

MatP is a small DNA-binding protein of about 18 kDa. In order to understand the DNA-compaction mechanism of MatP at an atomic level, the structures of apo MatP and of the nucleoprotein complex MatP–matS have been studied.
Keywords: MatP, matS, DNA-binding proteins, Escherichia coli
Abstract
The Escherichia coli chromosome is organized into four macrodomains which are found in the replication-origin region (Ori), at the terminus (Ter) and on both its sides (Right and Left). The localization of the macrodomains is subject to programmed changes during the cell cycle. The compaction of the 800 kb Ter macrodomain relies on the binding of the MatP protein to a 13 bp matS motif repeated 23 times. MatP is a small DNA-binding protein of about 18 kDa that shares homology in its C-terminal region with the ribbon–helix–helix (RHH) motifs present in regulatory DNA-binding proteins such as CopG. In order to understand the DNA-compaction mechanism of MatP at an atomic level, it was decided to study the structure of apo MatP and of the nucleoprotein complex MatP–matS by both X-ray diffraction and SAXS analysis. It was demonstrated that MatP forms dimers that bind a single matS motif. Complete native X-ray data sets were collected and phasing of the diffraction data is under way.
1. Introduction
If the bacterial chromosome were stretched, it would cover 1000 times the length of the bacterial cell. DNA supercoiling therefore folds and organizes the chromosome into a compact form called the nucleoid. Replication and transcription take place simultaneously in the nucleoid, even during chromosome partitioning. Nucleoid-associated proteins are abundant, often bind DNA with a low degree of sequence specificity, and impose constraints on DNA topology (Dame et al., 2006 ▶; van Noort et al., 2004 ▶; Cosgriff et al., 2010 ▶).
The subcellular localization of different DNA segments in Escherichia coli, the Ori domain in the replication-origin region and the Ter domain in the terminal region, was initially proposed by Niki et al. (2000 ▶) using fluorescence in situ hybridization. The organization of the chromosome of E. coli into four chromosomal macrodomains (Ori, Ter, and Right and Left on both its sides) was established by measurement of the frequency of recombination between phage λ att sites scattered throughout the E. coli genome (Valens et al., 2004 ▶). Through simultaneous differential fluorescent labelling of two specific chromosomal sites, it was demonstrated that the two right and left chromosomal arms are located in separate cell halves (Nielsen et al., 2006 ▶; Wang et al., 2006 ▶). Examination of the movement of chromosomal loci during a complete cell cycle by tracking fluorescent markers in time-lapse microscopy revealed that the localization and dynamics of chromosomal DNA vary according to the macrodomain topography (Espeli et al., 2008 ▶).
High-throughput analysis of DNA-binding events across bacterial genomes using chromatin-immunoprecipitation (ChIP) experiments (Grainger et al., 2006 ▶) has revealed that some major regulators of the cell have macrodomain-specific DNA-binding profiles. Three proteins that possess macrodomain-specific chromosome-binding properties (SeqA, SlmA and MatP) have recently been discovered (for a review, see Dame et al., 2011 ▶). MatP is a protein of about 18 kDa that is conserved in Enterobacteriaceae and Vibrionaceae and to a lesser extent in Pasteurellaceae species. MatP binds exclusively to the Ter macrodomain and has been shown to be its main organizer. In the absence of MatP, segregation of the Ter macrodomain occurs early in the cell cycle, no cohesion step between duplicated Ter macrodomains is observed, DNA is less compacted and the mobility of markers located in the macrodomain is increased (Mercier et al., 2008 ▶). A short motif of 13 bp called matS has been characterized as the specific recognition site for MatP. matS is repeated 23 times in a 800 kb domain located in the Ter region of the chromosome. The deletion of a matS site increases the mobility of the macrodomain.
In order to understand how MatP organizes the Ter macrodomain via binding to the matS sites, we set out to study the structure of the E. coli MatP–matS complex by both X-ray crystallography and small-angle X-ray scattering (SAXS) in solution.
2. Material and methods
2.1. Sequence amplification and cloning
DNA encoding full-length MatP was amplified by PCR from genomic DNA of E. coli using the following primers: 5′-GGTCCCATGGGCAAATATCAACAACTT-3′ as the forward primer and 5′-GGTCCTCGAGTTATTCCTTACCCAGCAATGC-3′ as the reverse primer. The resulting fragment was cloned into pET28 vector (Invitrogen). The construct did not contain any affinity-labelling tag.
2.2. Protein expression and purification, SeMet protein labelling, oligonucleotide labelling for cocrystallization and nucleoprotein-complex reconstitution
The plasmid was transformed into E. coli Rosetta (DE3) pLysS competent cells (Novagen) by electroporation. The cells were grown at 310 K in 2YT medium containing kanamycin (50 µg ml−1). Protein expression was induced using 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) when the OD600 reached about 0.8–1.0; the cells were grown at 310 K for 4 h after induction. The cells were harvested by centrifugation at 5000g for 15 min and were resuspended in lysis buffer consisting of 20 mM Tris–HCl pH 7.5, 200 mM sodium chloride. The cells were stored overnight at 253 K. Lysis was achieved by sonication at 277 K. The lysate was centrifuged at 13 000g at 281 K for 30 min. The supernatant was collected and loaded onto a 5 ml HiTrap Heparin HP column (GE Healthcare) previously equilibrated in buffer A (20 mM Tris–HCl pH 7.5, 200 mM sodium chloride), followed by a wash with five column volumes of buffer A. The protein was eluted with a linear gradient of buffer B (20 mM Tris–HCl pH 7.5, 1 M sodium chloride). Fractions containing MatP as indicated by SDS–PAGE were pooled and concentrated to 5 ml by ultrafiltration (Vivaspin) and further purified on a Superdex S75 HiLoad column (GE Healthcare) equilibrated in buffer A. Fractions containing MatP were pooled and concentrated to 1 mg ml−1 according to Nanodrop measurement at 280 nm. SeMet-labelled MatP was produced as described in Quevillon-Cheruel et al. (2007 ▶) and was purified as for the native protein.
The nucleoprotein complex was reconstituted in vitro by mixing equimolar amounts of MatP at 1 mg ml−1 and double-stranded native or iodo-labelled matS. No further purification steps were applied to the complex after mixing. Two different matS 23-mer oligonucleotides were used: one oligonucleotide had the specific 13-mer site in the centre, while the other had the matS site at the end (Fig. 1 ▶ a). The two types of nucleoprotein complex and the related crystals are named MatP–matS1 and MatP–matS2 in the following. The double-stranded DNA was prepared in water by hybridization of an equimolar mixture of the two complementary oligonucleotides (stock at 2 nmol µl−1 for each; 1 nmol µl−1 final concentration of the double-stranded DNA) by slow-cooling from 363 to 293 K in a water bath. The oligonucleotides were synthesized and purified (HPLC) by the company Eurogentec. Oligonucleotides substituted with one, two or three iodo-labelled bases (Eurogentec) were tested for phasing purposes.
Figure 1.

(a) Crystals of the MatP–matS1 and MatP–matS2 complexes. These crystals were used in data collection (Table 1 ▶). The sequences of the two double-stranded oligonucleotides used for cocrystallization are shown. The 13-mer matS sites are shown in green. (b) Typical 0.5° oscillation diffraction images of MatP–matS1 and MatP–matS2 crystals. Diffaction resolution circles are shown in pink.
2.3. Size-exclusion chromatography coupled to multi-angle laser light scattering (SEC-MALLS)
The determination of the molecular masses of MatP and the MatP–matS complex in solution was performed using Viscotek SEC–TDA equipment (Malvern Instruments). Elution was followed using a UV–visible spectrophotometer, a differential refractometer, a 7° low-angle light-scattering detector, a 90° right-angle light-scattering detector and a differential pressure viscometer. The OmniSEC software was used for the acquisition and analysis of the Viscotek data. 100 µl samples (1 mg ml−1) were injected at a flow rate of 0.5 ml min−1 onto a Superdex 75 HR 10/300 column (GE Healthcare) equilibrated in 20 mM Tris pH 7.5, 200 mM sodium chloride at 295 K. Lyophilized bovine serum albumin (BSA; Sigma–Aldrich catalogue No. A-7638) was used as the standard reference protein and was dissolved in the same buffer. The concentration was determined from the absorbance at 280 nm measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific). This reference protein of known molecular mass, concentration, refractive-index increment (dn/dc = 0.185 ml g−1) and intrinsic viscosity was used to determine the instrument-response factors of the detectors for the mobile phase being used.
2.4. Small-angle X-ray scattering (SAXS)
SAXS experiments on apo MatP were carried out on the SWING beamline at the SOLEIL synchrotron-radiation facility (Saint-Aubin, France). The incident beam energy was 12 keV and the sample-to-detector (Aviex CCD) distance was set to 1820 mm. The scattering-vector range was 0.01 < q < 0.50 Å−1, where q = 4πsinθ/λ and 2θ is the scattering angle. Several successive frames (typically 40) of 2 s each were recorded for both sample and pure solvent. We checked that the X-rays did not cause irradiation damage by comparing the successive frames before calculating the average intensity and subtracting the background. Resulting intensities were placed on an absolute scale using water scattering. Data were collected at various protein concentrations ranging from 0.27 to 0.71 mg ml−1. Smaller angle data at 0.27 mg ml−1 were merged with larger angle data at 0.71 mg ml−1 to yield a final composite curve free from any interparticle interaction contributions. Subsequent analysis and ab initio shape calculations were performed using the programs PRIMUS, GNOM, GASBOR and DAMAVER (Svergun et al., 1995 ▶, 2001 ▶; Konarev et al., 2003 ▶). Because concentrated samples of the MatP–matS1 and MatP–matS2 complexes tended to aggregate, the SAXS data could not be exploited.
2.5. Crystallization
Initial crystallization screenings of MatP or its complexes, either native, SeMet-labelled or iodo-labelled, stored in 20 mM Tris–HCl pH 7.5, 200 mM sodium chloride buffer were performed at 291 K using Greiner CrystalQuick sitting-drop vapour-diffusion plates. A Cartesian dispensing system (Genomic Solutions, Harvard Bioscience) was used to test 672 conditions from the following commercial screens from Qiagen: Classics, Classics II, JCSG+, PEGs, PEGs II, Nucleix and Protein Complex Suites. Drops consisted of 0.1 µl sample and 0.1 µl mother liquor. The reservoir volume was 100 µl. Crystals were obtained under various conditions for the two MatP–matS complexes (Fig. 1 ▶ a), but none were obtained for the apoprotein. The most promising crystallization conditions were optimized using automated two-dimensional screens (Leulliot et al., 2005 ▶) followed by transfer of the optimized conditions to a hanging-drop setup at 291 K. Amongst the numerous crystallization hits that we obtained, two were further optimized: condition No. 14 from Classics I Suite [25%(v/v) ethylene glycol] for MatP–matS1 and condition No. 78 from the JCSG+ Suite [240 mM disodium malonate, 20%(w/v) PEG 3350] for MatP–matS2. Conditions were optimized by screening the pH (between pH 4 and 6) and the ethylene glycol (18–24%) or PEG concentration (15–25%). The crystals were grown in droplets consisting of 1 or 2 µl protein or complex solution (1 mg ml−1) and 1 µl reservoir solution. The best crystals of MatP–matS1 were obtained using 20%(v/v) ethylene glycol in 100 mM sodium acetate pH 5. The crystals grew to their final dimensions in 2 d. The best crystals of MatP–matS2 were obtained using the commercial JCSG+ Suite solution in less than 1 d (Fig. 1 ▶). Crystals obtained in ethylene glycol could be directly flash-frozen in liquid nitrogen. Crystals that were obtained with PEG were cryoprotected using mother liquor complemented with 30%(v/v) ethylene glycol.
2.6. X-ray diffraction analysis, phasing approaches and molecular replacement
Crystals of MatP–matS1 and MatP–matS2 were tested on the ID23-2 beamline (225 mm MAR Mosaic CCD detector) at the ESRF, Grenoble, France or on the PROXIMA1 beamline (ADSC Q315r detector) at SOLEIL, Saclay, France (Fig. 1 ▶ b).
An initial complete data set was collected for native MatP–matS1 at the ESRF (λ = 0.87 Å; 2.7 Å resolution, space group P41212 or P43212, unit-cell parameters a = b = 75, c = 225 Å; Fig. 1 ▶ and Table 1 ▶). Four further data sets were collected from one MatP–matS1 crystal (2516 images in total, oscillation angle 0.5°) on PROXIMA-1 at a wavelength of 1.907 Å (3 Å resolution) in order to exploit the anomalous signal from S atoms (four S atoms per chain). One more data set was collected at λ = 0.98 Å from the same crystal.
Table 1. Data-collection and processing statistics.
| Data set | MatP–matS1 | MatP–matS2 |
|---|---|---|
| Beamline | ID23-2 | PROXIMA1 |
| Wavelength (Å) | 0.8726 | 1.2889 |
| Detector | MAR Mosaic CCD | ADSC Q315r |
| Temperature of data collection (K) | 93 | 93 |
| Crystal-to-detector distance (mm) | 346.56 | 367.14 |
| Rotation range per image (°) | 1 | 0.5 |
| Total rotation range (°) | 180 | 149 |
| Exposure time per image (s) | 1 | 0.4 |
| Resolution range (Å) | 47.96–2.69 (2.85–2.69) | 44.56–3.77 (4.00–3.77) |
| Unit-cell parameters (Å) | a = b = 75, c = 225 | a = 61, b = 149, c = 73 |
| Space group | P41212 or P43212 | C222 |
| Mosaicity (°) | 0.123 | 0.503 |
| No. of observations | 251894 (32116) | 19789 (3066) |
| No. of unique reflections | 18567 (2837) | 6239 (994) |
| Multiplicity | 13.5 (11.3) | 3.2 (3.1) |
| Completeness (%) | 99.5 (97.0) | 96.7 (94.8) |
| Mean I/σ(I) | 25.20 (2.92) | 9.17 (2.32) |
| Rmerge† (%) | 7.7 (56.0) | 14.2 (62.8) |
| Rmeas (%) | 8.9 (88.6) | 9.8 (55.7) |
| Overall B factor from Wilson plot (Å2) | 59.4 | 124.0 |
R
merge = 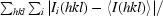
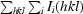 , where 〈I(hkl)〉 is the mean intensity of reflection hkl and I
i(hkl) is the intensity of the ith measurement of reflection hkl.
, where 〈I(hkl)〉 is the mean intensity of reflection hkl and I
i(hkl) is the intensity of the ith measurement of reflection hkl.
Complete data sets were also collected on the PROXIMA1 beamline for native MatP–matS2 (λ = 1.29 Å; 3.8 Å resolution, space group C222, unit-cell parameters a = 61, b = 149, c = 73 Å; Fig. 1 and Table 1 ▶) and for iodo-DNA-modified MatP–matS1 (λ = 1.91 Å; 8 Å resolution, space group C2221, unit-cell parameters a = 78, b = 127, c = 155 Å).
The images were integrated with the program XDS (Kabsch, 1993 ▶, 2010 ▶). The best X-ray data-collection statistics for each complex are gathered in Table 1 ▶. The unit-cell volume of the MatP–matS1 crystals was 1 243 669 Å3, which is compatible with one molecule of complex per asymmetric unit (molecular mass 50 kDa, calculated Matthews coefficient of 2.74 Å3 Da−1 and 60% solvent content). The MatP–matS2 crystals had a unit-cell volume of 658 199 Å3, corresponding to one copy of the complex per asymmetric unit (Matthews coefficient of 1.65 Å3 Da−1 and 33% solvent content) or to one copy of the dimer of MatP if we assumed that the complex dissociated during the crystallization trials (Matthews coefficient of 2.4 Å3 Da−1 and 47% solvent content).
3. Results and discussion
The recombinant expression of MatP in E. coli yielded about 10 mg protein per litre of culture. The purification process used standard protocols combining ion exchange and gel filtration. The purity of the final sample was checked by SDS–PAGE, and UV absorbance between 240 and 360 nm was used to verify the absence of contaminating nucleic acids. Gel filtration alone did not allow determination of the oligomeric state of MatP in solution since the elution volume was between those expected for a monomer and for a dimer. We therefore explored the quaternary structure of MatP using SEC-MALLS. The molecular mass of apo MatP determined by SEC-MALLS (Fig. 2 ▶ a) was 35 900 Da, which is compatible with the existence of a dimer [the calculated mass of a monomer is 17 693.3 Da; the hydrodynamic radius (Stokes radius) is 3.433 nm]. Analysis of the MatP–matS complex with an excess of matS DNA revealed a major peak centred at 48 100 Da, which is consistent with a complex composed of two copies of MatP plus one copy of double-stranded matS (calculated theoretical mass of 49 920 Da). A second peak eluted at 22 606 Da, which is compatible with the expected mass of the dsDNA (Fig. 2 ▶ a).
Figure 2.

(a) Oligomeric states of MatP and the MatP–matS complex in solution. SEC-MALLS curves of the MatP dimer (red line), the MatP–matS complex (blue line, first peak) and the matS oligonucleotide (blue line, second peak) are shown. (b) SAXS analysis of the MatP dimer. Left, distance distribution function P(r) of apo MatP. Right, experimental scattering curve (black dots) with the calculated curve (red line) from the model shown in (c). (c) Typical envelope for apo MatP obtained using the GASBOR program, shown in two orthogonal views. The crystal structure of the CopG dimer (PDB entry 1b01) was modelled into the envelope.
The oligomeric state of apo MatP was further confirmed by SAXS experiments (Fig. 2 ▶ b). The value of the scattered intensity at the origin I(0), which is directly proportional to the molecular mass of the scattering object, proved unequivocally that MatP is a dimer in solution. As shown in Fig. 2 ▶(b), the distance distribution function P(r) possesses two maxima that are characteristic of a dimer composed of two well separated monomers. The rather high values of the radius of gyration (R g = 31.5 ± 0.5 Å) and the maximum extension (D max = 105 ± 5 Å) suggested an elongated shape of the MatP dimer. This was confirmed by analysis of the molecular envelope obtained from the SAXS data using the GASBOR program, which describes the scattering MatP protein as a chain of (2 × 150) dummy residues. The data were interpreted by imposing P2 symmetry on the model. 50 envelopes were determined and then superimposed with the DAMAVER chain of programs. The various envelopes are rather similar since the NSD (normal spatial discrepancy) parameter used to quantify their difference was about 1.2, indicating good reliability of the solutions. The most typical solution (Fig. 2 ▶ c) corresponds to an envelope with an elongated shape and a small knob in the middle.
In order to find structural analogues of MatP, we used homology detection and structure prediction by HMM–HMM comparison using HHpred (Söding et al., 2005 ▶). Using the global alignment mode, three ribbon–helix–helix (RHH) proteins were found among the first ten HHpred hits [two PutA proteins (PDB entries 2gpe and 2ay0; Larson et al., 2006 ▶) and the ParD protein (PDB entry 2an7; Oberer et al., 2007 ▶)], with scores of 24.57, 21.4 and 19.84, respectively. The RHH motif is present in a class of prokaryotic transcription factors (for a review, see Schreiter & Drennan, 2007 ▶) and also in plasmid transcriptional repressors (Gomis-Rüth et al., 1998 ▶). The RHH motif is composed of one β-strand followed by two α-helices and usually associates as a dimer, forming a two-stranded central β-sheet that is involved in specific DNA recognition (Fig. 3 ▶). We then tested whether the central part of our SAXS envelope could accommodate the crystal structure of an RHH dimer. We selected the RHH dimer of CopG from Streptococcus agalactiae (PDB entry 1b01; Gomis-Rüth et al., 1998 ▶) because the quality of the sequence alignment between CopG and MatP was regular over the total length and no gaps were introduced (Fig. 3 ▶ a). The result is also shown in Fig. 2 ▶(c). The CopG RHH domain indeed fits nicely into this envelope, but the crystal structure of MatP will provide the definite answer.
Figure 3.

(a) Multiple alignment of the CopG and MatP sequences as obtained from ClustalW2 (Larkin et al., 2007 ▶). The figure was generated using ESPript (Gouet et al., 1999 ▶). The secondary-structure elements of CopG are shown at the top of the alignment. The four residues involved in DNA interaction in CopG are indicated by blue arrows. (b) Two 90° rotated views of the CopG dimer complexed with DNA.
We were unable to crystallize the apo MatP protein. However, crystals of various shapes and sizes (needles, sticks, cubes and lenses) were obtained of both the MatP–matS1 and MatP–matS2 complexes. Most crystals diffracted poorly, but complete native data sets were finally obtained for both complexes (Table 1 ▶). Unfortunately, molecular replacement using various crystal structures of RHH motifs [PutA (PDB entry 2gpe; Larson et al., 2006 ▶), Arc1 (PDB entry 1par; Raumann et al., 1994 ▶) and CopG (PDB entry 1bo1; Gomis-Rüth et al., 1998 ▶)] did not provide satisfactory solutions. We attempted to solve the phase problem using the sulfur anomalous signal. The anomalous signal extracted from the redundant data set collected at the sulfur-edge wavelength did not allow phasing of the structure. Crystals of complexes containing SeMet-labelled MatP or iodo-labelled matS all diffracted poorly. Heavy-atom soaking of native crystals is under way to solve the structure.
Our results show that MatP forms a dimer in solution and that it binds as a dimer to its matS recognition site. The final structure will reveal the molecular details of this interaction and will provide an important step in the understanding of this macrodomain-structuring protein.
Acknowledgments
We are grateful to F. Boccard, O. Espeli and P. Dupaigne for the gift of the E. coli genome and useful discussions. Structural data were collected on the PROXIMA1 beamline at Synchrotron SOLEIL (Saclay, France) and beamline ID23-2 at the ESRF (Grenoble, France). SAXS data were collected on the SWING beamline at Synchrotron SOLEIL (Saclay, France) or with a Bruker NanoSTAR located at IBBMC (Orsay, France). We thank the beamline staff for assistance and advice during data collection. This work was supported by funds from the CNRS, from the IFR115 Génomes, Transcriptomes, Protéomes and the University Paris-Sud 11 (UMR8619).
References
- Cosgriff, S., Chintakayala, K., Chim, Y. T. A., Chen, X., Allen, S., Lovering, A. L. & Grainger, D. C. (2010). Mol. Microbiol. 77, 1289–1300. [DOI] [PMC free article] [PubMed]
- Dame, R. T., Kalmykowa, O. J. & Grainger, D. C. (2011). PLoS Genet. 7, e1002123. [DOI] [PMC free article] [PubMed]
- Dame, R. T., Noom, M. C. & Wuite, G. J. L. (2006). Nature (London), 444, 387–390. [DOI] [PubMed]
- Espeli, O., Mercier, R. & Boccard, F. (2008). Mol. Microbiol. 68, 1418–1427. [DOI] [PubMed]
- Gomis-Rüth, F. X., Solá, M., Acebo, P., Párraga, A., Guasch, A., Eritja, R., González, A., Espinosa, M., del Solar, G. & Coll, M. (1998). EMBO J. 17, 7404–7415. [DOI] [PMC free article] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Métoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed]
- Grainger, D. C., Hurd, D., Goldberg, M. D. & Busby, S. J. (2006). Nucleic Acids Res. 34, 4642–4652. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (1993). J. Appl. Cryst. 26, 795–800.
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J. & Svergun, D. I. (2003). J. Appl. Cryst. 36, 1277–1282.
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Larson, J. D., Jenkins, J. L., Schuermann, J. P., Zhou, Y., Becker, D. F. & Tanner, J. J. (2006). Protein Sci. 15, 2630–2641. [DOI] [PMC free article] [PubMed]
- Leulliot, N., Trésaugues, L., Bremang, M., Sorel, I., Ulryck, N., Graille, M., Aboulfath, I., Poupon, A., Liger, D., Quevillon-Cheruel, S., Janin, J. & van Tilbeurgh, H. (2005). Acta Cryst. D61, 664–670. [DOI] [PubMed]
- Mercier, R., Petit, M. A., Schbath, S., Robin, S., El Karoui, M., Boccard, F. & Espéli, O. (2008). Cell, 135, 475–485. [DOI] [PubMed]
- Nielsen, H. J., Ottesen, J. R., Youngren, B., Austin, S. J. & Hansen, F. G. (2006). Mol. Microbiol. 62, 331–338. [DOI] [PubMed]
- Niki, H., Yamaichi, Y. & Hiraga, S. (2000). Genes Dev. 14, 212–223. [PMC free article] [PubMed]
- Noort, J. van, Verbrugge, S., Goosen, N., Dekker, C. & Dame, R. T. (2004). Proc. Natl Acad. Sci. USA, 101, 6969–6974. [DOI] [PMC free article] [PubMed]
- Oberer, M., Zangger, K., Gruber, K. & Keller, W. (2007). Protein Sci. 16, 1676–1688. [DOI] [PMC free article] [PubMed]
- Quevillon-Cheruel, S., Collinet, B., Trésaugues, L., Minard, P., Henckes, G., Aufrère, R., Blondeau, K., Zhou, C. Z., Liger, D., Bettache, N., Poupon, A., Aboulfath, I., Leulliot, N., Janin, J. & van Tilbeurgh, H. (2007). Methods Mol. Biol. 363, 21–37. [DOI] [PubMed]
- Raumann, B. E., Rould, M. A., Pabo, C. O. & Sauer, R. T. (1994). Nature (London), 367, 754–757. [DOI] [PubMed]
- Schreiter, E. R. & Drennan, C. L. (2007). Nature Rev. Microbiol. 5, 710–720. [DOI] [PubMed]
- Söding, J., Biegert, A. & Lupas, A. N. (2005). Nucleic Acids Res. 33, W244–W248. [DOI] [PMC free article] [PubMed]
- Svergun, D., Barberato, C. & Koch, M. H. J. (1995). J. Appl. Cryst. 28, 768–773.
- Svergun, D. I., Petoukhov, M. V. & Koch, M. H. J. (2001). Biophys. J. 80, 2946–2953. [DOI] [PMC free article] [PubMed]
- Valens, M., Penaud, S., Rossignol, M., Cornet, F. & Boccard, F. (2004). EMBO J. 23, 4330–4341. [DOI] [PMC free article] [PubMed]
- Wang, X., Liu, X., Possoz, C. & Sherratt, D. J. (2006). Genes Dev. 20, 1727–1731. [DOI] [PMC free article] [PubMed]