

Abstract
Outer membrane vesicles (OMVs) have been identified in a wide range of bacteria, yet little is known of their biogenesis. It has been proposed that OMVs can act as long-range toxin delivery vectors and as a novel stress response. We have found that the formation of OMVs in the Gram-negative opportunistic pathogen Serratia marcescens is thermoregulated, with a significant amount of OMVs produced at 22 or 30°C and negligible quantities formed at 37°C under laboratory conditions. Inactivation of the synthesis of the enterobacterial common antigen (ECA) resulted in a hypervesiculation phenotype, supporting the hypothesis that OMVs are produced in response to stress. We demonstrate that the phenotype can be reversed to wild-type (WT) levels upon the loss of the Rcs phosphorelay response regulator RcsB, but not RcsA, suggesting a role for the Rcs phosphorelay in the production of OMVs. MS fingerprinting of the OMVs provided evidence of cargo selection within wild-type cells, suggesting a possible role for Serratia OMVs in toxin delivery. In addition, OMV-associated cargo proved toxic upon injection into the haemocoel of Galleria mellonella larvae. These experiments demonstrate that OMVs are the result of a regulated process in Serratia and suggest that OMVs could play a role in virulence.
INTRODUCTION
Gram-negative bacteria, both pathogenic and nonpathogenic, release lipid bilayer vesicles from the outer membrane (OM) (3, 34). These outer membrane vesicles (OMVs), like the outer membrane from which they are generated, are composed of lipopolysaccharide (LPS), phospholipids, and outer membrane proteins, in addition to periplasmic components (17, 22, 24, 26, 30). Several studies suggest that vesiculation serves as an envelope stress response. Impairment of the σE or the Cpx stress control mechanisms in Escherichia coli was shown to significantly increase vesiculation, and hypervesiculation was associated with improved cell survival under stress-inducing conditions (46). OMVs also have the capacity to transport damaging agents such as antibiotics (52) or misfolded proteins (46) away from the bacterial cell. Stress caused by the host environment or by antibiotic treatment can also result in increased vesiculation in Pseudomonas aeruginosa and Shigella dysenteriae, respectively (10, 23). Within the context of the host environment, OMVs have been implicated as vectors for long-distance toxin delivery (45). This occurs either with the biogenesis of OMVs acting as a novel secretion mechanism or by shed vesicles associating with secreted proteins and acting as mediators for these exoproteins (12). Unlike the traditional, highly characterized type I to VI secretion systems, OMVs have the capacity to transport a mixture of soluble and insoluble material, which may be advantageous to the bacteria by protecting their cargo from proteases or by allowing for the assembly of multivalent complexes. Once released from bacteria, the vesicles are free to interact with the host, with considerable evidence that OMVs can be internalized into host cells (9, 16, 31) and have a role in modulating the host immune response (4, 49, 54, 61).
The mechanism for vesicle biogenesis remains elusive. In Porphyromonas gingivalis and in other species, OMVs are enriched in virulence factors (21, 22). It appears that these proteins are selectively packed during OMV biogenesis, in a process that also causes the enrichment of certain surface glycans and deacylated lipid A in the vesicles. Studies on P. aeruginosa OMVs have found that the quorum signaling molecule, PQS, is incorporated into the OM and can induce curvature (43, 44). This supports the theory that the breakdown of linkages between the peptidoglycan and the OM is the trigger for vesicle formation (35). Alternatively, a nanopod-like structure has also been implicated in vesicle formation in Delftia spp., but evidence for this process in other bacteria has yet to be found (55).
OMVs are also produced by Serratia marcescens, a Gram-negative bacillus commonly found in soil, water, air, plants, and animals. S. marcescens is an important nosocomial pathogen and has been implicated as a causative agent in a wide variety of infections, including respiratory and urinary tract infections, septicemia, meningitis, and pneumonia, with significantly increased morbidity associated with newborns (2). Serratia has become the subject of increased attention due to a rise in resistance to several antibiotics (41, 59) through a series of well-characterized multidrug efflux pumps (1) and porins (51), rendering the pathogen difficult to control and eradicate. Despite its prevalence, the mechanisms of Serratia pathogenesis remain poorly understood. Recently, a type VI secretion system has been described which displays an important role in bacterial killing (47), and the mechanism for survival within human epithelial cells postinternalization was described (14). The outer membrane vesicles of S. marcescens have been reported to be among the largest in Enterobacteriaceae (37), but little is known of their composition, their biogenesis, or a possible role in pathogenesis.
Enterobacterial common antigen (ECA) is a glycolipid found in the outer leaflet of the outer membrane in Gram-negative enteric bacteria. It has been shown that alterations to this pathway in S. marcescens induce the cell envelope stress-responsive Rcs phosphorelay system (8). The Rcs phosphorelay is a complex signaling pathway known to be activated upon alterations to lipopolysaccharide or membrane-derived oligosaccharides, with the outer membrane lipoprotein RcsF identified as playing an important role channeling selected input signals to the other components of the phosphorelay (11, 36, 40). The pathway has a number of components which require the transfer of a phosphate from the histidine kinase domain of RcsC to the phosphotransfer protein RcsD. RcsD can, in turn, transfer the phosphate group to the response regulator RcsB, which can homodimerize or form complexes with RcsA. The cascade is known to regulate up to 5% of the E. coli genome (15, 19), and in S. marcescens, it has been shown to control the expression of several genes, including flagellar biogenesis regulatory components (8). In this work, we have found that OMV biogenesis is thermoregulated and obtained evidence that, under certain conditions, RcsB controls an unknown mechanism which regulates vesiculation in S. marcescens.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Serratia marcescens RM66262 is a nonpigmented clinical isolate from a patient with a urinary tract infection (7). Strains were grown in Luria broth (LB) unless otherwise stated. Gentamicin and kanamycin were used at 10 μg/ml and 50 μg/ml, respectively. A description of the strains used in this study can be found in Table S1 in the supplemental material.
OMV preparation.
Cells corresponding to 55 OD600 units (OD600 is optical density at 600 nm) of overnight cultures of S. marcescens wild-type (WT) and mutant strains were removed from suspension by centrifugation at 6,000 × g. The supernatants were filtered through a 0.22-μm-pore-size polyvinylidene difluoride (PVDF) membrane (Millex GV; Millipore) to remove residual cells. OMVs were recovered from the resulting filtrates by ultracentrifugation at 100,000 × g for 3 h at 4°C (Optima L-90K ultracentrifuge; Beckman Coulter) and resuspended in 500 μl of phosphate-buffered saline (PBS) (24).
Kdo quantification.
3-Deoxy-d-manno-octulosonic acid (Kdo) was evaluated by the periodic acid-thiobarbituric acid method described by Hancock and Poxton (20). Briefly, after purification by ultracentrifugation, the vesicles are hydrolyzed by sulfuric acid for 8 min. Periodic acid (0.1 M) is added and the sample left at room temperature for 10 min before the addition of sodium arsenite resuspended in HCl. The samples are vortexed vigorously before the addition of thiobarbituric acid. After boiling for 10 min, butanol is added, followed by centrifugation for 4 min. The upper aqueous phase is read at 509 nm (OD), and the reading is subtracted from the values obtained at 552 nm and compared to a standard curve for quantification. Purified Kdo is obtained from Sigma-Aldrich.
Galleria mellonella killing assay.
Surface-sterilized G. mellonella worms were injected into the hind proleg with 5 μl of OMVs or LPS. LPS and OMVs were normalized to 2 ng/μl. Death was then determined after 12 h by responsiveness to touch. Worms were examined in triplicates of 30 with separate larval preparations. Non-OMV-attributable deaths were determined by the injection of worms with sterile PBS.
OM isolation.
The cells of overnight cultures were harvested by centrifugation at 10,000 × g for 10 min at 4°C. The pellets were gently resuspended in 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 50 mM MgCl2 containing complete EDTA-free protease inhibitor mixture (Roche Applied Science) and then lysed by sonication. The membranes were collected by ultracentrifugation at 100,000 × g for 1 h at 4°C. The OMs were isolated by differential extraction with the same buffer and 1.5% (vol/vol) Triton X-100 and incubated at 20°C for 1 h. The OM fractions were recovered by centrifugation at 100,000 × g for 1 h at 4°C.
Transmission electron microscopy (TEM).
Portions (3 μl) of the OMV preparations were adsorbed onto carbon-coated copper grids (3 min). Liquid excess was discarded, and the samples were negatively stained with 2% (wt/vol) uranyl acetate for 3 min and evaluated in a Morgagni (FEI) transmission electron microscope.
Proteomic analysis.
Two methods were used to screen for protein content of OMVs. The first involved resolving OMV samples by SDS-PAGE followed by the in-gel digestion of excised bands of interest using sequencing-grade modified trypsin (Promega) (56). Peptide fragments were eluted from the gel piece, desalted using ZipTipC18 (Millipore) according to the supplier's protocol, and dissolved in 0.1% formic acid. The second method involved the removal of attached lipids from lyophilized OMV pellets using trifluoroethanol. In-solution trypsin digestion was then carried out as before. A hybrid quadrupole orthogonal acceleration time-of-flight mass spectrometer, Q-TOF Premier (Waters), equipped with a nanoACQUITY ultraperformance liquid chromatography system (Waters) was used for tandem mass spectrometry (MS-MS) analyses of the peptides, and the resulting mass spectrums were used for the identification of the proteins by the Mascot search engine using the preliminary gene sequence of S. marcescens DB11 on the SEED server.
LPS analysis.
LPSs derived from the OMVs of all strains were prepared as described previously (42). The LPS was run on 12% SDS-PAGE and visualized by the silver staining method described by Tsai and Frasch (60).
RESULTS
OMV production in S. marcescens is thermoregulated.
Dry weight measurements are often used to compare relative vesiculation. The amount of extracellular material, including vesicles, recovered from the cell-free supernatant of cells grown at 37°C was approximately half that which was recovered from cells grown at lower temperatures (Fig. 1, inset). This technique is not accurate for OMV quantification, as it also measures nonvesicle content, like flagella, which often copurify with OMVs. We consequently measured the 3-deoxy-d-manno-octulosonic acid (Kdo) present in our OMV preparations. Kdo is an exclusive component of LPS and therefore better reflects the OMV content present in a sample. When OMV production was estimated measuring Kdo, vesiculation was diminished about 5-fold at 37°C compared to that at 22 or 30°C (Fig. 1). This indicates that in S. marcescens, OMV production is thermoregulated, being high at low temperatures and minimal at high temperatures. As an additional control, we determined that Kdo levels were not variable on the surface of whole cells at each of the temperatures (see Fig. S1 in the supplemental material).
Fig 1.

Vesiculation is thermoregulated in S. marcescens. Amounts of OMV were estimated by measuring Kdo content or dry weight (inset). Both methods show that production of OMV is higher at 22 or 30°C than at 37°C and that little difference is noted between 22°C and 30°C in terms of OMVs recovered. Average Kdo values were calculated from five separate experiments, and the error bars indicate standard deviations.
Disruption of the ECA synthesis pathway increases vesiculation at all temperatures.
In searching for genes that affect vesiculation in S. marcescens, elements of the ECA synthesis pathway were examined. WecG converts GlcNAc–PP-Und (lipid I) to ManNAc-GlcNAc–PP-Und (lipid II), with mutagenesis of the wecG gene resulting in the accumulation of lipid I at the inner membrane. Previous work has shown that bacteria carrying mutations to the ECA synthesis cluster express low levels of flagellin, lack phospholipase activity, and present reduced swimming and swarming compared to wild-type bacteria (7). We found that in the wecG mutant, vesiculation was induced at all temperatures (Fig. 2). The increase was approximately 2- to 3-fold at 22°C and 30°C and about 8-fold at 37°C. Interestingly, the wecG mutant produced at 37°C more vesicles than the wild-type strain at 22°C or 30°C. The complementation of the wecG mutant restored OMV production to levels comparable with those of the wild type. Investigation of the purified OMVs by TEM revealed that OMVs produced by the three strains at 37°C were similar in shape and averaged 150 nm in diameter (Fig. 2, inset). Though we acknowledge that larger OMVs may be absent from our preparations due to our harvesting method, the uniform size and appearance of the OMVs seen by TEM suggest that the majority of the OMVs are approximately 150 nm in size. Consistent with the data obtained via Kdo determination, the amounts of vesicles visualized in the wecG strain were higher than for the wild-type or the complemented strains.
Fig 2.

Interruption of the ECA synthesis pathway results in hypervesiculation. The amount of Kdo released from stationary-phase cells increased upon mutation of wecG. Reintroduction of wecG restored wild-type levels of vesiculation. Average Kdo values were calculated from five separate experiments, and the error bars indicate standard deviations. Also shown is TEM analysis of vesicles (indicated by arrows) in WT (inset A), wecG (inset B), and wecG (inset C) samples at 37°C. The scale bar present at the bottom left of each image corresponds to 200 nm.
ECA synthesis disruption at different stages increases vesiculation.
We analyzed if the hypervesiculation phenotype detected in the wecG mutant was also observable by disrupting other genes involved in ECA synthesis, as occurs for the other phenotypes previously described for ECA mutants (7, 8). wecA and wecD mutations of the ECA synthesis pathway result in the accumulation of undecaprenyl monophosphate (Und-P) and lipid II intermediates, respectively. Both mutations resulted in increased vesiculation at 30°C and 37°C, indicating that temperature-induced vesiculation and ΔECA-induced vesiculation may be different processes. The degree of increase varied, being more dramatic in the wecD mutant (Fig. 3). The approximately 20-fold increase of vesiculation apparent by comparison of the wild type and wecD mutant at 37°C and the 6- to 8-fold increase in the case of the wecA and wecG mutants are clear indications of the effect of altering the ECA synthesis pathway on vesiculation. TEM of the OMVs was carried out, with intact OMVs from the wecD and wecA strains appearing similar to the ones produced by the wild-type strain (Fig. 3, insets A and B). To rule out that cell lysis accounted for the apparent hypervesiculation phenotype observed, MS fingerprinting of the proteins present in our OMV preparations was carried out. The vast majority of the proteins found were outer membrane and periplasmic proteins (Tables 1 and 2). The cytoplasmic proteins alcohol dehydrogenase and EF-Tu were the only predicted cytoplasmic proteins present within both OM and OMV preparations. This finding, in addition to the absence of inner membrane proteins, argues against lysis. The LPSs of the OMVs of all strains were compared to ensure that the surface carbohydrate was present, a characteristic of all OMVs. As a final control to ensure that broken cells were not a contaminant of the OMV preparations, Western blot analysis of trichloroacetic acid (TCA)-precipitated OMV-free supernatants was carried out using α-RNA polymerase. The presence of cytoplasmic RNA polymerase in the supernatant would be indicative of cell lysis. No significant levels of RNA polymerase were detected in the supernatants, ruling out a contribution of cell lysis to the amounts of Kdo measured (data not shown).
Fig 3.

Interruption of the ECA synthesis pathway at various stages induced OMV formation. Kdo recovered from cell-free supernatants shows increased amounts of vesicles in wecA, wecD, and wecG strains. wecA (inset A) and wecD (inset B) OMVs recovered from 30°C cultures of wecA and wecD strains appear similar to OMVs isolated at 37°C from WT and wecG strains (Fig. 2). The scale bar present at the bottom left of each image corresponds to 200 nm. Average Kdo values were calculated from five separate experiments, and the error bars indicate standard deviations.
Table 1.
Proteins detected in the OM and OMVs of wild-type S. marcescensa
| Fraction | Systematic gene name | Annotation | MW | Score |
|---|---|---|---|---|
| OMV | 4161L | Lipase | 64.9 | 1,387 |
| 6253L | Flagellin | 34.6 | 1,103 | |
| 12577R | Serralysin A | 54 | 879 | |
| 2860L | OmpF | 41.7 | 719 | |
| 2408R | Surface layer protein A | 101.2 | 639 | |
| 2927L | OmpA | 39.2 | 439 | |
| 7297L | Alanine amidase | 28.4 | 351 | |
| 6014L | Porin | 39.6 | 227 | |
| 4103L | Lipoprotein precursor | 8.3 | 147 | |
| 7646L | OmpC | 41.4 | 161 | |
| 5558L | OmpW | 23.8 | 139 | |
| 6463R | Hcp effector | 16.8 | 138 | |
| 5731L | MipA protein | 28.5 | 127 | |
| 1470R | Peptidoglycan-associated lipoprotein | 19.4 | 123 | |
| 12753R | EF-Tu | 43.2 | 110 | |
| 2143R | OmpX | 18.4 | 110 | |
| 8985R | Phospholipase | 35.6 | 103 | |
| 5653R | Alcohol dehydrogenase | 96.2 | 86 | |
| OM | 2927L | OmpA | 39.2 | 1,494 |
| 2860L | OmpF | 41.7 | 1,034 | |
| 13014R | GroEL | 56.6 | 895 | |
| 6253L | Flagellin | 36.6 | 893 | |
| 7646L | OmpC | 41.4 | 832 | |
| 6014L | Porin | 39.6 | 498 | |
| 5558L | OmpW | 23.8 | 443 | |
| 1470R | Peptidoglycan-associated lipoprotein | 19.4 | 434 | |
| 6011L | LysM | 34.5 | 426 | |
| 5653R | Alcohol dehydrogenase | 96.2 | 329 | |
| 4176L | OM lipoprotein | 16.6 | 297 | |
| 4103L | Lipoprotein precursor | 8.3 | 292 | |
| 10953L | Ribosomal 30S | 23.6 | 220 | |
| 9153L | Omp factor YaeT | 89.6 | 193 | |
| 10162R | TolC | 54.5 | 190 | |
| 109L | LPS assembly protein LptD | 89.3 | 187 | |
| 6322L | Flagellar hook | 42.6 | 187 | |
| 12753R | EF-Tu | 43.2 | 165 | |
| 2143R | OmpX | 18.4 | 145 | |
| 2408R | Surface layer protein A | 101.2 | 141 | |
| 12670R | Ribosomal 50S | 24.6 | 124 | |
| 10954L | Ribosomal 30S | 17.6 | 122 | |
| 11000L | Ribosomal S7 | 13.9 | 117 | |
| 9916L | Dihydrolipoamide dehydrogenase | 43.9 | 107 |
Proteins detected by matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS). The score contributed to protein hits can also be used to estimate their prevalence within the sample. In bold, extracellular or hypothesized extracellular proteins are identified. MW, molecular weight.
Table 2.
Proteins detected in the OM and OMVs of WecD mutant strains of S. marcescensa
| Fraction | Systematic gene name | Annotation | MW | Score |
|---|---|---|---|---|
| OMV | 6253L | Flagellin | 34.6 | 920 |
| 5558L | OmpW | 23.8 | 697 | |
| 12753R | Ef-Tu | 43.2 | 624 | |
| 4161L | Lipase | 64.9 | 622 | |
| 10801L | Maltoporin | 48.2 | 596 | |
| 7646L | OmpC | 41.4 | 546 | |
| 6014L | Porin | 39.6 | 533 | |
| 8985R | Phospholipase | 35.6 | 523 | |
| 12577R | Serralysin A | 54.0 | 519 | |
| 5653R | Alcohol dehydrogenase | 96.2 | 317 | |
| 2860L | OmpF | 41.7 | 350 | |
| 2408R | SLA | 101.2 | 326 | |
| 7297L | Alanine amidase | 28.4 | 305 | |
| 12364R | Chitinase | 61.0 | 273 | |
| 5731L | MipA protein | 28.5 | 273 | |
| 1470R | Peptidoglycan-associated lipoprotein | 19.4 | 259 | |
| 9153L | Omp factor YaeT | 89.6 | 260 | |
| 2927L | OmpA | 39.2 | 250 | |
| 13014R | GroEL | 56.6 | 249 | |
| 3763R | Tsx | 33.4 | 234 | |
| 1467R | TolB | 46.0 | 227 | |
| 4103L | Lipoprotein precursor | 8.3 | 198 | |
| 10350R | Hypothetical protein | 19.9 | 191 | |
| 409R | Lipoprotein NlpD | 35.0 | 185 | |
| OM | 4161L | Lipase | 64.9 | 1,002 |
| 6253L | Flagellin | 34.6 | 929 | |
| 12577R | Serralysin A | 54 | 879 | |
| 2927L | OmpA | 39.2 | 798 | |
| 1467R | TolB | 46.0 | 629 | |
| 10814R | Putative membrane protein | 56.9 | 597 | |
| 7646L | OmpC | 41.4 | 537 | |
| 2408R | SLA | 101.2 | 530 | |
| 8612L | OM autotransporter | 102.3 | 476 | |
| 7297L | Alanine amidase | 28.4 | 474 | |
| 1283L | Glutamate transporter | 33.1 | 418 | |
| 2860L | OmpF | 41.7 | 354 | |
| 9153L | OM assembly protein | 89.6 | 349 | |
| 960L | Putative lipoprotein | 20.7 | 339 | |
| 5558L | OmpW | 23.8 | 334 | |
| 10801L | Maltoporin | 48.2 | 298 | |
| 1013R | Putative lipoprotein | 16.9 | 291 | |
| 2143R | OmpX | 18.4 | 289 | |
| 3535L | LolB | 25.0 | 281 | |
| 4561R | Serralysin B | 57.7 | 270 | |
| 4103L | Lipoprotein precursor | 8.3 | 268 | |
| 11524L | SSph2 | 107.5 | 261 | |
| 3763R | Tsx | 33.4 | 237 | |
| 5731L | MipA protein | 28.5 | 231 | |
| 9153L | Omp factor YaeT | 89.6 | 220 | |
| 6463R | Hcp1 | 16.8 | 213 | |
| 5643L | Periplasmic oligopeptide-binding protein | 61.5 | 204 | |
| 1473R | Tol-Pal protein YpgF | 28.1 | 201 | |
| 409R | Lipoprotein NlpD | 35.0 | 196 | |
| 3237L | YceI | 21.3 | 190 | |
| 1470R | Peptidoglycan-associated lipoprotein | 19.4 | 164 | |
| 4176L | OM lipoprotein | 16.6 | 162 | |
| 4456R | Peptidyl-dipeptidase Dcp | 82.4 | 156 | |
| 6014L | Porin | 39.6 | 151 | |
| 6011L | LysM | 34.5 | 151 | |
| 10162R | TolC | 54.5 | 150 | |
| 12369R | Extracellular solute-binding protein | 64.5 | 142 |
In bold, extracellular or hypothesized extracellular proteins are identified. Only proteins scoring above 100 are listed. The S. marcescens Db11 genome can be found at http://www.sanger.ac.uk/Projects/S_marcescens/. MW, molecular weight.
RcsB and not RcsA is necessary for hypervesiculation in the ECA-deficient background.
As mentioned earlier, mutating the ECA synthesis pathway has been previously shown to activate the Rcs phosphorelay (8). We analyzed if the hypervesiculation phenotype observed was dependent on the Rcs phosphorelay pathway by comparing the amounts of OMV produced by the wecD mutant and double wecD-rcsB and wecD-rcsA mutants. At 37°C, the hypervesiculation phenotype resulting from the introduction of a mutation into the ECA synthesis pathway was suppressed through the introduction of a secondary mutation in rcsB (Fig. 4A), with over 20 times the quantity of OMVs recovered in the wecD strain compared to the rcsB-wecD double mutant at this temperature. Conversely, vesiculation was not restored to WT levels in a wecD-rcsA double mutant. This indicated that within the Rcs phosphorelay, rcsB is the response regulator responsible for hypervesiculation at 37°C. At 30°C, however, the phenotype was not restored by the introduction of a secondary mutation in rcsB, suggesting that a factor unrelated to rcsB is involved in this process (Fig. 4A). To discount the effect of temperature on OMV production caused by disruption of ECA synthesis, we estimated vesiculation at 30°C and 37°C in terms of fold increase or decrease relative to their respective WT levels (Fig. 4B and C). This analysis revealed a 20-fold increase in vesiculation at 37°C, compared just a 3-fold increase at 30°C in the wecD mutant strain relative to WT levels (Fig. 4B and C). It is known that temperature has a marked effect on the lifestyle of the organism, with several genes already identified as being expressed differently at 30°C and 37°C (38, 62). At 30°C and 37°C, rcsB and rcsA single mutants display amounts of vesiculation comparable to that of the WT strain, suggesting that the loss of this Rcs response regulator does not result in stress-induced vesiculation.
Fig 4.

The loss of rcsB within an ECA mutant strain results in the reversion of the hypervesiculation phenotype. A comparison of the net amount of OMV recovered from the various strains is displayed in panel A. Panels B (37°C) and C (30°C) display the same data in terms of a fold difference in vesiculation,with WT levels set to 1. Note the difference in scale between panels B and C. These results demonstrate that introduction of a mutation into the rcsB gene reverts the hypervesiculation phenotype caused by the wecD mutation at 37°C. Average Kdo values were calculated from five separate experiments, and the error bars indicate standard deviations.
Proteomic analysis of S. marcescens OMVs.
It has been reported that protein cargo selection occurs in P. gingivalis upon the formation of OMV and that this process requires the integrity of the LPS (21). The enrichment or exclusion of outer membrane proteins from OMVs has also been reported for other bacteria, but a mechanism has not been identified (22, 28, 63). We tested if cargo selection in S. marcescens OMV takes place by comparing the protein compositions of OMV and OM at 30°C. This condition was chosen because at 30°C, vesiculation is activated in WT cells. The OMs and OMVs were harvested from overnight cultures and the protein profile analyzed by SDS-PAGE. The major bands present in Fig. 5 were identified by liquid chromatography-MS (LC-MS). MS analysis of the entire proteomic content of each fraction also showed that each fraction contained a different subset of proteins. For example, OmpA, OmpW, and OmpX seemed to be enriched in the OMV fraction (Fig. 5). The extracellular protease serralysin also appeared to be enriched in the OMV fraction. However, because this protein is secreted via a type I secretion system before its association to OMVs, its presence in the OMV fraction does not reflect a preferential sorting of this protein into the OMVs (29). To perform a more comprehensive comparison between the protein contents of OMVs and OMs, samples containing purified vesicles and OMs were subjected to trypsin digestion and fingerprinting by MS (Fig. 6 and Tables 1 and 2). This analysis can detect proteins present in minimal amounts but only indicates the presence or absence of proteins, without differentiating levels of proteins in the samples. Several outer membrane proteins, like TolC, LptD, maltoporin, and YaeT, were detected in the OM and not in the OMV fractions. It is tempting to speculate that these proteins are excluded from the vesicles during OMV biogenesis. A number of proteins were detected in OMV samples only. These are the outer membrane protein MipA and periplasmic alanine amidase and extracellular proteins like lipase A, phospholipase, and serralysin. Due to their localization to the periplasm, none of these proteins are expected to be found in OM fractions (29). Lipase A and phospholipase, like serralysin, likely associate with OMVs after their secretion into the media. Taken together, these data suggest that some proteins are enriched in OMVs and others are excluded from OMVs. These data support the hypothesis of cargo selection in OMVs that has been made for other organisms (21, 22).
Fig 5.
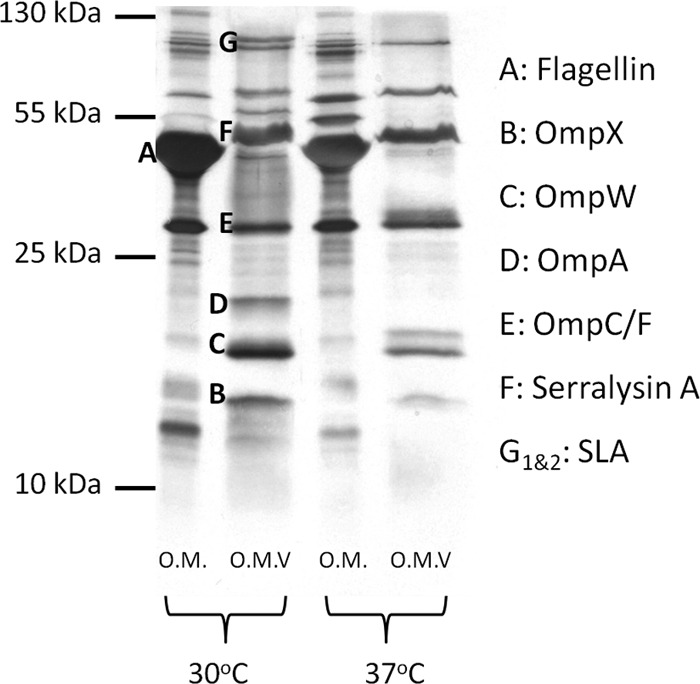
Protein profiles of the OM and OMVs appear different in WT S. marcescens. Purified OMs and OMVs were analyzed by silver staining and 15% glycine-PAGE. Cell fractions were collected from WT bacteria grown at 30°C and 37°C.
Fig 6.

Schematic of the compartmentalization of OM and OMV proteins. The Venn diagram indicates the location of proteins isolated from the OM and the OMVs of wild-type cells grown at 30°C. Proteomic content was determined by tryptic digestion of the entire fraction, followed by MS fingerprinting analysis.
OMVs from WT cells are toxic to Galleria mellonella independently of LPS.
It has been proposed that OMVs may play a role as vectors for the long-distance delivery of virulence determinants. To determine if the OMVs of S. marcescens carry active toxic components, we investigated their toxicity to larvae of the greater wax moth, Galleria mellonella. The immune response of the larvae has been well characterized, with haemocoel coagulation, cellular phagocytosis, and phenol oxidase-based melanization employed to defend from infection. Toll-like receptors and peptidoglycan receptors are also present for the recognition of invasive microbes (53). We investigated the effect of the injection of a defined amount of OMVs into the haemocoel of the larvae. Figure 7 shows that OMVs collected at both 30°C and 37°C, in an amount equivalent to 10 ng of Kdo and 500 to 750 ng of proteins, resulted in 100% larval death within 24 h. The toxicity was independent of LPS, as the same amount of LPS did not cause a similar effect, with more than 75% survival after 24 h. There was no significant difference between the toxicities of WT OMVs derived from cells grown at 30°C and at 37°C.
Fig 7.

OMVs have the ability to kill G. mellonella larvae. The effect of the injection of 10 ng of Kdo derived from OMVs and LPS into the haemocoel on survival is seen. The majority of the larvae died within 24 h, and this was independent of LPS toxicity, as injection of 10 ng of Kdo of LPS had no effect on survival. A PBS control is included in gray. The data points represent the average for 60 worms. The error corresponds to the standard deviation.
DISCUSSION
For several decades, OMVs were considered to be microscopy artifacts and/or cell debris. However, in the last years several reports have shown that OMV biogenesis is a fine-tuned process in a number of Gram-negative bacteria. In this work we showed that S. marcescens produces large amounts of OMVs at 22 or 30°C, while negligible quantities of OMVs are produced at 37°C. To our knowledge, the identification of thermoregulation control in OMV biogenesis in Gram-negative bacteria is unprecedented, and this finding adds to the evidence that vesiculation is an active, and a highly regulated, process.
We observed an increase in vesiculation by mutating genes involved in the ECA synthesis pathway. Despite the presence of ECA on the surface of all Gram-negative enteric bacteria, its role is relatively unknown. ECA mutant strains of Salmonella enterica were found to have lower resistance to bile and were attenuated in oral and interperitoneal mouse models of infection, though it remains possible that this was a secondary result of the accumulation of undecaprenyl monophosphate or lipid II intermediates (18, 50). The mutation of wecA, wecD, and wecG eliminates all forms of ECA, of which there are three forms, cyclic periplasmic, LPS anchored, and linked to phosphatidylglycerol (13, 25). Figure 4 shows that at both 37°C and 30°C, the mutations to wecA, wecD, and wecG, despite resulting in the accumulation of different lipid intermediates, all have upregulated vesiculation levels, suggesting that it is the loss of one or all forms of ECA rather than the accumulation of undecaprenol monophosphate, lipid I, or lipid II intermediates that causes the hypervesiculation. The mutation of these genes results in the activation of rcsB at 37°C (8), but the loss of rcsB in the ECA-deficient background results in low levels of vesiculation, comparable to those of the wild type, at 37°C (Fig. 5). A wide variety of other stimuli that affect the integrity of the cell envelope are also known to induce the Rcs phosphorelay, including high osmolarity and the overexpression of some membrane proteins (39). We have also noticed an increase in vesiculation in WT cells grown in high-osmolarity media, suggesting that OMV production through RcsB is not specific to ECA loss but a more general mechanism for coping with OM stress (data not shown). The deletion of genes involved in LPS synthesis is known to activate the Rcs phosphorelay in other bacteria (50), and recently, it was shown that in Vibrio fischeri, the overexpression of a sensor kinase which regulates polysaccharide production also contributed to an increase in vesiculation (57). We could consequently speculate that the cell surface carbohydrates not only are important for cell recognition and interactions but also play an important role in maintaining the integrity of the cell envelope and triggering vesiculation as a stress response.
Thermoregulation within the Rcs phosphorelay has been reported before, as the unstable effector rcsA is thought to be more readily degraded at higher temperatures, rendering Rcs-dependent expression of cps genes greatly reduced (58). However, we have shown that rcsA does not contribute to the increase in vesiculation in this case (Fig. 5). Little is known about thermoregulation in S. marcescens. The production of the surfactant serrawettin is known to occur only at 30°C (38, 62). The ability of S. marcescens to swarm, swim, and express flagellin is also differentially affected by growth at 30°C and at 37°C (7). The model that we propose is that at 37°C, significant vesiculation is not detected, but upon disruption of the ECA synthesis pathway, rcsB is activated and rcsB-regulated gene or genes encoding enzymes involved in OMV biogenesis are induced, resulting in hypervesiculation. At 30°C, a non-rcsB-dependent mechanism moderately induces vesiculation in wild-type cells. The rcsB DNA recognition sequence has been identified, and we have found the recognition site upstream of a number of important LPS-modifying genes, for example, that for the lipid A acyltransferase PagP (5). In a recent study, it was determined that lipid A had a different acetylation profile in OMVs than in the OM in P. gingivalis (21). It is tempting to speculate that Rcs-controlled genes like the PagP gene would modify lipid A in S. marcescens, contributing to an increase in vesicle production.
As previously observed for the OMVs of Xenorhabdus nematophilus (32), the OMVs of S. marcescens were toxic to G. mellonella, supporting the theory that bacterial OMVs can act as vectors for delivery of virulence factors. The extracellular proteases of S. marcescens were shown to be toxic to G. mellonella (27), and these are likely the same proteases identified within the OMVs (Tables 1 and 2). A large number of other virulence-associated factors (lipases, phospholipases, and chitinases) are present in these OMVs, and therefore, it is unsurprising that they have the ability to induce larval death; however, the toxicity of S. marcescens OMV preparations to G. mellonella has not been demonstrated before, and this simple model provides a useful tool in the characterization of the role of the OMVs in pathogenesis.
As study into OMV biogenesis progresses, it is likely that further characterization of the complex interactions between the outer membrane and peptidoglycan will be critical to our understanding of the mechanistic requirements of the process. The identification of an organism which upregulates and downregulates vesiculation through the well-characterized Rcs phosphorelay could be an important step toward our understanding of the dynamic process of vesiculation. Identification of the key components of the process could allow for the design of novel antibiotics to inhibit this key step in pathogenesis for many enteric bacteria. Alternatively, the process could be exploited for the improvement of the OMV-based vaccines (6, 48), which may impact global vaccination strategies.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Florencia Haurat for critical reading of the manuscript, Jennifer Ng for technical assistance, and Bela Reiz (Department of Chemistry, University of Alberta) for assistance with mass spectrometry.
This work was supported by grants from the Alberta Glycomics Centre. E.G.V. and M.E.C. are supported by grants from Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) and from Consejo Nacional de Investigaciones Científicas y Tecnológicas (CONICET), Argentina. E.G.V. and M.E.C. are career investigators of CONICET, Argentina. M.F.F. is an Alberta Heritage Foundation for Medical Research (AHFMR) scholar and a Canadian Institute for Health Research (CIHR) New Investigator.
Footnotes
Published ahead of print 6 April 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Begic S, Worobec EA. 2008. The role of the Serratia marcescens SdeAB multidrug efflux pump and TolC homologue in fluoroquinolone resistance studied via gene-knockout mutagenesis. Microbiology 154:454–461 [DOI] [PubMed] [Google Scholar]
- 2. Berthelot P, et al. 1999. Investigation of a nosocomial outbreak due to Serratia marcescens in a maternity hospital. Infect. Control Hosp. Epidemiol. 20:233–236 [DOI] [PubMed] [Google Scholar]
- 3. Beveridge TJ. 1999. Structures of gram-negative cell walls and their derived membrane vesicles. J. Bacteriol. 181:4725–4733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bielig H, Dongre M, Zurek B, Wai SN, Kufer TA. 2011. A role for quorum sensing in regulating innate immune responses mediated by Vibrio cholerae outer membrane vesicles (OMVs). Gut Microbes 2:274–279 [DOI] [PubMed] [Google Scholar]
- 5. Bishop RE. 2005. The lipid A palmitoyltransferase PagP: molecular mechanisms and role in bacterial pathogenesis. Mol. Microbiol. 57:900–912 [DOI] [PubMed] [Google Scholar]
- 6. Caron F, et al. 2011. From tailor-made to ready-to-wear meningococcal B vaccines: longitudinal study of a clonal meningococcal B outbreak. Lancet Infect. Dis. 11:455–463 [DOI] [PubMed] [Google Scholar]
- 7. Castelli ME, et al. 2008. Enterobacterial common antigen integrity is a checkpoint for flagellar biogenesis in Serratia marcescens. J. Bacteriol. 190:213–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Castelli ME, Vescovi EG. 2011. The Rcs signal transduction pathway is triggered by enterobacterial common antigen structure alterations in Serratia marcescens. J. Bacteriol. 193:63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Demuth DR, James D, Kowashi Y, Kato S. 2003. Interaction of Actinobacillus actinomycetemcomitans outer membrane vesicles with HL60 cells does not require leukotoxin. Cell. Microbiol. 5:111–121 [DOI] [PubMed] [Google Scholar]
- 10. Dutta S, et al. 2004. Release of Shiga toxin by membrane vesicles in Shigella dysenteriae serotype 1 strains and in vitro effects of antimicrobials on toxin production and release. Microbiol. Immunol. 48:965–969 [DOI] [PubMed] [Google Scholar]
- 11. Ebel W, Vaughn GJ, Peters HK, III, Trempy JE. 1997. Inactivation of mdoH leads to increased expression of colanic acid capsular polysaccharide in Escherichia coli. J. Bacteriol. 179:6858–6861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ellis TN, Kuehn MJ. 2010. Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol. Mol. Biol. Rev. 74:81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Erbel PJ, et al. 2004. Cyclic enterobacterial common antigen: potential contaminant of bacterially expressed protein preparations. J. Biomol. NMR 29:199–204 [DOI] [PubMed] [Google Scholar]
- 14. Fedrigo GV, Campoy EM, Di Venanzio G, Colombo MI, Garcia Vescovi E. 2011. Serratia marcescens is able to survive and proliferate in autophagic-like vacuoles inside non-phagocytic cells. PLoS One 6:e24054 doi:10.1371/journal.pone.0024054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferrières L, Clarke DJ. 2003. The RcsC sensor kinase is required for normal biofilm formation in Escherichia coli K-12 and controls the expression of a regulon in response to growth on a solid surface. Mol. Microbiol. 50:1665–1682 [DOI] [PubMed] [Google Scholar]
- 16. Furuta N, et al. 2009. Porphyromonas gingivalis outer membrane vesicles enter human epithelial cells via an endocytic pathway and are sorted to lysosomal compartments. Infect. Immun. 77:4187–4196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gankema H, Wensink J, Guinee PA, Jansen WH, Witholt B. 1980. Some characteristics of the outer membrane material released by growing enterotoxigenic Escherichia coli. Infect. Immun. 29:704–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gilbreath JJ, et al. 2012. Enterobacterial common antigen mutants of Salmonella enterica serovar Typhimurium establish a persistent infection and provide protection against subsequent lethal challenge. Infect. Immun. 80:441–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hagiwara D, et al. 2003. Genome-wide analyses revealing a signaling network of the RcsC-YojN-RcsB phosphorelay system in Escherichia coli. J. Bacteriol. 185:5735–5746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hancock I, Poxton I. 1988. Appendix 1, p 273 Hancock I, Poxton I. (ed), Bacterial cell surface techniques, John Wiley & Sons, Chichester, United Kingdom [Google Scholar]
- 21. Haurat MF, et al. 2011. Selective sorting of cargo proteins into bacterial membrane vesicles. J. Biol. Chem. 286:1269–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horstman AL, Kuehn MJ. 2000. Enterotoxigenic Escherichia coli secretes active heat-labile enterotoxin via outer membrane vesicles. J. Biol. Chem. 275:12489–12496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Irazoqui JE, et al. 2010. Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus. PLoS Pathog. 6:e1000982 doi:10.1371/journal.ppat.1000982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kadurugamuwa JL, Beveridge TJ. 1995. Virulence factors are released from Pseudomonas aeruginosa in association with membrane vesicles during normal growth and exposure to gentamicin: a novel mechanism of enzyme secretion. J. Bacteriol. 177:3998–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kajimura J, Rahman A, Rick PD. 2005. Assembly of cyclic enterobacterial common antigen in Escherichia coli K-12. J. Bacteriol. 187:6917–6927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaparakis M, et al. 2010. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell. Microbiol. 12:372–385 [DOI] [PubMed] [Google Scholar]
- 27. Kaska M, Lysenko O, Chaloupka J. 1976. Exocellular proteases of Serratia marcescens and their toxicity to larvae of Galleria mellonella. Folia Microbiol. (Praha) 21:465–473 [DOI] [PubMed] [Google Scholar]
- 28. Kato S, Kowashi Y, Demuth DR. 2002. Outer membrane-like vesicles secreted by Actinobacillus actinomycetemcomitans are enriched in leukotoxin. Microb. Pathog. 32:1–13 [DOI] [PubMed] [Google Scholar]
- 29. Kawai E, Akatsuka H, Idei A, Shibatani T, Omori K. 1998. Serratia marcescens S-layer protein is secreted extracellularly via an ATP-binding cassette exporter, the Lip system. Mol. Microbiol. 27:941–952 [DOI] [PubMed] [Google Scholar]
- 30. Kesty NC, Kuehn MJ. 2004. Incorporation of heterologous outer membrane and periplasmic proteins into Escherichia coli outer membrane vesicles. J. Biol. Chem. 279:2069–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kesty NC, Mason KM, Reedy M, Miller SE, Kuehn MJ. 2004. Enterotoxigenic Escherichia coli vesicles target toxin delivery into mammalian cells. EMBO J. 23:4538–4549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khandelwal P, Banerjee-Bhatnagar N. 2003. Insecticidal activity associated with the outer membrane vesicles of Xenorhabdus nematophilus. Appl. Environ. Microbiol. 69:2032–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reference deleted.
- 34. Kuehn MJ, Kesty NC. 2005. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 19:2645–2655 [DOI] [PubMed] [Google Scholar]
- 35. Kulp A, Kuehn MJ. 2010. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu. Rev. Microbiol. 64:163–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leverrier P, et al. 2011. Crystal structure of the outer membrane protein RcsF, a new substrate for the periplasmic protein-disulfide isomerase DsbC. J. Biol. Chem. 286:16734–16742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li Z, Clarke AJ, Beveridge TJ. 1998. Gram-negative bacteria produce membrane vesicles which are capable of killing other bacteria. J. Bacteriol. 180:5478–5483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lindum PW, et al. 1998. N-Acyl-l-homoserine lactone autoinducers control production of an extracellular lipopeptide biosurfactant required for swarming motility of Serratia liquefaciens MG1. J. Bacteriol. 180:6384–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Majdalani N, Gottesman S. 2005. The Rcs phosphorelay: a complex signal transduction system. Annu. Rev. Microbiol. 59:379–405 [DOI] [PubMed] [Google Scholar]
- 40. Majdalani N, Heck M, Stout V, Gottesman S. 2005. Role of RcsF in signaling to the Rcs phosphorelay pathway in Escherichia coli. J. Bacteriol. 187:6770–6778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maragakis LL, et al. 2008. Outbreak of multidrug-resistant Serratia marcescens infection in a neonatal intensive care unit. Infect. Control Hosp. Epidemiol. 29:418–423 [DOI] [PubMed] [Google Scholar]
- 42. Marolda CL, Lahiry P, Vines E, Saldias S, Valvano MA. 2006. Micromethods for the characterization of lipid A-core and O-antigen lipopolysaccharide. Methods Mol. Biol. 347:237–252 [DOI] [PubMed] [Google Scholar]
- 43. Mashburn LM, Whiteley M. 2005. Membrane vesicles traffic signals and facilitate group activities in a prokaryote. Nature 437:422–425 [DOI] [PubMed] [Google Scholar]
- 44. Mashburn-Warren L, et al. 2008. Interaction of quorum signals with outer membrane lipids: insights into prokaryotic membrane vesicle formation. Mol. Microbiol. 69:491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mashburn-Warren LM, Whiteley M. 2006. Special delivery: vesicle trafficking in prokaryotes. Mol. Microbiol. 61:839–846 [DOI] [PubMed] [Google Scholar]
- 46. McBroom AJ, Kuehn MJ. 2007. Release of outer membrane vesicles by Gram-negative bacteria is a novel envelope stress response. Mol. Microbiol. 63:545–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Murdoch SL, et al. 2011. The opportunistic pathogen Serratia marcescens utilizes type VI secretion to target bacterial competitors. J. Bacteriol. 193:6057–6069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oster P, et al. 2007. Immunogenicity and safety of a strain-specific MenB OMV vaccine delivered to under 5-year olds in New Zealand. Vaccine 25:3075–3079 [DOI] [PubMed] [Google Scholar]
- 49. Park KS, et al. 2010. Outer membrane vesicles derived from Escherichia coli induce systemic inflammatory response syndrome. PLoS One 5:e11334 doi:10.1371/journal.pone.0011334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ramos-Morales F, Prieto AI, Beuzon CR, Holden DW, Casadesus J. 2003. Role for Salmonella enterica enterobacterial common antigen in bile resistance and virulence. J. Bacteriol. 185:5328–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ruiz N, Montero T, Hernandez-Borrell J, Vinas M. 2003. The role of Serratia marcescens porins in antibiotic resistance. Microb. Drug Resist. 9:257–264 [DOI] [PubMed] [Google Scholar]
- 52. Schooling SR, Beveridge TJ. 2006. Membrane vesicles: an overlooked component of the matrices of biofilms. J. Bacteriol. 188:5945–5957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Seitz V, et al. 2003. Identification of immunorelevant genes from greater wax moth (Galleria mellonella) by a subtractive hybridization approach. Dev. Comp. Immunol. 27:207–215 [DOI] [PubMed] [Google Scholar]
- 54. Sharpe SW, Kuehn MJ, Mason KM. 2011. Elicitation of epithelial cell-derived immune effectors by outer membrane vesicles of nontypeable Haemophilus influenzae. Infect. Immun. 79:4361–4369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shetty A, Chen S, Tocheva EI, Jensen GJ, Hickey WJ. 2011. Nanopods: a new bacterial structure and mechanism for deployment of outer membrane vesicles. PLoS One 6:e20725 doi:10.1371/journal.pone.0020725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68:850–858 [DOI] [PubMed] [Google Scholar]
- 57. Shibata S, Visick KL. 2012. Sensor kinase RscS induces the production of antigenically distinct outer membrane vesicles that depend on the symbiosis polysaccharide locus in Vibrio fischeri. J. Bacteriol. 194:185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sledjeski DD, Gottesman S. 1996. Osmotic shock induction of capsule synthesis in Escherichia coli K-12. J. Bacteriol. 178:1204–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Suh B, Bae IK, Kim J, Jeong SH, Yong D, Lee K. 2010. Outbreak of meropenem-resistant Serratia marcescens comediated by chromosomal AmpC beta-lactamase overproduction and outer membrane protein loss. Antimicrob. Agents Chemother. 54:5057–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tsai CM, Frasch CE. 1982. A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal. Biochem. 119:115–119 [DOI] [PubMed] [Google Scholar]
- 61. Vidakovics ML, et al. 2010. B cell activation by outer membrane vesicles—a novel virulence mechanism. PLoS Pathog. 6:e1000724 doi:10.1371/journal.ppat.1000724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wei JR, Lai HC. 2006. N-acylhomoserine lactone-dependent cell-to-cell communication and social behavior in the genus Serratia. Int. J. Med. Microbiol. 296:117–124 [DOI] [PubMed] [Google Scholar]
- 63. Wensink J, Witholt B. 1981. Outer-membrane vesicles released by normally growing Escherichia coli contain very little lipoprotein. Eur. J. Biochem. 116:331–335 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.