

The amino-terminal domain of the Hsp70 co-chaperone Bag2 from M. musculus has been crystallized in native and selenomethionyl forms diffracting to 2.27 and 3.1 Å resolution, respectively.
Keywords: Bag2, selenomethionine, SAD phasing
Abstract
Bag2, an atypical member of the Bag family of Hsp70 co-chaperones, acts as both an Hsp70 nucleotide-exchange factor and an inhibitor of the Hsp70-binding E3 ubiquitin ligase CHIP (carboxyl-terminus of Hsp70-interacting protein). The amino-terminal domain of Bag2 (Bag2-NTD), which is required for inhibition of CHIP, has no sequence homologs in the PDB. Native and selenomethionyl (SeMet) forms of Bag2-NTD were crystallized by hanging-drop vapor diffusion. Native Bag2-NTD crystals diffracted to 2.27 Å resolution and belonged to space group P212121, with unit-cell parameters a = 75.5, b = 84.7, c = 114.1 Å. SeMet Bag2-NTD crystals diffracted to 3.10 Å resolution and belonged to space group P212121, with unit-cell parameters a = 37.2, b = 53.3, c = 86.7 Å. Phases for the SeMet Bag2-NTD crystal were solved by single-wavelength anomalous diffraction. Initial phasing and model building using the 3.10 Å resolution SeMet Bag2-NTD data set suggested that Bag2-NTD forms a dimer and adopts a fold distinct from those of any domains annotated in the Pfam or SMART domain databases.
1. Introduction
The prevention of protein misfolding is a crucial aspect of cellular homeostasis. Protein quality-control systems in the distinct cellular compartments counter protein misfolding through substrate refolding or degradation (Chen et al., 2011 ▶). Protein refolding is carried out by molecular chaperones (Young et al., 2004 ▶), while protein degradation typically utilizes the ubiquitin/proteasome system (Hershko & Ciechanover, 1998 ▶) or the lysosome system (Cuervo & Dice, 1998 ▶). The cytosolic chaperone Hsp70 conducts protein refolding in an ATP-dependent manner through an iterative process of ATP hydrolysis, nucleotide exchange and action upon misfolded proteins directly bound to the chaperone (Bukau & Horwich, 1998 ▶; Hartl & Hayer-Hartl, 2002 ▶). As Hsp70-mediated refolding is limited by an inherently low rate of nucleotide exchange, efficient in vivo refolding requires interaction between Hsp70 and a nucleotide-exchange factor (NEF). Hsp70 NEFs such as the Bag-domain-containing family of proteins increase the rate of nucleotide exchange, thus enhancing the rate of chaperone-mediated protein refolding (Sondermann et al., 2001 ▶). Within the context of Hsp70-mediated protein refolding, proteasomal degradation is promoted by the E3 ubiquitin ligase CHIP (carboxyl-terminus of Hsp70-interacting protein; Connell et al., 2001 ▶; Meacham et al., 2001 ▶). CHIP binds to the C-terminal tail of Hsp70 and ubiquitinates misfolded clients bound to the chaperone.
Recent studies have identified the 23 kDa Bag-family member Bag2 as a regulator of the protein refolding/degradation balance for Hsp70 clients (Carrettiero et al., 2009 ▶; Dai et al., 2005 ▶; Arndt et al., 2005 ▶). Bag2 directly and indirectly promotes protein refolding via multiple mechanisms (Dai et al., 2005 ▶) that are associated with its two distinct domains: a predicted coiled-coil amino-terminal domain (Bag2-NTD) and a carboxy-terminal Bag domain. We have solved the structure of the Bag2 carboxy-terminal domain (murine Bag2 residues 107–189), showing it to be a noncanonical dimeric Bag domain that we termed the Brand New Bag (BNB) domain (Bag2-BNB; Xu et al., 2008 ▶). Structural and mechanistic studies show that Bag2-BNB directly accelerates Hsp70-mediated refolding by acting as an Hsp70 NEF (Dai et al., 2005 ▶; Takayama & Reed, 2001 ▶; Xu et al., 2008 ▶). A second pro-folding activity of Bag2, inhibition of CHIP-mediated ubiquitination, requires the Bag2-NTD (Dai et al., 2005 ▶) but is otherwise poorly understood. There are currently no structures of Bag2-NTD or homologous domains in the Protein Data Bank (PDB). Here, we report the crystallization and preliminary X-ray crystallographic analysis of selenomethionyl (SeMet) and native Bag2-NTD (murine Bag2 residues 21–103). Structural characterization of Bag2-NTD will facilitate further studies to probe the mechanisms underlying the Bag2-mediated inhibition of CHIP.
2. Materials and methods
2.1. Cloning, expression and purification of murine Bag2-NTD
The DNA sequences for mouse Bag2 residues 21–90, 21–103 and 21–108 (GenBank accession No. BC016230) were amplified by PCR. The PCR primers introduced BamHI and SpeI restriction sites at the 5′ and 3′ ends, respectively. PCR products were ligated into a pGST-parallel-2 or pGST-parallel-3 expression vector (Sheffield et al., 1999 ▶). The resulting plasmids, pGST-parallel-Bag2-NTD, code for glutathione-S-transferase, a tobacco etch virus (TEV) protease-cleavage site and Bag2 residues 21–90, 21–103 or 21–108. Sequence-verified constructs were transformed into Escherichia coli Rosetta2 (DE3) cells (EMD Chemicals) for expression. Freshly transformed E. coli cells were used to inoculate 100 ml Luria–Bertani (LB) media overnight starter cultures supplemented with 100 µg ml−1 ampicillin and 34 µg ml−1 chloramphenicol. 4 l Terrific Broth (TB) expression cultures were inoculated with the starter cultures and grown until the OD600 reached 1.0. Protein expression was induced by addition of 500 µM isopropyl β-d-1-thiogalactopyranoside (IPTG) and culture growth continued for 20 h at 293 K. SeMet Bag2-NTD was expressed in E. coli B834 (DE3) cells (EMD Chemicals). Starter cultures were grown as described above and used to inoculate 4 l non-autoinducing selenomethionine minimal medium (Studier, 2005 ▶) with 40 mg l−1 seleno-l-methione (Sigma); protein expression was carried out in the same manner as for the native protein.
Bag2-NTD expression cultures were harvested by centrifugation at 4000g and resuspended in 25 mM Tris–HCl, 150 mM NaCl, 1 mM DTT, 1 mM MgCl2 pH 8.0, 1 mg ml−1 lysozyme (Sigma), 20 µg ml−1 DNaseI (MP Biomedicals), 100 µg ml−1 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF; Amresco). Resuspended cells were flash-frozen in liquid nitrogen and stored at 193 K. Cells were thawed and lysed overnight with slow agitation at 277 K. Insoluble cell debris was removed by centrifugation at 20 000g for 45 min. The supernatant was loaded onto a 5 ml glutathione Sepharose column (GE Healthcare) pre-equilibrated with wash buffer (25 mM Tris–HCl, 150 mM NaCl, 1 mM DTT pH 8.0). Nonspecifically bound proteins were washed from the column with 15 column volumes of wash buffer. Bag2-NTD was cleaved from the glutathione Sepharose-bound GST-fusion protein by circulating 40 ml wash buffer supplemented with 1 mM EDTA and 0.02 mg ml−1 His6-tagged tobacco etch virus (TEV) protease (Kapust et al., 2002 ▶). The TEV cleavage buffer was circulated overnight at 277 K and subsequently washed from the column with three column volumes of wash buffer. The TEV cleavage reaction and buffer washes were loaded onto a 5 ml HisTrap HP nickel-affinity column (GE Healthcare) to remove TEV protease. Nickel-affinity flowthrough fractions were analyzed by SDS–PAGE and fractions containing pure target protein were concentrated using 3 kDa molecular-weight cutoff centrifugal concentrators (Sartorius Stedim). Final protein concentrations were determined using the Pierce 660 nm protein assay (Thermo). Purified and concentrated Bag2-NTD, typically at 1 mM, was flash-frozen in liquid nitrogen and stored at 193 K. Typical purifications of native protein yielded approximately 4 mg purified Bag2-NTD per litre of expression medium.
2.2. Crystallization
Crystallization trials were carried out in 96-well Intelli-Plate vapour-diffusion plates (Art Robbins Instruments) at 293 K using a Crystal Gryphon liquid-handling robot (Art Robbins Instruments). The 0.4 µl sitting drops consisted of a 1:1 ratio of 1 mM protein solution (∼10 mg ml−1) and reservoir solution. Initial screening was conducted using the sparse-matrix crystallization screens JCSG+, JCSG Core I–IV and ProComplex from Qiagen and Classics, Index and SaltRx from Hampton Research. Optimization of the initial hits was performed using hanging-drop vapour diffusion at 293 K in 1 µl drops. Native and SeMet crystals both appeared after 24 h and grew to their final size within 3 d. Optimization of the initial SeMet MmBag2(21–103) hits identified an optimal reservoir solution composed of 100 mM sodium cacodylate pH 6.8, 7% PEG 400, 200 mM calcium chloride, 10 mM magnesium chloride, 1 mM hexamminecobalt(III) chloride, 1% dimethylsulfoxide. Optimization of the initial screening hits for native MmBag2(21–103) yielded an optimal reservoir solution composed of 1.9 M ammonium sulfate, 10 mM magnesium chloride, 100 mM HEPES pH 7.5, 2% PEG 400. SeMet and native MmBag2(21–103) crystals were cryoprotected by a brief transfer into their respective reservoir solutions supplemented with PEG 400 to a final concentration of 30%, followed by flash-cooling in liquid nitrogen.
2.3. X-ray diffraction data collection and processing
Single-wavelength anomalous diffraction (SAD) data were collected from a single SeMet MmBag2(21–103) crystal on beamline 4.2.2 at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory. SAD data were collected at the Se K edge (0.979 Å wavelength). Data reduction was performed with d*TREK (Pflugrath, 1999 ▶). The final SAD data set, in space group P212121, was processed to a cutoff of 3.1 Å based on significant drops in the unaveraged I/σ(I) (<2.0) and completeness (<90%) in higher resolution shells. Native diffraction data were collected from a single MmBag2(21–103) crystal at 1.000 Å wavelength. The final native data set, in space group P212121, was processed to a 2.27 Å cutoff. No evidence of twinning was evident for either data set from the results of twin-law tests, including the L test (Padilla & Yeates, 2003 ▶) and N(Z) test, as calculated by phenix.xtriage within PHENIX (Adams et al., 2010 ▶). An anomalous measurability cutoff of 3.9 Å, as reported by PHENIX, was chosen for heavy-atom searches and phasing of the SeMet data set using the HySS (Grosse-Kunstleve & Adams, 2003 ▶) and SOLVE (Terwilliger, 2003 ▶) components of the PHENIX AutoSol wizard (Terwilliger et al., 2009 ▶). Initial model building was performed with RESOLVE (Terwilliger, 2003 ▶) in PHENIX followed by model building in Coot (Emsley et al., 2010 ▶).
3. Results and discussion
Expression screening of murine Bag2 constructs in the pGST-parallel family of expression vectors yielded a number of well expressing constructs in the residue ranges 21–90 to 21–108. Constructs that expressed as soluble GST fusions remained stable throughout the purification process; murine constructs that included residues 1–20, or similar Bag2-NTD constructs from Homo sapiens or Danio rerio, exhibited lower expression levels and suboptimal solution behavior. The removal of residues 1–20 had no discernable effect upon the ability of Bag2-NTD constructs to inhibit CHIP-mediated ubiquitination (data not shown).
Crystallization trials with MmBag2(21–90), MmBag2(21–103), MmBag2(21–106) and MmBag2(21–108) were performed by screening against the sparse-matrix crystallization screens mentioned above. Initial hits against screens containing PEG 400 or PEG 3350 consistently produced thin needle-shaped crystals within 24–48 h. Optimization of the initial hits and crystal screening using a Rigaku MicroMax-007 HF X-ray generator identified the best diffracting MmBag2(21–103) crystals as those grown in ammonium sulfate, magnesium chloride and PEG 400 in the pH range 7–8. The optimized condition produced rod-shaped crystals with a significantly increased thickness, with typical dimensions of approximately 60 µm in width and 300 µm in length (Fig. 1 ▶ a). These crystals diffracted to 2.27 Å resolution on beamline 4.2.2 at the Advanced Light Source and resulted in a high-quality data set (Table 1 ▶). The crystals belonged to space group P212121 and exhibited a Matthews coefficient of 2.35 Å3 Da−1 and a solvent content of 48%, corresponding to eight MmBag2(21–103) molecules per asymmetric unit.
Figure 1.
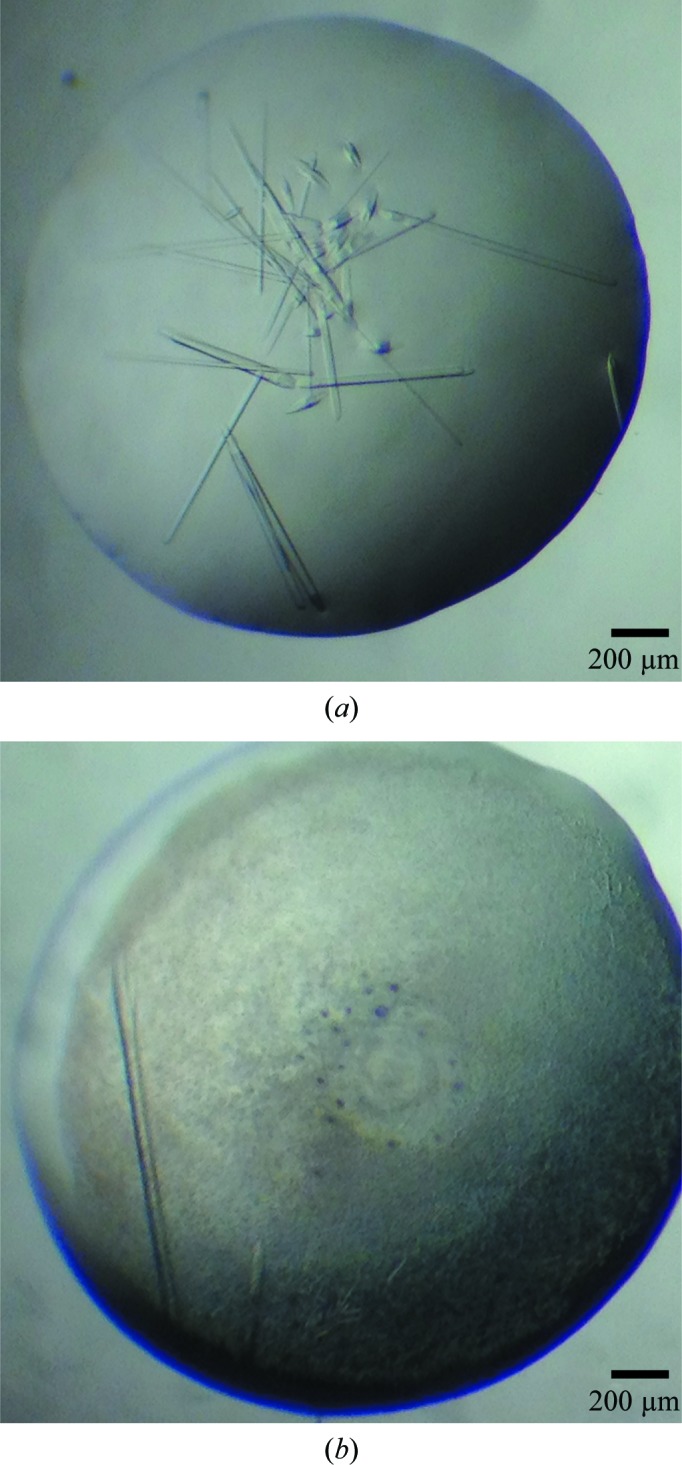
Crystals of native MmBag2(21–103) and SeMet MmBag2(21–103). (a) Crystals of native mBag2(21–103) exhibited a range of sizes. The best diffracting crystals were approximately 60 µm in width and 300 µm in length. (b) Crystals of SeMet MmBag2(21–103) were approximately 30 µm in width and 800 µm in length.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| SeMet MmBag2(21103) | Native MmBag2(21103) | |
|---|---|---|
| X-ray source | ALS 4.2.2 | ALS 4.2.2 |
| Wavelength () | 0.9790 | 1.0000 |
| Space group | P212121 | P212121 |
| Unit-cell parameters () | a = 37.2, b = 53.3, c = 86.7 | a = 75.5, b = 84.7, c = 114.1 |
| Resolution () | 45.393.10 (3.213.10) | 32.992.27 (2.352.27) |
| R merge † | 0.099 (0.477) | 0.071 (0.281) |
| R r.i.m. ‡ | 0.104 (0.497) | 0.076 (0.303) |
| I/(I)§ | 13.4 (2.8) | 15.0 (4.8) |
| Completeness (%) | 99.3 (99.7) | 98.5 (96.3) |
| Multiplicity | 11.3 (12.0) | 6.9 (6.8) |
| Wilson B factor (2) | 96.5 | 32.8 |
| No. of reflections | 39264 | 236359 |
| No. of unique reflections | 3373 | 34124 |
The merging R factor is defined as R
merge = 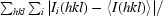
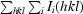 .
.
The redundancy-independent merging R factor R
r.i.m. is defined as R
r.i.m. = 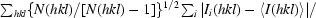 (Weiss, 2001 ▶).
(Weiss, 2001 ▶).
I/(I) denotes the averaged signal-to-noise ratio.
Phasing options for calculating an initial electron-density map from the 2.27 Å resolution native data set were limited, as there are currently no structures deposited in the PDB for proteins with significant homology to Bag2-NTD. A BLAST search (Altschul et al., 1990 ▶) of current PDB structures yielded only a single low-scoring hit, the Sec15 carboxy-terminal domain (PDB entry 2a2f; Wu et al., 2005 ▶), with an E value of 2.32 and a score of 28. Molecular-replacement trials with Sec15 carboxy-terminal domain search models were unsuccessful. Secondary-structure prediction tools, including Jpred (Cole et al., 2008 ▶), PSIPRED (Jones, 1999 ▶; Buchan et al., 2010 ▶) and JUFO (Meiler & Baker, 2003 ▶; Meiler et al., 2001 ▶), predicted a helix–turn–helix motif, possibly followed by a short β-strand (Fig. 2 ▶). These predictions were consistent with the initial classification of Bag2-NTD as a coiled-coil domain. Molecular-replacement models generated using ROSETTA (Rigden et al., 2008 ▶; Rohl et al., 2004 ▶) and iTASSER (Roy et al., 2010 ▶) failed to produce molecular-replacement solutions, necessitating phasing by heavy-atom methods. The majority of the ROSETTA and iTASSER models were bundles of three or more α-helices. None of the molecular-replacement search models correctly predicted the boundaries of helices α1 or α2 or the presence of a short β-strand (Fig. 2 ▶).
Figure 2.
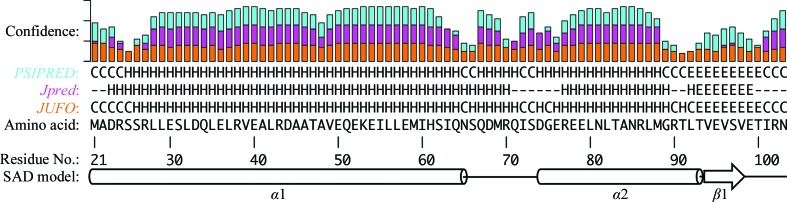
Bag2-NTD amino-acid sequence and secondary-structure prediction. The MmBag2(21–103) amino-acid sequence was used as input into the PSIPRED (Jones, 1999 ▶; Buchan et al., 2010 ▶), Jpred (Cole et al., 2008 ▶) and JUFO (Meiler & Baker, 2003 ▶; Meiler et al., 2001 ▶) secondary-structure prediction programs. The predicted secondary structures (C, coil; H, helix; E, strand) are shown along with the confidence for each prediction. The secondary structure observed in the 3.1 Å resolution model from SeMet SAD phasing is shown for comparison.
Heavy-atom soaks of native MmBag2(21–103) crystals with either conventional metal salts or 5-amino-2,4,6-triiodoisophthalic acid (I3C; Beck et al., 2008 ▶) were unsuccessful. Initial efforts to crystallize SeMet MmBag2(21–103) using the optimized native crystallization condition produced crystals that diffracted to only 8 Å resolution. However, a complete rescreen of SeMet MmBag2(21–103) and subsequent optimization efforts produced long rod-shaped crystals of approximately 30 µm in width and 800 µm in length (Fig. 1 ▶ b) that diffracted to 3.10 Å resolution. Analysis of these crystals, which belonged to space group P212121, indicated a solvent content of 44.5% and a Matthews coefficient of 2.22 Å3 Da−1, corresponding to two SeMet MmBag2(21–103) molecules per asymmetric unit.
A 3.10 Å resolution data set collected at 0.9790 Å wavelength contained suitable anomalous scattering for phasing using single-wavelength anomalous diffraction (SAD) methods. The positions of eight of the ten Se atoms in the asymmetric unit and a subsequent electron-density map were determined using the PHENIX AutoSol wizard (Terwilliger et al., 2009 ▶). Electron density for the amino-terminal selenomethionine residue was not observed for both copies of SeMet MmBag2(21–103). Quality statistics for SAD phasing and an initial refined model (Table 2 ▶) suggest that the structure solution is correct. Automated model building into the density-modified map (Fig. 3 ▶) in PHENIX with RESOLVE (Terwilliger, 2003 ▶) produced an initial model of MmBag2(21–103). Additional model building in Coot (Emsley et al., 2010 ▶) and refinement in PHENIX (Adams et al., 2010 ▶) yielded a model with R work and R free values of 31.9% and 36.3%, respectively. This refined model from SAD phasing was used as a molecular-replacement search model for the 2.27 Å resolution P212121 native MmBag2(21–103) data set. A molecular-replacement search using Phaser (McCoy et al., 2007 ▶) found a single solution that placed eight copies of MmBag2(21–103) in the asymmetric unit. Quality statistics for the molecular-replacement solution indicate the correct placement of MmBag2(21–103) molecules in the asymmetric unit (Table 2 ▶).
Table 2. Quality statistics for SAD phasing using SeMet MmBag2(21103) and initial model building.
| SAD phasing and model building | |
| No. of Se atoms in protein | 5 |
| No. of protein copies in asymmetric unit | 2 |
| No. of placed Se atoms | 8 |
| Figure of merit | 0.373 |
| No. of residues built† | 98 |
| Bayesian map correlation coefficient‡ | 0.72 |
| Final No. of residues built and refined§ | 138 |
| Final R work/R free | 0.319/0.363 |
| Molecular-replacement statistics¶ | |
| No. of protein copies in asymmetric unit | 8 |
| Translation-function Z score (TFZ) | 29.2 |
| No. of C clashes (PAK) | 0 |
| Log-likelihood gain (LLG) | 1469 |
Number of residues built by the PHENIX AutoBuild wizard out of 166 theoretically possible residues.
Bayesian estimate of map quality determined by the PHENIX AutoSol wizard (Terwilliger et al., 2009 ▶).
Total number of residues, out of a theoretically possible 166 residues, built by a combined effort of automated building by PHENIX (Adams et al., 2010 ▶) followed by manual rebuilding in Coot (Emsley et al., 2010 ▶).
Scores for the best Phaser (McCoy et al., 2007 ▶) molecular-replacement solution using the model built from the 3.1 resolution SAD data set.
Figure 3.
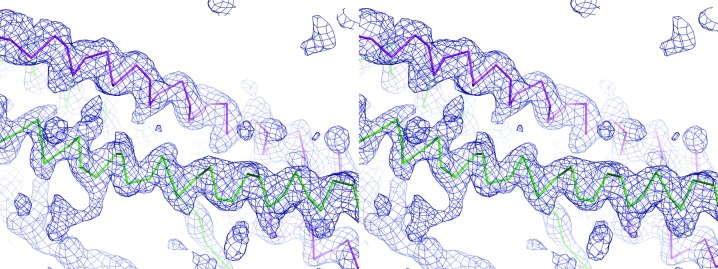
Stereo image of the density-modified map and Cα-backbone trace from SAD phasing. An initial density-modified map (dark blue) was produced by PHENIX AutoSol from a 3.1 Å resolution data set collected at the Se K edge. The Cα-backbone trace of the final model from the SAD-phased data for both copies of SeMe MmBag2(21–103) within the asymmetric unit (chain A, magenta; chain B, green) demonstrates the coiled-coil topology of Bag2-NTD. 2F o − F c density is contoured at 1.3σ.
Interestingly, a DALI search (Holm & Sander, 1995 ▶; Holm & Rosenström, 2010 ▶) with our partly refined 3.1 Å resolution model found no hits with a Z score higher than 6.3; the median Z score of the 1606 hits was 3.1 and the median sequence identity was 8%. Additionally, the SAD-phased MmBag2(21–103) model is significantly different in structure from the ten best DALI hits and the single low-scoring hit from a PDB BLAST search. Model building and refinement of the 2.27 Å resolution P212121 native Bag2-NTD data set with eight molecules per asymmetric unit are now in progress. The final Bag2-NTD structure will contribute a novel sequence and may contribute a novel domain structure to the PDB, in addition to providing insights into the role of this domain in the inhibition of CHIP by Bag2.
Acknowledgments
The authors acknowledge financial support from the US National Institutes of Health (grant RO1-GM080271 to SM). RCP was supported by National Institutes of Health Public Health Service Kirschstein National Research Service Award Postdoctoral Fellowship T32 HL007914. The Advanced Light Source is supported by the US Department of Energy under contract No. DE-AC03-76SF00098 at Lawrence Berkeley National Laboratory.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990). J. Mol. Biol. 215, 403–410. [DOI] [PubMed]
- Arndt, V., Daniel, C., Nastainczyk, W., Alberti, S. & Höhfeld, J. (2005). Mol. Biol. Cell, 16, 5891–5900. [DOI] [PMC free article] [PubMed]
- Beck, T., Krasauskas, A., Gruene, T. & Sheldrick, G. M. (2008). Acta Cryst. D64, 1179–1182. [DOI] [PubMed]
- Buchan, D. W., Ward, S. M., Lobley, A. E., Nugent, T. C., Bryson, K. & Jones, D. T. (2010). Nucleic Acids Res. 38, W563–W568. [DOI] [PMC free article] [PubMed]
- Bukau, B. & Horwich, A. L. (1998). Cell, 92, 351–366. [DOI] [PubMed]
- Carrettiero, D. C., Hernandez, I., Neveu, P., Papagiannakopoulos, T. & Kosik, K. S. (2009). J. Neurosci. 29, 2151–2161. [DOI] [PMC free article] [PubMed]
- Chen, B., Retzlaff, M., Roos, T. & Frydman, J. (2011). Cold Spring Harb. Perspect. Biol. 3, a004374. [DOI] [PMC free article] [PubMed]
- Cole, C., Barber, J. D. & Barton, G. J. (2008). Nucleic Acids Res. 36, W197–W201. [DOI] [PMC free article] [PubMed]
- Connell, P., Ballinger, C. A., Jiang, J., Wu, Y., Thompson, L. J., Höhfeld, J. & Patterson, C. (2001). Nature Cell Biol. 3, 93–96. [DOI] [PubMed]
- Cuervo, A. M. & Dice, J. F. (1998). J. Mol. Med. 76, 6–12. [DOI] [PubMed]
- Dai, Q., Qian, S.-B., Li, H.-H., McDonough, H., Borchers, C., Huang, D., Takayama, S., Younger, J. M., Ren, H. Y., Cyr, D. M. & Patterson, C. (2005). J. Biol. Chem. 280, 38673–38681. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Grosse-Kunstleve, R. W. & Adams, P. D. (2003). Acta Cryst. D59, 1966–1973. [DOI] [PubMed]
- Hartl, F. U. & Hayer-Hartl, M. (2002). Science, 295, 1852–1858. [DOI] [PubMed]
- Hershko, A. & Ciechanover, A. (1998). Annu. Rev. Biochem. 67, 425–479. [DOI] [PubMed]
- Holm, L. & Rosenström, P. (2010). Nucleic Acids Res. 38, W545–W549. [DOI] [PMC free article] [PubMed]
- Holm, L. & Sander, C. (1995). Trends Biochem. Sci. 20, 478–480. [DOI] [PubMed]
- Jones, D. T. (1999). J. Mol. Biol. 292, 195–202. [DOI] [PubMed]
- Kapust, R. B., Tözsér, J., Copeland, T. D. & Waugh, D. S. (2002). Biochem. Biophys. Res. Commun. 294, 949–955. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Meacham, G. C., Patterson, C., Zhang, W., Younger, J. M. & Cyr, D. M. (2001). Nature Cell Biol. 3, 100–105. [DOI] [PubMed]
- Meiler, J. & Baker, D. (2003). Proc. Natl Acad. Sci. USA, 100, 12105–12110. [DOI] [PMC free article] [PubMed]
- Meiler, J., Müller, M., Zeidler, A. & Schmäschke, F. (2001). J. Mol. Model. 7, 360–369.
- Padilla, J. E. & Yeates, T. O. (2003). Acta Cryst. D59, 1124–1130. [DOI] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Rigden, D. J., Keegan, R. M. & Winn, M. D. (2008). Acta Cryst. D64, 1288–1291. [DOI] [PubMed]
- Rohl, C. A., Strauss, C. E., Misura, K. M. & Baker, D. (2004). Methods Enzymol. 383, 66–93. [DOI] [PubMed]
- Roy, A., Kucukural, A. & Zhang, Y. (2010). Nature Protoc. 5, 725–738. [DOI] [PMC free article] [PubMed]
- Sheffield, P., Garrard, S. & Derewenda, Z. (1999). Protein Expr. Purif. 15, 34–39. [DOI] [PubMed]
- Sondermann, H., Scheufler, C., Schneider, C., Hohfeld, J., Hartl, F. U. & Moarefi, I. (2001). Science, 291, 1553–1557. [DOI] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif. 41, 207–234. [DOI] [PubMed]
- Takayama, S. & Reed, J. C. (2001). Nature Cell Biol. 3, E237–E241. [DOI] [PubMed]
- Terwilliger, T. C. (2003). Methods Enzymol. 374, 22–37. [DOI] [PubMed]
- Terwilliger, T. C., Adams, P. D., Read, R. J., McCoy, A. J., Moriarty, N. W., Grosse-Kunstleve, R. W., Afonine, P. V., Zwart, P. H. & Hung, L.-W. (2009). Acta Cryst. D65, 582–601. [DOI] [PMC free article] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst. 34, 130–135.
- Wu, S., Mehta, S. Q., Pichaud, F., Bellen, H. J. & Quiocho, F. A. (2005). Nature Struct. Mol. Biol. 12, 879–885. [DOI] [PubMed]
- Xu, Z., Page, R. C., Gomes, M. M., Kohli, E., Nix, J. C., Herr, A. B., Patterson, C. & Misra, S. (2008). Nature Struct. Mol. Biol. 15, 1309–1317. [DOI] [PMC free article] [PubMed]
- Young, J. C., Agashe, V. R., Siegers, K. & Hartl, F. U. (2004). Nature Rev. Mol. Cell Biol. 5, 781–791. [DOI] [PubMed]