distinct pilus structures on the surface of Corynebacterium diphtheriae. J. Bacteriol. 188:1526–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gomes DL, et al. 2009. Corynebacterium diphtheriae as an emerging pathogen in nephrostomy catheter-related infection: evaluation of traits associated with bacterial virulence. J. Med. Microbiol. 58:1419–1427 [DOI] [PubMed] [Google Scholar]
- 20. Gordon D, Abajian C, Green P. 1998. Consed: a graphical tool for sequence finishing. Genome Res. 8:195–202 [DOI] [PubMed] [Google Scholar]
- 21. Griffiths E. 1991. Iron and bacterial virulence—a brief overview. Biol. Met. 4:7–13 [DOI] [PubMed] [Google Scholar]
- 22. Grissa I, Bouchon P, Pourcel C, Vergnaud G. 2008. On-line resources for bacterial micro-evolution studies using MLVA or CRISPR typing. Biochimie 90:660–668 [DOI] [PubMed] [Google Scholar]
- 23. Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35:W52–W57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hadfield TL, McEvoy P, Polotsky Y, Tzinserling VA, Yakovlev AA. 2000. The pathology of diphtheria. J. Infect. Dis. 181:S116–S120 [DOI] [PubMed] [Google Scholar]
- 25. Halachev MR, Loman NJ, Pallen MJ. 2011. Calculating orthologs in bacteria and archaea: a divide and conquer approach. PLoS One 6:e28388 doi:10.1371/journal.pone.0028388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hiller NL, et al. 2007. Comparative genomic analyses of seventeen Streptococcus pneumoniae strains: insights into the pneumococcal supragenome. J. Bacteriol. 189:8186–8195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hirata R, Jr, et al. 2004. Patterns of adherence to HEp-2 cells and actin polymerisation by toxigenic Corynebacterium diphtheriae strains. Microb. Pathog. 36:125–130 [DOI] [PubMed] [Google Scholar]
- 28. Holmes RK. 2000. Biology and molecular epidemiology of diphtheria toxin and the tox gene. J. Infect. Dis. 181:S156–S167 [DOI] [PubMed] [Google Scholar]
- 29. Honma Y, et al. 2009. A case of afebrile pneumonia caused by non-toxigenic Corynebacterium diphtheriae. J. Infect. Dis. 62:327–329 [PubMed] [Google Scholar]
- 30. Husemann P, Stoye J. 2010. r2cat: synteny plots and comparative assembly. Bioinformatics 26:570–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iwaki M, et al. 2010. Genome organization and pathogenicity of Corynebacterium diphtheriae C7(−) and PW8 strains. Infect. Immun. 78:3791–3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jolley KA, Chan MS, Maiden MC. 2004. mlstdbNet—distributed multi-locus sequence typing (MLST) databases. BMC Bioinformatics 5:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kalinowski J, et al. 2003. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J. Biotechnol. 104:5–25 [DOI] [PubMed] [Google Scholar]
- 34. Kitchin NR. 2011. Review of diphtheria, tetanus and pertussis vaccines in clinical development. Expert Rev. Vaccines 10:605–615 [DOI] [PubMed] [Google Scholar]
- 35. Klebs E. 1883. Ueber Diphtherie. Verh. Kongr. Innere Med. 2:139–154 [Google Scholar]
- 36. Kolodkina V, Denisevich T, Titov L. 2011. Identification of Corynebacterium diphtheriae gene involved in adherence to epithelial cells. Infect. Genet. Evol. 11:518–521 [DOI] [PubMed] [Google Scholar]
- 37. Kunkle CA, Schmitt MP. 2005. Analysis of a DtxR-regulated iron transport and siderophore biosynthesis gene cluster in Corynebacterium diphtheriae. J. Bacteriol. 187:422–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kunkle CA, Schmitt MP. 2003. Analysis of the Corynebacterium diphtheriae DtxR regulon: identification of a putative siderophore synthesis and transport system that is similar to the Yersinia high-pathogenicity island-encoded yersiniabactin synthesis and uptake system. J. Bacteriol. 185:6826–6840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Larkin MA, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 40. Leong D, Murphy JR. 1985. Characterization of the diphtheria tox transcript in Corynebacterium diphtheriae and Escherichia coli. J. Bacteriol. 163:1114–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lerat E, Daubin V, Moran NA. 2003. From gene trees to organismal phylogeny in prokaryotes: the case of the gamma-Proteobacteria. PLoS Biol. 1:E19 doi:10.1371/journal.pbio.0000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Linke B, McHardy AC, Neuweger H, Krause L, Meyer F. 2006. REGANOR: a gene prediction server for prokaryotic genomes and a database of high quality gene predictions for prokaryotes. Appl. Bioinformatics 5:193–198 [DOI] [PubMed] [Google Scholar]
- 43. Loeffler F. 1884. Untersuchung über die Bedeutung der Mikroorganismen für die Entstehung der Diphtherie. Mitt. Kaiserl. Gesundheitsamt 2:421–499 [Google Scholar]
- 44. Lukjancenko O, Wassenaar TM, Ussery DW. 2010. Comparison of 61 sequenced Escherichia coli genomes. Microb. Ecol. 60:708–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mandlik A, Das A, Ton-That H. 2008. The molecular switch that activates the cell wall anchoring step of pilus assembly in gram-positive bacteria. Proc. Natl. Acad. Sci. U. S. A. 105:14147–14152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mandlik A, Swierczynski A, Das A, Ton-That H. 2007. Corynebacterium diphtheriae employs specific minor pilins to target human pharyngeal epithelial cells. Mol. Microbiol. 64:111–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mattos-Guaraldi AL, Cappelli EA, Previato JO, Formiga LC, Andrade AF. 1999. Characterization of surface saccharides in two Corynebacterium diphtheriae strains. FEMS Microbiol. Lett. 170:159–166 [DOI] [PubMed] [Google Scholar]
- 48. Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. 2005. The microbial pan-genome. Curr. Opin. Genet. Dev. 15:589–594 [DOI] [PubMed] [Google Scholar]
- 49. Meyer F, et al. 2003. GenDB—an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 31:2187–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mishra B, Dignan RJ, Hughes CF, Hendel N. 2005. Corynebacterium diphtheriae endocarditis—surgery for some but not all! Asian Cardiovasc. Thorac. Ann. 13:119–126 [DOI] [PubMed] [Google Scholar]
- 51. Mokrousov I, Limeschenko E, Vyazovaya A, Narvskaya O. 2007. Corynebacterium diphtheriae spoligotyping based on combined use of two CRISPR loci. Biotechnol. J. 2:901–906 [DOI] [PubMed] [Google Scholar]
- 52. Nagarkar PP, Ravetkar SD, Watve MG. 2002. The amino acid requirements of Corynebacterium diphtheriae PW 8 substrain CN 2000. J. Appl. Microbiol. 92:215–220 [DOI] [PubMed] [Google Scholar]
- 53. Nyström T. 2004. Growth versus maintenance: a trade-off dictated by RNA polymerase availability and sigma factor competition? Mol. Microbiol. 54:855–862 [DOI] [PubMed] [Google Scholar]
- 54. Oram DM, Avdalovic A, Holmes RK. 2004. Analysis of genes that encode DtxR-like transcriptional regulators in pathogenic and saprophytic corynebacterial species. Infect. Immun. 72:1885–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ott L, et al. 2010. Corynebacterium diphtheriae invasion-associated protein (DIP1281) is involved in cell surface organization, adhesion and internalization in epithelial cells. BMC Microbiol. 10:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Park WH, Williams AW. 1896. The production of diphtheria toxin. J. Exp. Med. 1:164–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8:785–786 [DOI] [PubMed] [Google Scholar]
- 58. Pimenta FP, Hirata R, Jr, Rosa AC, Milagres LG, Mattos-Guaraldi AL. 2008. A multiplex PCR assay for simultaneous detection of Corynebacterium diphtheriae and differentiation between non-toxigenic and toxigenic isolates. J. Med. Microbiol. 57:1438–1439 [DOI] [PubMed] [Google Scholar]
- 59. Rappuoli R, Michel JL, Murphy JR. 1983. Integration of corynebacteriophages βtox+, ωtox+, and γtox− into two attachment sites on the Corynebacterium diphtheriae chromosome. J. Bacteriol. 153:1202–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rappuoli R, Michel JL, Murphy JR. 1983. Restriction endonuclease map of corynebacteriophage γctox+ isolated from the Park-Williams no. 8 strain of Corynebacterium diphtheriae.. J. Virol. 45:524–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ratti G, Covacci A, Rappuoli R. 1997. A tRNA2Arg gene of Corynebacterium diphtheriae is the chromosomal integration site for toxinogenic bacteriophages. Mol. Microbiol. 25:1179–1181 [DOI] [PubMed] [Google Scholar]
- 62. Rogers EA, Das A, Ton-That H. 2011. Adhesion by pathogenic corynebacteria. Adv. Exp. Med. Biol. 715:91–103 [DOI] [PubMed] [Google Scholar]
- 63. Salyers AA, Amabile-Cuevas CF. 1997. Why are antibiotic resistance genes so resistant to elimination? Antimicrob. Agents Chemother. 41:2321–2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schneider J, et al. 2010. CARMEN—Comparative Analysis and in silico Reconstruction of organism-specific MEtabolic Networks. Genet. Mol. Res. 9:1660–1672 [DOI] [PubMed] [Google Scholar]
- 65. Schröder J, et al. 2012. Complete genome sequence, lifestyle, and multi-drug resistance of the human pathogen Corynebacterium resistens DSM 45100 isolated from blood samples of a leukemia patient. BMC Genomics 13:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shapiro JA. 1979. Molecular model for the transposition and replication of bacteriophage Mu and other transposable elements. Proc. Natl. Acad. Sci. U. S. A. 76:1933–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Skarstad K, Bernander R, Boye E. 1995. Analysis of DNA replication in vivo by flow cytometry. Methods Enzymol. 262:604–613 [DOI] [PubMed] [Google Scholar]
- 68. Soares SC, et al. 2012. PIPS: pathogenicity island prediction software. PLoS One 7:e30848 doi:10.1371/journal.pone.0030848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Suzek BE, Ermolaeva MD, Schreiber M, Salzberg SL. 2001. A probabilistic method for identifying start codons in bacterial genomes. Bioinformatics 17:1123–1130 [DOI] [PubMed] [Google Scholar]
- 70. Swaminathan A, et al. 2007. Housekeeping sortase facilitates the cell wall anchoring of pilus polymers in Corynebacterium diphtheriae. Mol. Microbiol. 66:961–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Swierczynski A, Ton-That H. 2006. Type III pilus of corynebacteria: pilus length is determined by the level of its major pilin subunit. J. Bacteriol. 188:6318–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599 [DOI] [PubMed] [Google Scholar]
- 73. Tao X, Schiering N, Zeng HY, Ringe D, Murphy JR. 1994. Iron, DtxR, and the regulation of diphtheria toxin expression. Mol. Microbiol. 14:191–197 [DOI] [PubMed] [Google Scholar]
- 74. Terns MP, Terns RM. 2011. CRISPR-based adaptive immune systems. Curr. Opin. Microbiol. 14:321–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tettelin H, et al. 2005. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc. Natl. Acad. Sci. U. S. A. 102:13950–13955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tettelin H, et al. 2001. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293:498–506 [DOI] [PubMed] [Google Scholar]
- 77. Tettelin H, Riley D, Cattuto C, Medini D. 2008. Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11:472–477 [DOI] [PubMed] [Google Scholar]
- 78. Ton-That H, Schneewind O. 2003. Assembly of pili on the surface of Corynebacterium diphtheriae. Mol. Microbiol. 50:1429–1438 [DOI] [PubMed] [Google Scholar]
- 79. Trost E, et al. 2011. Comparative analysis of two complete Corynebacterium ulcerans genomes and detection of candidate virulence factors. BMC Genomics 12:383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Trost E, et al. 2010. The complete genome sequence of Corynebacterium pseudotuberculosis FRC41 isolated from a 12-year-old girl with necrotizing lymphadenitis reveals insights into gene-regulatory networks contributing to virulence. BMC Genomics 11:728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ventura M, et al. 2007. Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol. Mol. Biol. Rev. 71:495–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Viguetti SZ, et al. 2012. Multilocus sequence types of invasive Corynebacterium diphtheriae isolated in the Rio de Janeiro urban area, Brazil. Epidemiol. Infect. 140:617–620 [DOI] [PubMed] [Google Scholar]
- 83. Vitek CR. 2006. Diphtheria. Curr. Top. Microbiol. Immunol. 304:71–94 [DOI] [PubMed] [Google Scholar]
- 84. von Behring EA. 1893. Zur Behandlung der Diphtherie mit Diphtherieheilserum. Dtsch. Med. Wochenschr. 23:543–547 [Google Scholar]
- 85. von Graevenitz A, Bernard K. 2006. The genus Corynebacterium—medical, p 819–842. In Dworkin M, Falkow F, Rosenberg E, Schleifer KH, Stackebrandt E. (ed), The prokaryotes, 3rd ed, vol 3 Springer, New York, NY [Google Scholar]
- 86. Yukawa H, et al. 2007. Comparative analysis of the Corynebacterium glutamicum group and complete genome sequence of strain R. Microbiology 153:1042–1058 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.