

Abstract
Clostridium difficile infection is the leading cause of antibiotic- and healthcare-associated diarrhea, and its containment and treatment imposes a significant financial burden, estimated to be over $3 billion in the USA alone. Since the year 2000, CDI epidemics/outbreaks have occurred in North America, Europe and Asia. These outbreaks have been variously associated with, or attributed to, the emergence of Clostridium difficile strains with increased virulence, an increase in resistance to commonly used antimicrobials such as the fluoroquinolones, or host susceptibilities, including the use of gastric acid suppressants, to name a few. Efforts to elucidate C. difficile pathogenic mechanisms have been hampered by a lack of molecular tools, manipulatable animal models, and genetic intractability of clinical C. difficile isolates. However, in the past 5 y, painstaking efforts have resulted in the unraveling of multiple C. difficile virulence-associated pathways and mechanisms. We have recently reviewed the disease, its associated risk factors, transmission and interventions (Viswanathan, Gut Microbes 2010). This article summarizes genetics, non-toxin virulence factors, and host-cell biology associated with C. difficile pathogenesis as of 2011, and highlights those findings/factors that may be of interest as future intervention targets.
Introduction
Clostridium difficile is a Gram-positive, spore-forming obligate anaerobe, belonging to the phylum Firmicutes. Both toxin-producing (toxigenic) and non-toxigenic strains exist naturally, and both can colonize their hosts (humans and non-human mammals), although only toxigenic strains are associated with disease. Though not traditionally considered to be part of the human microbiome, it is estimated that C. difficile may comprise 1–3% of commensal flora in adult humans.1C. difficile spores are ubiquitous in the environment, and widely distributed in healthcare settings. Spores are ingested via contact with contaminated surfaces and, under favorable conditions (in susceptible hosts), will germinate to a vegetative morphotype that can produce toxins, if a toxigenic strain is involved (Fig. 1). Antibiotic-mediated suppression of normal human gut microbiota is strongly associated with colonization and proliferation of C. difficile, and germination of spores is dependent on specific bile salts produced in the small intestine.2 Toxigenic C. difficile encode one or two glucosyltransferase toxins [Toxin A (TcdA) or Toxin B (TcdB)] within a 19.6 kb genomic island known as the Pathogenicity Locus (PaLoc). These toxins inactivate Rho, Rac or Cdc42-family molecules in host epithelial cells to cause signaling alterations and, ultimately, apoptosis. A third ADP-ribosylating toxin [Binary toxin (CDT)] is also produced by some, but not all, C. difficile strains. Understandably, much of the focus has been placed on intoxication mechanisms, since TcdA/B and binary toxin cause profound alterations in host-cell biology, and are ultimately responsible for the diarrheagenic phenotype of CDI. However, it is increasingly appreciated that non-toxin virulence factors likely play equally important roles in C. difficile colonization, proliferation and maintenance in the host GI tract. This is underscored by the increased rates of recurrent disease in patients infected with the newer epidemic-associated strains of C. difficile; it is estimated that up to 33% of patients will recur after a first episode of CDI, and 45% after a second episode.3 These observations argue that recently emerged strains of C. difficile have evolved mechanisms to persist in the GI tract even after repeated antimicrobial regimens; this long-term establishment is likely mediated via newly-evolved colonization strategies. Functional and mechanistic elucidation of the contribution of non-toxin factors to CDI is therefore of paramount importance if a comprehensive view of C. difficile-induced disease is envisaged.

Figure 1. Schematic of human C. difficile infection. Spores, vegetative cells, bacterial factors and host factors that impact/modulate disease are depicted.
Epidemiology and Molecular Typing of Clostridium difficile Clinical Isolates
C. difficile isolates are classified or typed using a number of molecular methodologies.4 The most commonly used methods classify strains on the basis of tcdA/tcdB sequence polymorphisms (toxinotyping), 16s-23s rDNA intergenic spacer region patterns (ribotyping), whole genome restriction pattern polymorphisms [pulse-field typing and restriction endonuclease analysis (REA) typing], multi-locus repeat- or non-repeat-based sequence variations (MLST, MLVA), and surface-protein variations (SLP typing). The newer epidemic- and severe disease-associated C. difficile strains that have emerged in the past 10 years in North America and Europe are most frequently identified as belonging to ribotypes 001, 017, 027, 078 and 106 (toxinotypes 0, VIII, III, V and 0, and pulse-field types NAP2, NAP9, NAP1, NAP7/8 and NAP11 respectively).5
Multiple phylogenetic studies have also revealed that most epidemic-associated strains described above can be grouped into specific clades (HY, HA1, A-B+, HA-2).6 However, intra- and inter-clade variation does occur, and the analysis of scores of C. difficile genomes has revealed a remarkable degree of chromosomal plasticity as well as the propensity for horizontal DNA transfer.7-10
Clostridium difficile Toxins
The diarrheagenic phenotype of CDI is attributed to toxins produced by the organism. Toxigenic strains produce up to three toxins (TcdA, TcdB and the binary toxin CDT). All naturally occurring diarrheagenic strains of C. difficile produce TcdB, and most strains also produce TcdA; there are no reports of naturally occurring TcdA+TcdB- C. difficile strains.
TcdA and TcdB belong to the same family of large clostridial glucosylating toxins, and harbor a receptor binding domain, a transmembrane domain and a glucosyl-transferase domain. These toxins glucosylate and inactivate host GTPases, including Rac, Rho and Cdc42, leading to alterations in the actin cytoskeleton, disruption of barrier function and apoptosis.11 Interestingly, some variants of TcdB have altered substrate specificity, and target Rap and Ras proteins instead of Rac.12
In addition to their direct cytotoxic effects, TcdA and TcdB also provoke inflammatory responses that contribute to tissue damage, and may lead to serious clinical sequelae such as pseudomembranous colitis. Both toxins induce production of the pro-inflammatory cytokine IL-1β in tissue culture models. Ng et al. recently demonstrated that this induction depends on intracellular recognition of the toxins and subsequent assembly of a multi-protein complex, the inflammasome, involved in activating inflammation.13 Inhibiting inflammasome formation prevented TcdA/TcdB-induced tissue damage in a mouse model of infection. In a different study, the two toxins were shown to induce HIF-1α, a transcription factor that regulates genes involved in immune function, inflammation and intestinal barrier function. TcdA/TcdB-treated HIF-1α knockout mice display more severe tissue damage, implicating a protective role for HIF-1α during CDI.14
The actual contributions of the two toxins, particularly TcdA, to disease progression remain a matter of debate. Two laboratories have independently constructed C. difficile mutants deficient in production of TcdA, TcdB or both toxins, and assessed their virulence in the hamster model of infection.15,16 Both groups found that TcdA-negative mutants remained virulent, and that mutants lacking both toxins were avirulent. However, the results for the TcdB-negative mutant were more varied, with one study showing that these strains were nearly as virulent as the wild-type parent, and the other study finding them to be significantly less virulent. Several explanations have been proposed for the apparent discrepancy between the two studies, including differences in the end points at which animals were euthanized, as well as differences in the parent C. difficile strains used to create the mutants.16
Within the PaLoc is a gene, tcdC, that encodes an anti-sigma factor speculated to play a role in TcdA/TcdB expression. Many epidemic-associated C. difficile isolates of the 027 ribotype/HY clade harbor a missense mutation in tcdC, and it has been proposed that the consequent loss of function contributes to the enhanced toxin production and virulence of 027 strains. Consistent with this hypothesis, Carter et. al demonstrated that an 027 strain complemented with full length tcdC (from a non-027 strain VPI10463) produced less toxin, and was less virulent, than the parental strain in a hamster model of infection.17 There are, however, many naturally-occurring C. difficile strains harboring the tcdC mutation, but not associated with severe disease,18 as well as strains that lack this mutation, but produce copious amounts of toxin without being associated with severe disease in hamsters (e.g., strain VPI10463).19 Therefore, further exploration of specific mechanistic contribution(s) of TcdC to C. difficile disease is warranted.
In addition to TcdA and TcdB, some C. difficile isolates, notably 027 and 078 ribotype strains, produce a binary toxin, also called Clostridium difficile transferase (CDT). A retrospective study in Sweden concluded that human infection with CDT-producing strains of C. difficile was associated with greater mortality compared with infection with strains that did not produce CDT, regardless of ribotype, suggesting a role for CDT in virulence.20 CDT irreversibly ADP-ribosylates actin, leading to disruption or rearrangement of the host cell cytoskeleton. Schwan et al. showed that CDT-treated Caco-2 cells elaborate long protrusions to which C. difficile can bind, suggesting that this toxin may enhance bacterial attachment to host cells.21 Using a genetic screen in a haploid human cell line, Papatheodorou et al. identified the receptor of CDT, a protein called lipolysis-stimulated lipoprotein receptor.22 It has also been demonstrated that entry of CDT into the host cell cytosol requires endosome acidification, and that this process depends on the host cell proteins Hsp90 and cyclophilin A, as is the case for the C2 toxin of C. botulinum.23
Sporulation and Germination of C. difficile
Spores, the etiological agents of CDI, are easily spread between healthcare workers and patients.24 Further, spores are metabolically dormant and highly resistant to standard disinfection procedures, allowing them to persist for long periods in the environment. Spores ingested by susceptible hosts can reactivate (or germinate) in response to specific bile acids in the small intestine, and return to an active lifestyle to produce toxins and cause disease.3 Therefore, an increased ability to form spores in the host or, perhaps, the ability to form more resilient spores might account for a change in the ability of C. difficile to spread more easily. Could the rapid spread of the recently-emerged epidemic-associated strains be due to an increased efficiency of sporulation, compared with the clinical isolates recovered prior to the year 2000? Multiple studies suggest that some,25,26 but not all,18,27,28 epidemic-associated, and ribotype 027 strains, do produce more spores in vitro. However, the efficiency of sporulation in vivo, and its contribution to the prevalence of specific C. difficile strains, is still an open question. For these reasons, spores produced by C. difficile, the process by which they are formed (sporulation), their associated surface proteins and structures such as the exosporium (Fig. 2, Mallozzi M et al., unpublished), as well as their reactivation kinetics in the host, are active areas of research.
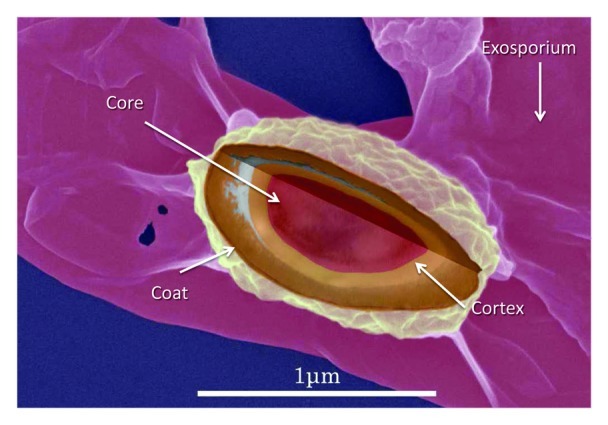
Figure 2. Spore surface of C. difficile clinical isolates. High-resolution, scanning electron micrograph overlayed with transmission-electron micrograph data of spore morphology of C. difficile strain VPI10463 immobilized on a solid surface. The orderly arrangement of spore protein complexes is clearly visible, as is an exosporial structure (diffusely-shaped and surrounding the spore. Micrograph collection, Vedantam laboratory.
Sporulation in the Clostridia
Sporulation in all spore-forming organisms is thought to occur in response to stress, especially starvation. The genes and genetic regulation of spore formation have been well studied in Bacillus subtilis (the archetypal organism for sporulation studies).29 In their review, Paredes et al. used genome sequence analyses to compare and contrast the putative sporulation signaling pathways and regulatory proteins in Bacillus spp. and the clostridia.30 While this analysis revealed that almost all regulators and signaling molecules downstream of the master-regulator of sporulation (SpoOA) were conserved, the signaling molecules upstream of SpoOA (which contribute to SpoOA phosphorylation and activation) were conspicuously absent from clostridial genomes.30 Therefore, the factor(s) influencing the sporulation checkpoint, and the corresponding signaling pathways, remain to be established for the clostridia. In contrast, the view that the orthologous sigma factors known to control sporulation in Bacillus sp likely function similarly in clostridia has been buoyed by recent studies showing that mutants lacking these sigma factors have an asporogeneous phenotype in multiple clostridial species.31-35
Nevertheless, studies in other clostridia have shed light on the signaling events that help initiate sporulation in C. difficile. Steiner et al. showed that two orphan histidine kinases of Clostridium acetobutylicum were responsible for the phosphorylation of SpoOA in vivo.36 Interestingly, Underwood et al. showed that a mutation in the orthologous orphan histidine kinase in C. difficile (CD2492) reduced the capacity to sporulate, suggesting functional conservation with the identified C. acetobutylicum sporulation proteins.37 This work also revealed that another C. difficile histidine kinase, CD1579, could phosphorylate SpoOA in vitro; however, its function in vivo could not be determined. While quorum sensing influences the decision to sporulate in Bacillus sp,38 and plays a role in sporulation initiation in C. botulinum and C. perfringens39,40 (via the AgrB quorum sensing peptide), its role in C. difficile sporulation is currently unknown. Understanding the signaling events involved in C. difficile sporulation could potentially yield novel targets for therapeutic intervention.
Germination
While the signaling and regulatory cascades controlling sporulation have been well-elucidated in many bacteria, events that lead to the reactivation of spores (germination) are much less clear. Here too, findings from Bacillus spp have helped identify factors that activate and control germination in the clostridia. In Bacillus spp, germination begins when a small molecule (usually a nutrient) binds to a cognate receptor (homologs of the canonical GerA receptor in B. subtilis) in the spore’s inner membrane. Detection of the germinant leads to a metabolism-independent transformation of the spore, and consequent release of stored mono- and di-valent cations, as well as dipicolinic acid. This is followed by the degradation of the spore peptidoglycan (via specific lytic enzymes), concomitant spore rehydration, and the eventual reactivation of metabolism in a process defined as outgrowth.41 In a recent comparative analysis, Paredes-Sabja et al. noted the apparent conservation of many critical protein components of germination between the bacilli and clostridia.42 Further, several studies suggest that the clostridial germination-protein orthologs function similarly to their counterparts in Bacillus spp.43-48 The absence of a homolog of known germination receptors in C. difficile suggests that signaling events contributing to germination may differ in some clostridia. Nevertheless, germinants for C. difficile have been identified (the bile acid taurocholate and the amino acid glycine),49 and they appear to act specifically in a sequential and kinetically similar fashion to germinants with known cognate receptors.50,51 These findings have led to a search for an antagonist to block C. difficile germination and, thereby, prevent infection. Indeed, chenodeoxycholic acid, a primary bile acid, as well as stable chemical analogs of this molecule competitively inhibit C. difficile spore germination in vitro.2,52 These studies suggest that a therapeutic agent aimed at preventing C. difficile spore germination in vivo may be an effective strategy for preventing infections in health care settings. Clearly, many questions remain regarding the biology of clostridial spores in general, and C. difficile spores in particular, suggesting that spore-centered investigations will be a major focus of CDI research for the foreseeable future.
C. difficile Colonization
Colonization resistance
Following ingestion, C. difficile spores germinate in the gut to produce vegetative cells that express the toxins. While the toxins are responsible for the observed pathologies, the requirement of antibiotic-mediated clearance of the gut microbiota for CDI establishment suggests that colonization of host mucosal surfaces is an essential first step (CDI; Fig. 3). The collective mechanisms by which the native flora help to prevent infection is known as colonization resistance.53,54 Correspondingly, successful resolution of CDI likely requires the concomitant reestablishment of a protective flora. Failure to reestablish the protective flora is likely a contributing factor to recurrent CDI, and the high success rate of fecal transplantation in resolving these cases supports this notion.55

Figure 3.C. difficile spores and vegetative cells adhere to host intestinal epithelium. False-color, high resolution, scanning electron micrograph of C. difficile spore (blue) and vegetative cell (red) adhering to human intestinal epithelial cells (green) grown in culture. Microvilli are clearly visible (feather-like green protrusions). Micrograph collection, Vedantam laboratory.
In the case of CDI, colonization resistance is thought to include the inactivation of germinant molecules that induce C. difficile germination,52,56 the production of toxic or inhibitory small molecules which prevent C. difficile growth and/or kill vegetative cells,56-58 and competitive exclusion from a microenvironment;59-61 other mechanisms, such as occlusion of host receptors required for C. difficile colonization, and the stimulation of host responses that prevent C. difficile establishment, may also play a role. These processes likely act concomitantly and/or synergistically to prevent CDI.
Due to the technical difficulty of culturing most gut bacteria, attempts at identifying a single bacterial species, or a defined subset of species, among the normal microbiota capable of preventing CDI have largely been unsuccessful. Several recent studies have used high-throughput technologies to probe the antibiotic-mediated alterations in the gut microbial consortium, as well as the replenished flora following fecal transplantation.62-65 Chang et al. performed 16S rRNA gene sequence analysis to compare the gut microbiota of patients with initial CDI, recurrent CDI and healthy controls.62 The fecal microbiome of patients with recurrent CDI displayed a marked and consistent decrease in diversity (phylotype richness) compared with healthy controls or even those with initial CDI; a marked decrease in the phylum Bacteroidetes was evident in the recurrent CDI patients.
Khoruts et al. used fecal transplantation to treat a 63 year-old woman with C. difficile diarrhea, and concomitantly monitored the restoration of the protective microbiota in this patient.64 CDI in this patient had initially resulted from cephalosporin/quinolone treatment; prior to her admission for fecal transplantation, she had had 8 mo of diarrhea that was managed by repeated administration of metronidazole and vancomycin. Prior to, and after fecal transplantation, the fecal flora of the patient and the donor were analyzed by restriction fragment length polymorphism and 16S rRNA gene sequence analyses. Before fecal transplantation, the patient’s fecal/intestinal flora was dominated by Clostridium spp and Veilonella sp, and also contained Streptococcus sp and Lachnospiraceae incertae sedis; in contrast, these organisms were not detected in the (“normal”) donor stool sample. Importantly, the patient’s flora lacked Bacteriodes spp prior to treatment. Fourteen days after treatment, the patient showed a dramatic improvement, and her fecal flora, dominated by Bacteroides spp, was very similar to that of the donor.
What predisposes a subset of the patient population to recurrent CDI? A recent study (not involving CDI) showed that antibiotic treatment has distinct effects on different subjects. While exploring the shift in the gut microbial population induced by the experimental administration of ciprofloxacin, Dethelefsen and Relman observed rapid and pronounced alterations, with a reduction in diversity, and a shift in community composition.66 While cessation of antibiotic treatment gradually restored the microbiota, the community structure stabilized to a state that was distinct from the initial composition. Moreover, the dynamics of these changes, as well as the reestablishment of a stable community after antibiotic cessation, was distinct between the different subjects. By extension, the starting flora of CDI patients, as well as the (precipitating) antibiotic regimen and other factors, may dictate the dynamics of flora recovery during the treatment phase. The effectiveness of fecal transplantation against recurrent CDI suggests that the host itself is not resistant to the establishment of a protective flora.
C. difficile Colonization Factors
Relatively little is known about the factors that contribute to C. difficile colonization, and the following sections provide an overview of the forays that have been made to understand this process.
Fibronectin-binding proteins
Many bacterial species use surface proteins, usually bound to the cell wall either by a sortase-dependent, or non-covalent mechanisms, to recognize adhesive matrix molecules of host cells. Receptors include host cell surface proteins such as fibronectin, laminin, collagen and fibrinogen.67 A search of C. difficile published genome sequences identified multiple proteins with putative and demonstrated abilities to bind host extracellular matrix components (Table 1).
Table 1. Non-toxin virulence factors of C. difficile and their putative or experimentally-determined functions.
| Function |
Gene Identifiers |
Description |
References |
|---|---|---|---|
| Motility and Secretion | |||
| Putative Type IV pilus |
CD3505–3513 |
Putative type IV pilus biosynthesis and function |
68 |
| Capsule |
CD3253, CD0775, CD2769 |
Poly-gamma-glutamate biosynthesis |
54 |
| Flagella | CD0226–0271 | Flagellar biosynthesis operon and flagellin glycosylation | 69 8 |
| Adhesion and Immune Evasion | |||
|---|---|---|---|
| Collagen-binding proteins |
CD2831, CD3392, CD0386 |
Putative recognition of extracellular matrix collagen |
70 |
| Fibronectin-binding proteins |
FbpA, CD2797 |
Putative recognition of extracellular matrix fibronectin |
71-73 |
| Thrombospondin-domain containing protein |
CD3145 |
Putative recognition of extracellular matrix fibrinogen |
70 |
| von-Willebrand Factor binding proteins |
CD3038 CD2248 CD0323 |
Putative von-Willebrand Factor binding |
70 |
| Sortase |
CD2718 |
Class B sortase |
70 |
| Major surface layer protein |
slpA |
Cleaved into high and low molecular weight S-layer proteins, phase variable |
74 |
| Cysteine protease |
cwp84 |
Cleaves SlpA, possible degradation of host extracellular matrix proteins |
75,76 |
| Adhesin |
cwp66 |
Putative adherence to host cells |
77 |
| Hemagglutinin/Adhesin |
CD0514 |
Putative hemagglutinin |
70 |
| Phase-variable cell wall protein | cwpV | Bacterial aggregation, putative immune evasion | 78 |
The C-terminus of C. difficile Fbp68, a 68kD magnesium-containing protein, interacts with fibronectin.73 A peptide consisting of the fibronectin-binding domain of Fbp68 partially blocked C. difficile adhesion to Caco-2 cells, supporting a role for Fbp68 in C. difficile intestinal colonization.72 Curiously, deletion of another fibronectin-binding protein, FbpA, resulted in increased adherence to Caco-2 cells. Interestingly, the FbpA-deficient strains were impaired for colonization of cecal tissues in a monoxenic mouse model; overall bacterial shedding, however, was not significantly different from the isogenic parent strain in both monoxenic and dixenic mouse model systems.71 The contribution of fibronectin binding to colonization, therefore, requires further evaluation.
Surface layer and cell-wall proteins (SLPs and CWPs)
Various structures and proteins on Gram-positive cell surfaces contribute to pathogenic mechanisms such as adhesion, tissue invasion, regulation of bacterial growth and metabolism, and stimulation of the host immune response. Some Gram-positive organisms, including C. difficile, possess a proteinaceous cell surface layer, or S-layer, which has been implicated in bacterial adhesion to host tissues.79 The C. difficile S-layer is composed of numerous proteins arranged in a crystalline lattice, with surface-layer protein A (SlpA) being the predominant species. Additional surface layer proteins have been identified within the C. difficile S-layer, and may have unique functions with respect to protein processing, adhesion, and cell wall regulation. SlpA and other surface-layer proteins (SLPs) are also a subset of a larger class of molecules referred to as cell-wall proteins (CWPs). SlpA has been implicated in C. difficile adhesion by either directly or indirectly aiding in bacterial attachment to intestinal epithelial cells. We have recently demonstrated that SlpA is indeed the major contributor to C. difficile attachment in vitro, as pre-treatment of host cells with purified SlpA, or SlpA subunits, abrogates C. difficile attachment in a dose-dependent manner. Conversely, pre-treatment of viable C. difficile bacteria with anti-SlpA antibodies also abrogates adherence.25
Two cysteine proteases, Cwp84 and Cwp13, are involved in assembling the S-layer of C. difficile.75,80 Both Cwp84 and Cwp13 are pro-enzymes that are activated by proteolytic cleavage (Cwp13 undergoes autocatalytic cleavage). Cwp84 is a dynamic molecule that not only functions at the cell surface to process proteins for incorporation into the cell wall and S-layer, but also has enzymatic activity against host proteins and may function as an exo-enzyme facilitating pathogenesis. Two forms of Cwp84 are produced by C. difficile, a secreted 80kD high molecular weight (HMW) form and a 47kD low molecular weight (LWM) form, considered to be the active form of the enzyme, which is maintained at the cell wall.80 Cwp84 cleaves and processes SlpA and plays an essential role in S-layer assembly. Mutants lacking Cwp84 secrete full-length SlpA, resulting in an aberrant S-layer architecture. This, and the inability to retain proteins normally localized to the cell wall and S-layer, results in altered colony morphology. Incorrect processing of the S-layer may also limit the ability of C. difficile to compete with other gut bacteria. Cwp13 has sequence similarity to Cwp84, but different activities; in fact Cwp13 is responsible for the cleavage and correct processing of Cwp84. Therefore, both proteins are important in the correct assembly of the S-layer. An in-depth discussion of SLP/CWP processing and display on the C. difficile cell surface is beyond the scope of this article, but has been elegantly reviewed elsewhere.74,81-83
C. difficile cell wall proteins may have additional functions that facilitate bacterial establishment in the gut. CwpV harbors a C-terminal repeat-containing domain that is variable in the number of repeats, as well as antigenicity.78 Interestingly, the repetitive domain, although divergent in sequence between C. difficile strains, has been demonstrated to be uniformly important in bacterial aggregation. This auto-aggregation phenotype likely has implications for host colonization, since bacteria that are able to precipitate out of solution could adhere more robustly to gut epithelial tissues that are normally covered by mucus.84,85 CwpV expression is controlled in a phase-dependent manner by the site-specific recombinase, RecV. Site-specific recombination in the cwpV promoter region regulates the transcription of the gene via a DNA sequence inversion event. Sequence inversion to the “phase-OFF” state results in the formation of a transcriptional terminator that prevents CwpV expression.78 The relevance of phase-regulated CwpV expression to C. difficile adhesion dynamics is currently unknown.
Flagella
The enteric environment can be both an accommodating and/or harsh landscape for many colonizing bacteria. Since the microbial content of the gut is so high, many innate defenses against opportunistic pathogens are in place to prevent overgrowth of the microbiota and protect against disease. Mucus production by goblet cells in the gut is one mechanism by which intestinal epithelial cells are protected, and the constant turnover of chemical and proteinaceous constituents of mucus prevent potentially pathogenic microbes from gaining a foothold. Not surprisingly, many bacterial pathogens have developed strategies to circumvent these innate defenses, including the expression of flagella for enhanced motility in mucus-rich environments.
Flagella are important colonization factors for a number of enteropathogenic species including Escherichia coli, Salmonella sp and Listeria monocytogenes.86-89 C. difficile strains express peritrichous flagella (Fig. 4; Clark A et al., unpublished), although flagellar expression and its associated motility phenotype is variable among different strains.90 C. difficile flagella were utilized in early attempts at classifying C. difficile isolates based on slide agglutination assays.91,92 Much of the preliminary molecular characterization of the C. difficile flagellar locus was performed by Tasteyre and colleagues who identified the genes encoding the flagellin monomer (fliC), and the flagellar cap protein (fliD) examined the genetic diversity within these loci and used molecular techniques to examine relationships between gene sequence and serological classifications.93-95 Tasteyre et al. also completed one of the first phenotypic studies examining the FliD cap protein and presented preliminary evidence that the FliD cap protein may play a role in adhesion to mucus, as is reported in other organisms,96 and that both FliC and FliD are implicated in C. difficile binding to cecal tissue.97

Figure 4.C. difficile clinical isolates produce flagella. False-color, high-resolution, scanning electron micrograph of a C. difficile ribotype 001 strain. Flagella (red) are clearly visible on vegetative cells (purple) after growth to exponential phase (OD600 = 0.6) in Brain-Heart infusion medium supplemented with yeast and cysteine. Biophysical measurements indicate that flagella have a diameter of 17–22 nm, consistent with those described on other enteric bacteria. Micrograph collection, Vedantam laboratory.
The C. difficile flagellin has been analyzed by mass spectrometry to examine structural elements which may potentially influence both flagellar function and immune activation.69 A discrepancy between the predicted mass of C. difficile flagellin based on gene sequence and the mass observed in top-down proteomic analysis prompted further investigation by Twine et al. C. difficile flagellin was found to be glycosylated at multiple locations accounting for the discrepancy in mass calculations of FliC protein. Moreover, it was determined that FliC is differentially glycosylated in different strains of C. difficile and that the FliC glycosylation is dependent on the composition of genes immediately downstream from the fliC locus. Flagellar glycosylation has been reported in other organisms98,99 and may play a yet undermined role in the adherence and colonization of C. difficile vegetative cells.
A recent study examined the effects of inactivation of both the C. difficile 630ΔErm fliC and fliD genes in the hamster model of infection and adhesion to cultured epithelial cells. Insertional inactivation of fliC and fliD genes, respectively, using ClosTron-based methods resulted in aflagellate mutants.100 The authors reported an enhanced ability of both mutants to adhere to cultured Caco-2 cells in vitro and suggested that flagella may not play a role in adhesion, contradictory to previous reports.93 C. difficile fliC and fliD mutant strains were reported to be more virulent in hamsters, causing the authors to conclude that flagella were either unnecessary for virulence or were downregulated during the course of CDI in the hamster model.100 However, the parent 630ΔErm strain exhibits reduced motility,8 and differs greatly in genetic content when compared with other (especially epidemic-associated) C. difficile isolates,69,101 suggesting that this strain may not be ideal for drawing comprehensive conclusions about the role of flagella in C. difficile pathogenesis.
Thus there are still a number of unanswered questions regarding the function of C. difficile flagella. Although it has been suggested that flagella may not be necessary for virulence in hamster CDI, what of other animal models? The hamster model is exquisitely sensitive to C. difficile toxin, with death from C. difficile challenge often coming as soon as 2–3 d post infection, perhaps not allowing enough time for the observation of phenotypic differences between flagellated and non-flagellated strains. Additionally, since different C. difficile strain backgrounds exhibit variations in flagellar expression levels and motility phenotypes,90 findings regarding C. difficile flagella may need to be evaluated on a strain-to-strain basis in multiple C. difficile backgrounds. Finally, with the availability of techniques to generate isogenic mutants in C. difficile now widely available, mutagenic analyses of additional genes responsible for flagellar glycosylation, flagellar gene expression and regulation and chemotaxis are warranted in order to better understand flagellar function and its contribution to C. difficile pathogenesis.
Cell-surface polysaccharides
Two cell-surface polysaccharides, PS-I and PS-II, have been characterized in C. difficile. Of the three strains analyzed so far, one was from the ribotype 027 (NAP1) clade and two from a non-027 strain. PS-I was found only on the NAP1 strain, whereas PS-II was found on all strains tested.102-104 This suggests that PS-II is a conserved antigen among different clades of C. difficile. Recently, both PS-I and PS-II have been chemically synthesized, and both now serve as potential candidates for vaccination against CDI. Though the exact function of these exopolysaccharides in CDI development/establishment is currently unknown, the contribution of similar molecules to bacterial infection is well described.105,106
Another important polysaccharide, peptidoglycan, represents an important virulence factor in many pathogenic bacteria including C. difficile. Variations in peptidoglycan structure can provide resistance to β-lactam antibiotics and lysozyme.107 C. difficile is known to be highly-resistant to lysozyme,108 and the structural analysis of its peptidoglycan revealed a high level of N-deacetylated GlcNAc residues in the glycan strand.109 N-deacetylation of the peptidoglycan has been shown to mediate lysozyme resistance in pathogenic Listeria and Streptococcus species,110,111 suggesting it may also play an important role in the resistance of C. difficile to host innate immunity.
Host Immune Response(s) to C. difficile Surface Molecules
The host innate and adaptive responses to C. difficile attachment remain largely uncharacterized. C. difficile SLPs are immunodominant antigens, and anti-SLP antibodies are readily recovered from CDI patient serum.112 SLPs from different strains of C. difficile induced the production of immunomodulatory cytokines IL-1β, IL-6 and IL-10 in cultured monocytes and stimulated the maturation of monocyte-derived dendritic cells.113 SLPs from epidemic-associated 027 and 001 and non-epidemic associated 012 ribotypes did not reveal any differences in their ability to activate monocytes and monocyte-derived dendritic cells.113 Since SLPs have been identified in all C. difficile isolates to date, and since SlpA plays a critical role in C. difficile adherence, it is not surprising that these proteins modulate the inflammatory response during CDI.
SLPs have also been recently investigated for their ability to activate pro-inflammatory signaling, including those utilizing toll-like receptor (TLR) signaling. TLR recognition of microbe-associated molecular patterns results in the engagement of adaptor molecules and subsequent signaling to activate inflammatory responses. TLR4, for instance, plays a role in the recognition of lipopolysaccharide from Gram-negative organisms, as well as lipoteichoic acid from Gram-positive bacteria.114 TLR4 engagement results in signaling through the adaptor molecule MyD88, leading to NFκB and IRF3 activation and subsequent production of inflammatory cytokines and interferon. In mice, SLPs induced dendritic cell maturation, as well as the production of inflammatory cytokines and cell surface markers. SLPs were also found to activate NFκB signaling in a TLR4-dependent manner, but IRF-3 was not activated. Correspondingly, TLR4-/- and MyD88-/- mice were more susceptible to CDI, confirming a role for TLR-dependent protection against C. difficile.115 Flagellin, the structural component of the flagellar shaft, is also a potent activator of the innate immune response. Flagellin engages TLR5 on host cells, resulting in NFκB activation and the production of pro-inflammatory cytokines. Stimulation of the mouse innate immune system with peritoneally-administered Salmonella Typhimurium flagellin inhibited subsequent (antibiotic-precipitated) C. difficile infection by slowing bacterial growth and toxin production and protected the animals from colitis and death.116
Other Host Response(s) to C. difficile Infection: Antimicrobial Peptides, Stress-response Factors and S-Nitrosylation
Antimicrobial peptides and stress-response factors
In order to establish infection, C. difficile must withstand a variety of host innate immune defenses, including the presence of antimicrobial peptides produced by host tissues in the gut. McBride et al. recently demonstrated that C. difficile employs a well-characterized antimicrobial peptide resistance mechanism, d-alanylation of teichoic acid, in response to antimicrobial peptide exposure.117 This group also identified a set of genes, encoding a histidine kinase and an ABC transporter, that confer resistance to multiple antimicrobial peptides in C. difficile.118 Extracytoplasmic function (ECF) sigma factors that sense and respond to antimicrobial peptides may also play a role in resistance.108 ECF sigma factors in other gram-positive bacteria are known to alter transcription in response to extracellular stresses, including antimicrobial peptide treatment. Compared with the isogenic parental strains, C. difficile mutants depleted for the ECF sigma factors CsfT and CsfU are more susceptible to antimicrobial peptides and are significantly reduced in virulence in the hamster CDI model. While the ECF sigma factor-induced transcriptional alterations contributing to antimicrobial peptide resistance have not been identified, these studies suggest that innate immune invasion likely plays a role in C. difficile pathogenesis.
C. difficile may stimulate chemoattractant cytokine production by intestinal epithelial cells. Sibartie et al. found that treatment of cultured intestinal epithelial cells with C. difficile supernatants increases the production of both CCL20, which recruits various immune effector cells and IL-1β.119 Interestingly, pre-exposure of intestinal epithelial cells to the gut commensal Bifidobacterium infantis blocks C.difficile-induced cytokine production. This suggests that the presence of the normal flora in a healthy gut prevents inappropriate inflammation in response to a low burden of potential pathogens.
S-nitrosylation
While the mechanism of action of the large C. difficile toxins on intestinal epithelial cells has been rigorously elucidated, relatively little is known about the host response to C. difficile toxins. C. difficile toxins are immunogenic, since antibodies against both TcdA and TcdB have been recovered in patient sera,120,121 and it is now recognized that both TcdA and TcdB inflammatory effects are mediated through inflammasome activation.13 Injection of TcdA in a rabbit ileal loop model stimulates an inflammatory response, including the upregulation of interlukin-1β (IL-1β), nitric oxide synthase (Nos2) and tumor necrosis factor α (TNFα).13,122 Interestingly, Nos2 has been implicated as a limiting factor during C. difficile intoxication, blocking the intestinal secretory effects of TcdA in the ileal loop model of infection.123 Many of the effects of nitric oxide, including its role in innate immunity, are mediated via S-nitrosylation of proteins. Savidge et al. demonstrated the presence of high levels of S-nitrosylated proteins in TcdA-exposed sections of ileal loops and in human biopsies from patients with active intestinal inflammation.122 Curiously, global proteomic analysis of TcdA-treated ileal mucosal extracts revealed TcdA itself to be among the proteins displaying increased S-nitrosylation. Further, Nos3-transfected epithelial cells were protected against TcdA-mediated intoxication, and TcdA isolated from these samples was S-nitrosylated. In vitro-nitrosylation of TcdA reduced its cytotoxicity relative to the native toxin. Mapping studies revealed that the catalytic cysteine residues involved in self-cleavage of TcdA and TcdB, respectively, were targets for S-nitrosylation; the modification prevented toxin self-processing and host entry. Finally, recovery of S-nitrosylated TcdA from patient stool specimens confirmed that this modification occurred in vivo.122
Recent Advances in Treatment for Single-Episode and Recurrent CDI
Since the year 2000, the incidence of severe, recurrent and/or intransigent CDI has confounded acceptable standards of care for the disease, since there is still a reliance on broad-spectrum antibiotics that further suppresses the normal microbiota, leaving patients susceptible to future (re-)infection. A variety of therapeutic strategies are therefore being investigated to either prevent CDI and/or to treat CDI in a manner that does not leave patients as susceptible to re-infection or recurrent disease. These strategies have included the use of both toxin and non-toxin based vaccines, bacteriotherapy and “designer” antibiotics which are more highly targeted and, therefore, reduce collateral damage to normal (protective) microbiota.
Immunotherapy: Active and passive vaccination targeting C. difficile toxins
A major focus of preventing recurrent CDI is the development of a vaccine that will stimulate a mucosal immune response and protect against C. difficile colonization. Past studies explored the toxins as antigens for vaccination against CDI, but their ability to prevent bacterial colonization was uncertain.124 However, a variety of vaccines strategies are being investigated, including those that target toxins. Recently, Permpoonpattana et al. assessed the C-terminal repeat domains of TcdA and TcdB (associated with host-cell binding), using B. subtilis spores as a delivery vehicle, as possible protective antigens.125 Hamsters immunized with B. subtilis spores expressing the TcdA repeat domain were protected from infection. Hamsters immunized with spores expressing the repeat domains of both C. difficile TcdA and TcdB produced high titers of IgG and secretory IgA antibodies against the toxins, and these antibodies neutralized TcdA and TcdB in vitro. Interestingly, surviving hamsters (immunized with B. subtilis spores expressing the A26–39 domain of TcdA) were fully protected from re-challenge with C. difficile.
A recent double-blind placebo-controlled phase II study explored the efficacy and safety of a monoclonal antibody against C. difficile TcdA (CDA1).126 Forty-six patients were evaluated; 29 patients received CDA1, and 17 received a placebo. Both groups had a similar number of recurrences [17.2% in the CDA1 group (5 recurrences) and 17.7% in the placebo group (3 recurrences)]. Further, since patients were only given monoclonal antibodies to TcdA, there were low levels of anti-TcdB antibodies in the serum. Thus it was concluded that lower serum levels of anti-TcdB antibodies are associated with, and may be predictive of, CDI recurrence. Antibodies to both toxins would then likely prevent CDI recurrence. One human monoclonal antibody (mAb) combination, MDX-066 (MBL CDA-1) and MDX-1388 (MBL CDA-2), has recently been licensed to Merck, Inc. MDX-066 and MDX-1388 target and neutralize toxins A and B respectively.127 In a phase II clinical trial involving 200 patients, an intravenous treatment of a combination of MDX-066 and MDX-1388 reduced CDI recurrence by 70%.
Immunotherapy: Vaccines targeting C. difficile non-toxin molecules
The Cwp84 protease may represent a promising non-toxin protein candidate for the development of a C. difficile vaccine.128 In their study, Pechine et al. delivered the protein to hamsters via three routes: rectal (mucosal), intragastric (mucosal) and subcutaneous (parenteral) immunization. Immunized hamsters were treated with clindamycin and then challenged with C. difficile spores. Subcutaneous immunization generated the highest serum antibody titer, although rectal immunization provided the greatest protection. To further assess the post-challenge survival of hamsters, and to measure C. difficile colonization, a second experiment was performed with a larger number of hamsters immunized through the rectal route. Hamsters immunized with Cwp84 survived longer; 90% of hamsters (15 of 16) died in the control group, while 33% of the Cwp84-immunized hamsters (6 of 18) survived the challenge. C. difficile colonization was evident only in 66% of the rectally-immunized hamsters, in contrast to 90% of the non-immunized control animals.
In a related study on a Cwp84-based vaccine, Sandolo et al. explored the use of pectin bead-encapsulation as an alternate mechanism to deliver the protein to the colon and confer protective immunity.124 While hamsters immunized with unloaded beads died in two days, 40% of the hamsters given Cwp84-loaded bead survived 10 d post-infection. Thus oral administration of Cwp84 via pectin-encapsulated beads was partially protective. However, this study did not address whether Cwp84 was able to stimulate a secretory IgA response. Taken together, Pechine et al. and Sandolo et al., have demonstrated that vaccination with Cwp84 in the colon (either through the rectal route or oral administration of pectin beads) provides a higher level of protection from CDI compared with more traditional routes of immunization. Given these promising initial results, additional work to explore the use of Cwp84 as a vaccine candidate is warranted.
The recently characterized surface polysaccharide, PS-II, is another possible non-toxin candidate for vaccination against C. difficile. The antigenic and immunogenic properties of PS-II were assessed in a recent study involving the chemical synthesis of a PS-II hapten.129 The chemically synthesized PS-II, denoted as hexasaccharide 2, was conjugated to the diphtheria toxoid CRM197 (as a protein carrier) and used to immunize female C57BL/6 mice to test its immunogenicity. Glycan microarray analysis was used to monitor the antibody titers to the PS-II hapten. Immunized mice produced IgG specific to the PS-II hapten. It was also shown that some patients infected with C. difficile make antibodies against the native glycopolymer, since 3/10 C. difficile-infected patients had high titers of IgA in their stool which recognized the hexasaccharide 2 hapten.
Bacterio-therapy: The fecal transplant
Recurrent CDI has been reported to occur in 15–30% of patients after initial infection with CDI. Up to 65% of those patients who have one recurrence will continue to have recurrences after each round of antibiotic therapy.3 Until recently, antibiotics have been a standard treatment option for CDI; however, because they invariably have a broad spectrum of anti-bacterial activity, they (irrevocably) alter the intestinal flora. Thus fecal transplantation has been proposed as an alternative method to treat recurrent CDI, with the major goal of reconstituting lost gut microbiota in patients.130
In a meta-analysis of 27 studies involving a total of 317 patients, intestinal microbiota transplantation (IMT) was shown to be successful when other commonly used treatments failed, with CDI resolution in 92% of cases.55 A similar analysis of published studies of patients receiving fecal transplants (n = 239), also revealed that the treatment was effective in 145 out of 166 patients.131 Although further studies are necessary to demonstrate the kinetics and efficiency of microbiota restoration (or a lack thereof in those patients who did not respond), this approach holds promise as biologically robust and non-drug based intervention for recalcitrant CDI.
Chemotherapy: Fidaxomicin
Fidaxomicin is a new macrocycle antibiotic approved to treat CDI in 2011.132 This drug appears to be more active than vancomycin against clinical isolates of ribotype 027/NAP1 strains of C. difficile in vitro.133 It has narrow spectrum antibiotic activity against C. difficile and is not effective against gram-negative bacteria, fungi or protozoa and may, therefore, help preserve the normal microbiota.132,134 Fidaxomicin may also be more effective at clearing the infection since it is bacteriocidal, whereas vancomycin is bacteriostatic. In a study done by Louie et al.,133 596 CDI patients were evaluated; 287 patients received fidaxomicin, with 253 patients experiencing diarrhea resolution, and 309 patients received vancomycin, with 265 patients experiencing cessation of diarrhea. A significantly reduced rate of CDI recurrence in fidaxomicin-treated patients was also observed (15.4%), compared with those on vancomycin (25.3%). Fidaxomicin may be more efficacious than vancomycin in treating CDI patients who are also taking other antibiotics for concomitant infections (90% of patients receiving fidaxomicin had a resolution of diarrhea vs. 79.4% for those receiving vancomycin.135
Future Perspectives
It is clear that advances in C. difficile biology and C. difficile-host interactions have exponentially increased in just the past two years, leading to a much more refined and mechanistically-oriented approach to dissecting C. difficile pathogenesis. The ability to rapidly genetically manipulate this once intractable pathogen will necessarily focus attention on traditional and non-traditional virulence factors, including those associated with the spore, colonization factors and immune evasion mechanisms. Explorations in the areas of C. difficile-host innate immunity, host gut mucosal biology and C. difficile spore germination dynamics, will continue to further our understanding of how C. difficile rapidly and robustly colonizes a susceptible host. Such studies will also identify bacterial and host-specific molecules and processes that, along with vaccine development, can serve as appropriate intervention targets for the future management of CDI.
Acknowledgments
Research in the Vedantam and Viswanathan laboratories is supported by the US Dept. of Veterans Affairs (G.V.; BX1I01BX001183), NIH (V.K.V.; 1R01AI081742), USDA (CSREES Hatch Program; G.V. and V.K.V.; Nos. ARZT-570410-A-02-139 and ARZT-570410-A-02-140 respectively), and the College of Agriculture and Life Sciences, University of Arizona (G.V. and V.K.V.). Sincere thanks to Al Agellon, Kevin Lewis, Bryan Roxas and Dr. Scott Wilbur for help with electron microscopy and image processing. We also thank the members of the G.V. and V.K.V. laboratories for helpful discussions.
Footnotes
Previously published online: www.landesbioscience.com/journals/gutmicrobes/article/19399
References
- 1.Barbut F, Jones G, Eckert C. Epidemiology and control of Clostridium difficile infections in healthcare settings: an update. Curr Opin Infect Dis. 2011;24:370–6. doi: 10.1097/QCO.0b013e32834748e5. [DOI] [PubMed] [Google Scholar]
- 2.Sorg JA, Sonenshein AL. Chenodeoxycholate is an inhibitor of Clostridium difficile spore germination. J Bacteriol. 2009;191:1115–7. doi: 10.1128/JB.01260-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Viswanathan VK, Mallozzi MJ, Vedantam G. Clostridium difficile infection: An overview of the disease and its pathogenesis, epidemiology and interventions. Gut Microbes. 2010;1:234–42. doi: 10.4161/gmic.1.4.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Killgore G, Thompson A, Johnson S, Brazier J, Kuijper E, Pepin J, et al. Comparison of seven techniques for typing international epidemic strains of Clostridium difficile: restriction endonuclease analysis, pulsed-field gel electrophoresis, PCR-ribotyping, multilocus sequence typing, multilocus variable-number tandem-repeat analysis, amplified fragment length polymorphism, and surface layer protein A gene sequence typing. J Clin Microbiol. 2008;46:431–7. doi: 10.1128/JCM.01484-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tenover FC, Akerlund T, Gerding DN, Goering RV, Boström T, Jonsson AM, et al. Comparison of strain typing results for Clostridium difficile isolates from North America. J Clin Microbiol. 2011;49:1831–7. doi: 10.1128/JCM.02446-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stabler RA, Gerding DN, Songer JG, Drudy D, Brazier JS, Trinh HT, et al. Comparative phylogenomics of Clostridium difficile reveals clade specificity and microevolution of hypervirulent strains. J Bacteriol. 2006;188:7297–305. doi: 10.1128/JB.00664-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dingle KE, Griffiths D, Didelot X, Evans J, Vaughan A, Kachrimanidou M, et al. Clinical Clostridium difficile: clonality and pathogenicity locus diversity. PLoS One. 2011;6:e19993. doi: 10.1371/journal.pone.0019993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, et al. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 2009;10:R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brouwer MS, Warburton PJ, Roberts AP, Mullany P, Allan E. Genetic organisation, mobility and predicted functions of genes on integrated, mobile genetic elements in sequenced strains of Clostridium difficile. PLoS One. 2011;6:e23014. doi: 10.1371/journal.pone.0023014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valiente E, Dawson LF, Cairns MD, Stabler RA, Wren BW. Emergence of new PCR ribotypes from the hypervirulent Clostridium difficile 027 lineage. J Med Microbiol. 2012;61:49–56. doi: 10.1099/jmm.0.036194-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies AH, Roberts AK, Shone CC, Acharya KR. Super toxins from a super bug: structure and function of Clostridium difficile toxins. Biochem J. 2011;436:517–26. doi: 10.1042/BJ20110106. [DOI] [PubMed] [Google Scholar]
- 12.Soehn F, Wagenknecht-Wiesner A, Leukel P, Kohl M, Weidmann M, von Eichel-Streiber C, et al. Genetic rearrangements in the pathogenicity locus of Clostridium difficile strain 8864--implications for transcription, expression and enzymatic activity of toxins A and B. Mol Gen Genet. 1998;258:222–32. doi: 10.1007/s004380050726. [DOI] [PubMed] [Google Scholar]
- 13.Ng J, Hirota SA, Gross O, Li Y, Ulke-Lemee A, Potentier MS, et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology. 2010;139:542–52, 552, e1-3. doi: 10.1053/j.gastro.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 14.Hirota SA, Fines K, Ng J, Traboulsi D, Lee J, Ihara E, et al. Hypoxia-inducible factor signaling provides protection in Clostridium difficile-induced intestinal injury. Gastroenterology. 2010;139:259–69, e3. doi: 10.1053/j.gastro.2010.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lyras D, O’Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, et al. Toxin B is essential for virulence of Clostridium difficile. Nature. 2009;458:1176–9. doi: 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuehne SA, Cartman ST, Minton NP. Both, toxin A and toxin B, are important in Clostridium difficile infection. Gut Microbes. 2011;2:252–5. doi: 10.4161/gmic.2.4.16109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carter GP, Douce GR, Govind R, Howarth PM, Mackin KE, Spencer J, et al. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog. 2011;7:e1002317. doi: 10.1371/journal.ppat.1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sirard S, Valiquette L, Fortier LC. Lack of association between clinical outcome of Clostridium difficile infections, strain type, and virulence-associated phenotypes. J Clin Microbiol. 2011;49:4040–6. doi: 10.1128/JCM.05053-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merrigan M, Venugopal A, Mallozzi M, Roxas B, Viswanathan VK, Johnson S, et al. Human hypervirulent Clostridium difficile strains exhibit increased sporulation as well as robust toxin production. J Bacteriol. 2010;192:4904–11. doi: 10.1128/JB.00445-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bacci S, Mølbak K, Kjeldsen MK, Olsen KE. Binary toxin and death after Clostridium difficile infection. Emerg Infect Dis. 2011;17:976–82. doi: 10.3201/eid1706.101483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwan C, Stecher B, Tzivelekidis T, van Ham M, Rohde M, Hardt WD, et al. Clostridium difficile toxin CDT induces formation of microtubule-based protrusions and increases adherence of bacteria. PLoS Pathog. 2009;5:e1000626. doi: 10.1371/journal.ppat.1000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papatheodorou P, Carette JE, Bell GW, Schwan C, Guttenberg G, Brummelkamp TR, et al. Lipolysis-stimulated lipoprotein receptor (LSR) is the host receptor for the binary toxin Clostridium difficile transferase (CDT) Proc Natl Acad Sci U S A. 2011;108:16422–7. doi: 10.1073/pnas.1109772108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaiser E, Kroll C, Ernst K, Schwan C, Popoff M, Fischer G, et al. Membrane translocation of binary actin-ADP-ribosylating toxins from Clostridium difficile and Clostridium perfringens is facilitated by cyclophilin A and Hsp90. Infect Immun. 2011;79:3913–21. doi: 10.1128/IAI.05372-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerding DN, Muto CA, Owens RC., Jr Measures to control and prevent Clostridium difficile infection. Clin Infect Dis. 2008;46(Suppl 1):S43–9. doi: 10.1086/521861. [DOI] [PubMed] [Google Scholar]
- 25.Merrigan M. Hypervirulent Clostridium difficile strains: Adherence, toxin production and sporulation. Microbiology and Immunology. 2010;178 [Google Scholar]
- 26.Akerlund T, Persson I, Unemo M, Nore´n T, Svenungsson B, Wullt M, et al. Increased sporulation rate of epidemic Clostridium difficile Type 027/NAP1. J Clin Microbiol. 2008;46:1530–3. doi: 10.1128/JCM.01964-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burns DA, Heap JT, Minton NP. The diverse sporulation characteristics of Clostridium difficile clinical isolates are not associated with type. Anaerobe. 2010;16:618–22. doi: 10.1016/j.anaerobe.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 28.Burns DA, Heeg D, Cartman ST, Minton NP. Reconsidering the sporulation characteristics of hypervirulent Clostridium difficile BI/NAP1/027. PLoS One. 2011;6:e24894. doi: 10.1371/journal.pone.0024894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Hoon MJ, Eichenberger P, Vitkup D. Hierarchical evolution of the bacterial sporulation network. Curr Biol. 2010;20:R735–45. doi: 10.1016/j.cub.2010.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paredes CJ, Alsaker KV, Papoutsakis ET. A comparative genomic view of clostridial sporulation and physiology. Nat Rev Microbiol. 2005;3:969–78. doi: 10.1038/nrmicro1288. [DOI] [PubMed] [Google Scholar]
- 31.Harry KH, Zhou R, Kroos L, Melville SB. Sporulation and enterotoxin (CPE) synthesis are controlled by the sporulation-specific sigma factors SigE and SigK in Clostridium perfringens. J Bacteriol. 2009;191:2728–42. doi: 10.1128/JB.01839-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, McClane BA. Evaluating the involvement of alternative sigma factors SigF and SigG in Clostridium perfringens sporulation and enterotoxin synthesis. Infect Immun. 2010;78:4286–93. doi: 10.1128/IAI.00528-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones SW, Tracy BP, Gaida SM, Papoutsakis ET. Inactivation of σF in Clostridium acetobutylicum ATCC 824 blocks sporulation prior to asymmetric division and abolishes σE and σG protein expression but does not block solvent formation. J Bacteriol. 2011;193:2429–40. doi: 10.1128/JB.00088-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saujet L, Monot M, Dupuy B, Soutourina O, Martin-Verstraete I. The key sigma factor of transition phase, SigH, controls sporulation, metabolism, and virulence factor expression in Clostridium difficile. J Bacteriol. 2011;193:3186–96. doi: 10.1128/JB.00272-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tracy BP, Jones SW, Papoutsakis ET. Inactivation of σE and σG in Clostridium acetobutylicum illuminates their roles in clostridial-cell-form biogenesis, granulose synthesis, solventogenesis, and spore morphogenesis. J Bacteriol. 2011;193:1414–26. doi: 10.1128/JB.01380-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steiner E, Dago AE, Young DI, Heap JT, Minton NP, Hoch JA, et al. Multiple orphan histidine kinases interact directly with Spo0A to control the initiation of endospore formation in Clostridium acetobutylicum. Mol Microbiol. 2011;80:641–54. doi: 10.1111/j.1365-2958.2011.07608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Underwood S, Guan S, Vijayasubhash V, Baines SD, Graham L, Lewis RJ, et al. Characterization of the sporulation initiation pathway of Clostridium difficile and its role in toxin production. J Bacteriol. 2009;191:7296–305. doi: 10.1128/JB.00882-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solomon JM, Magnuson R, Srivastava A, Grossman AD. Convergent sensing pathways mediate response to two extracellular competence factors in Bacillus subtilis. Genes Dev. 1995;9:547–58. doi: 10.1101/gad.9.5.547. [DOI] [PubMed] [Google Scholar]
- 39.Cooksley CM, Davis IJ, Winzer K, Chan WC, Peck MW, Minton NP. Regulation of neurotoxin production and sporulation by a Putative agrBD signaling system in proteolytic Clostridium botulinum. Appl Environ Microbiol. 2010;76:4448–60. doi: 10.1128/AEM.03038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Chen J, Vidal JE, McClane BA. The Agr-like quorum-sensing system regulates sporulation and production of enterotoxin and beta2 toxin by Clostridium perfringens type A non-food-borne human gastrointestinal disease strain F5603. Infect Immun. 2011;79:2451–9. doi: 10.1128/IAI.00169-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Setlow P. Spore germination. Curr Opin Microbiol. 2003;6:550–6. doi: 10.1016/j.mib.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 42.Paredes-Sabja D, Setlow P, Sarker MR. Germination of spores of Bacillales and Clostridiales species: mechanisms and proteins involved. Trends Microbiol. 2011;19:85–94. doi: 10.1016/j.tim.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 43.Paredes-Sabja D, Setlow P, Sarker MR. GerO, a putative Na+/H+-K+ antiporter, is essential for normal germination of spores of the pathogenic bacterium Clostridium perfringens. J Bacteriol. 2009;191:3822–31. doi: 10.1128/JB.00158-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paredes-Sabja D, Setlow P, Sarker MR. SleC is essential for cortex peptidoglycan hydrolysis during germination of spores of the pathogenic bacterium Clostridium perfringens. J Bacteriol. 2009;191:2711–20. doi: 10.1128/JB.01832-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paredes-Sabja D, Setlow P, Sarker MR. Role of GerKB in germination and outgrowth of Clostridium perfringens spores. Appl Environ Microbiol. 2009;75:3813–7. doi: 10.1128/AEM.00048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paredes-Sabja D, Torres JA, Setlow P, Sarker MR. Clostridium perfringens spore germination: characterization of germinants and their receptors. J Bacteriol. 2008;190:1190–201. doi: 10.1128/JB.01748-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alberto F, Botella L, Carlin F, Nguyen-The C, Broussolle V. The Clostridium botulinum GerAB germination protein is located in the inner membrane of spores. FEMS Microbiol Lett. 2005;253:231–5. doi: 10.1016/j.femsle.2005.09.037. [DOI] [PubMed] [Google Scholar]
- 48.Burns DA, Heap JT, Minton NP. SleC is essential for germination of Clostridium difficile spores in nutrient-rich medium supplemented with the bile salt taurocholate. J Bacteriol. 2010;192:657–64. doi: 10.1128/JB.01209-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sorg JA, Sonenshein AL. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol. 2008;190:2505–12. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Howerton A, Ramirez N, Abel-Santos E. Mapping interactions between germinants and Clostridium difficile spores. J Bacteriol. 2011;193:274–82. doi: 10.1128/JB.00980-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramirez N, Liggins M, Abel-Santos E. Kinetic evidence for the presence of putative germination receptors in Clostridium difficile spores. J Bacteriol. 2010;192:4215–22. doi: 10.1128/JB.00488-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sorg JA, Sonenshein AL. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J Bacteriol. 2010;192:4983–90. doi: 10.1128/JB.00610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stecher B, Hardt WD. Mechanisms controlling pathogen colonization of the gut. Curr Opin Microbiol. 2011;14:82–91. doi: 10.1016/j.mib.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 54.Stecher B, Hardt WD. The role of microbiota in infectious disease. Trends Microbiol. 2008;16:107–14. doi: 10.1016/j.tim.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 55.Gough E, Shaikh H, Manges AR. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Dis. 2011;53:994–1002. doi: 10.1093/cid/cir632. [DOI] [PubMed] [Google Scholar]
- 56.He X, Tian Y, Guo L, Lux R, Zusman DR, Shi W. Oral-derived bacterial flora defends its domain by recognizing and killing intruders--a molecular analysis using Escherichia coli as a model intestinal bacterium. Microb Ecol. 2010;60:655–64. doi: 10.1007/s00248-010-9708-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Borriello SP, Barclay FE. An in-vitro model of colonisation resistance to Clostridium difficile infection. J Med Microbiol. 1986;21:299–309. doi: 10.1099/00222615-21-4-299. [DOI] [PubMed] [Google Scholar]
- 58.McFarland L. Normal flora: diversity and functions. Microb Ecol Health Dis. 2000;12:193–207. doi: 10.1080/08910600050216183. [DOI] [Google Scholar]
- 59.Merrigan MM, Sambol SP, Johnson S, Gerding DN. New approach to the management of Clostridium difficile infection: colonisation with non-toxigenic C. difficile during daily ampicillin or ceftriaxone administration. Int J Antimicrob Agents. 2009;33(Suppl 1):S46–50. doi: 10.1016/S0924-8579(09)70017-2. [DOI] [PubMed] [Google Scholar]
- 60.Sambol SP, Merrigan MM, Tang JK, Johnson S, Gerding DN. Colonization for the prevention of Clostridium difficile disease in hamsters. J Infect Dis. 2002;186:1781–9. doi: 10.1086/345676. [DOI] [PubMed] [Google Scholar]
- 61.Songer JG, Jones R, Anderson MA, Barbara AJ, Post KW, Trinh HT. Prevention of porcine Clostridium difficile-associated disease by competitive exclusion with nontoxigenic organisms. Vet Microbiol. 2007;124:358–61. doi: 10.1016/j.vetmic.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 62.Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, et al. Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile-associated diarrhea. J Infect Dis. 2008;197:435–8. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- 63.De La Cochetière MF, Durand T, Lalande V, Petit JC, Potel G, Beaugerie L. Effect of antibiotic therapy on human fecal microbiota and the relation to the development of Clostridium difficile. Microb Ecol. 2008;56:395–402. doi: 10.1007/s00248-007-9356-5. [DOI] [PubMed] [Google Scholar]
- 64.Khoruts A, Sadowsky MJ. Therapeutic transplantation of the distal gut microbiota. Mucosal Immunol. 2011;4:4–7. doi: 10.1038/mi.2010.79. [DOI] [PubMed] [Google Scholar]
- 65.Manges AR, Labbe A, Loo VG, Atherton JK, Behr MA, Masson L, et al. Comparative metagenomic study of alterations to the intestinal microbiota and risk of nosocomial Clostridum difficile-associated disease. J Infect Dis. 2010;202:1877–84. doi: 10.1086/657319. [DOI] [PubMed] [Google Scholar]
- 66.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4554–61. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vengadesan K, Narayana SV. Structural biology of Gram-positive bacterial adhesins. Protein Sci. 2011;20:759–72. doi: 10.1002/pro.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varga JJ, Nguyen V, O’Brien DK, Rodgers K, Walker RA, Melville SB. Type IV pili-dependent gliding motility in the Gram-positive pathogen Clostridium perfringens and other Clostridia. Mol Microbiol. 2006;62:680–94. doi: 10.1111/j.1365-2958.2006.05414.x. [DOI] [PubMed] [Google Scholar]
- 69.Twine SM, Reid CW, Aubry A, McMullin DR, Fulton KM, Austin J, et al. Motility and flagellar glycosylation in Clostridium difficile. J Bacteriol. 2009;191:7050–62. doi: 10.1128/JB.00861-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38:779–86. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 71.Barketi-Klai A, Hoys S, Lambert-Bordes S, Collignon A, Kansau I. Role of fibronectin-binding protein A in Clostridium difficile intestinal colonization. J Med Microbiol. 2011;60:1155–61. doi: 10.1099/jmm.0.029553-0. [DOI] [PubMed] [Google Scholar]
- 72.Lin YP, Kuo CJ, Koleci X, McDonough SP, Chang YF. Manganese binds to Clostridium difficile Fbp68 and is essential for fibronectin binding. J Biol Chem. 2011;286:3957–69. doi: 10.1074/jbc.M110.184523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hennequin C, Janoir C, Barc MC, Collignon A, Karjalainen T. Identification and characterization of a fibronectin-binding protein from Clostridium difficile. Microbiology. 2003;149:2779–87. doi: 10.1099/mic.0.26145-0. [DOI] [PubMed] [Google Scholar]
- 74.Calabi E, Ward S, Wren B, Paxton T, Panico M, Morris H, et al. Molecular characterization of the surface layer proteins from Clostridium difficile. Mol Microbiol. 2001;40:1187–99. doi: 10.1046/j.1365-2958.2001.02461.x. [DOI] [PubMed] [Google Scholar]
- 75.de la Riva L, Willing SE, Tate EW, Fairweather NF. Roles of cysteine proteases Cwp84 and Cwp13 in biogenesis of the cell wall of Clostridium difficile. J Bacteriol. 2011;193:3276–85. doi: 10.1128/JB.00248-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Janoir C, Pe´chine´ S, Grosdidier C, Collignon A. Cwp84, a surface-associated protein of Clostridium difficile, is a cysteine protease with degrading activity on extracellular matrix proteins. J Bacteriol. 2007;189:7174–80. doi: 10.1128/JB.00578-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Waligora AJ, Hennequin C, Mullany P, Bourlioux P, Collignon A, Karjalainen T. Characterization of a cell surface protein of Clostridium difficile with adhesive properties. Infect Immun. 2001;69:2144–53. doi: 10.1128/IAI.69.4.2144-2153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reynolds CB, Emerson JE, de la Riva L, Fagan RP, Fairweather NF. The Clostridium difficile cell wall protein CwpV is antigenically variable between strains, but exhibits conserved aggregation-promoting function. PLoS Pathog. 2011;7:e1002024. doi: 10.1371/journal.ppat.1002024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Calabi E, Calabi F, Phillips AD, Fairweather NF. Binding of Clostridium difficile surface layer proteins to gastrointestinal tissues. Infect Immun. 2002;70:5770–8. doi: 10.1128/IAI.70.10.5770-5778.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chapeto´nMontes D, Candela T, Collignon A, Janoir C. Localization of the Clostridium difficile cysteine protease Cwp84 and insights into its maturation process. J Bacteriol. 2011;193:5314–21. doi: 10.1128/JB.00326-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fagan RP, Janoir C, Collignon A, Mastrantonio P, Poxton IR, Fairweather NF. A proposed nomenclature for cell wall proteins of Clostridium difficile. J Med Microbiol. 2011;60:1225–8. doi: 10.1099/jmm.0.028472-0. [DOI] [PubMed] [Google Scholar]
- 82.Fagan RP, Albesa-Jove´ D, Qazi O, Svergun DI, Brown KA, Fairweather NF. Structural insights into the molecular organization of the S-layer from Clostridium difficile. Mol Microbiol. 2009;71:1308–22. doi: 10.1111/j.1365-2958.2009.06603.x. [DOI] [PubMed] [Google Scholar]
- 83.Karjalainen T, Waligora-Dupriet AJ, Cerquetti M, Spigaglia P, Maggioni A, Mauri P, et al. Molecular and genomic analysis of genes encoding surface-anchored proteins from Clostridium difficile. Infect Immun. 2001;69:3442–6. doi: 10.1128/IAI.69.5.3442-3446.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Swidsinski A, Sydora BC, Doerffel Y, Loening-Baucke V, Vaneechoutte M, Lupicki M, et al. Viscosity gradient within the mucus layer determines the mucosal barrier function and the spatial organization of the intestinal microbiota. Inflamm Bowel Dis. 2007;13:963–70. doi: 10.1002/ibd.20163. [DOI] [PubMed] [Google Scholar]
- 85.Swidsinski A, Loening-Baucke V, Theissig F, Engelhardt H, Bengmark S, Koch S, et al. Comparative study of the intestinal mucus barrier in normal and inflamed colon. Gut. 2007;56:343–50. doi: 10.1136/gut.2006.098160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O’Neil HS, Marquis H. Listeria monocytogenes flagella are used for motility, not as adhesins, to increase host cell invasion. Infect Immun. 2006;74:6675–81. doi: 10.1128/IAI.00886-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Crawford RW, Reeve KE, Gunn JS. Flagellated but not hyperfimbriated Salmonella enterica serovar Typhimurium attaches to and forms biofilms on cholesterol-coated surfaces. J Bacteriol. 2010;192:2981–90. doi: 10.1128/JB.01620-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stecher B, Hapfelmeier S, Müller C, Kremer M, Stallmach T, Hardt WD. Flagella and chemotaxis are required for efficient induction of Salmonella enterica serovar Typhimurium colitis in streptomycin-pretreated mice. Infect Immun. 2004;72:4138–50. doi: 10.1128/IAI.72.7.4138-4150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Erdem AL, Avelino F, Xicohtencatl-Cortes J, Giro´n JA. Host protein binding and adhesive properties of H6 and H7 flagella of attaching and effacing Escherichia coli. J Bacteriol. 2007;189:7426–35. doi: 10.1128/JB.00464-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pituch H, Obuch-Woszczatyñski P, van den Braak N, van Belkum A, Kujawa M, Luczak M, et al. Variable flagella expression among clonal toxin A-/B+Clostridium difficile strains with highly homogeneous flagellin genes. Clin Microbiol Infect. 2002;8:187–8. doi: 10.1046/j.1469-0691.2002.00394.x. [DOI] [PubMed] [Google Scholar]
- 91.Delme´e M, Avesani V, Delferriere N, Burtonboy G. Characterization of flagella of Clostridium difficile and their role in serogrouping reactions. J Clin Microbiol. 1990;28:2210–4. doi: 10.1128/jcm.28.10.2210-2214.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Martirosian G, Wize J, Soko´ł-Leszczyn´ska B, Zielin´ska U, Meisel-Mikołajczyk F, Masálin´ski S. Comparison of Delme´e and Polish serogroup-specific Clostridium difficile strains. Acta Microbiol Pol. 1993;42:251–7. [PubMed] [Google Scholar]
- 93.Tasteyre A, Barc MC, Collignon A, Boureau H, Karjalainen T. Role of FliC and FliD flagellar proteins of Clostridium difficile in adherence and gut colonization. Infect Immun. 2001;69:7937–40. doi: 10.1128/IAI.69.12.7937-7940.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tasteyre A, Karjalainen T, Avesani V, Delme´e M, Collignon A, Bourlioux P, et al. Phenotypic and genotypic diversity of the flagellin gene (fliC) among Clostridium difficile isolates from different serogroups. J Clin Microbiol. 2000;38:3179–86. doi: 10.1128/jcm.38.9.3179-3186.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tasteyre A, Barc MC, Karjalainen T, Dodson P, Hyde S, Bourlioux P, et al. A Clostridium difficile gene encoding flagellin. Microbiology. 2000;146:957–66. doi: 10.1099/00221287-146-4-957. [DOI] [PubMed] [Google Scholar]
- 96.Arora SK, Ritchings BW, Almira EC, Lory S, Ramphal R. The Pseudomonas aeruginosa flagellar cap protein, FliD, is responsible for mucin adhesion. Infect Immun. 1998;66:1000–7. doi: 10.1128/iai.66.3.1000-1007.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tasteyre A, Karjalainen T, Avesani V, Delme´e M, Collignon A, Bourlioux P, et al. Molecular characterization of fliD gene encoding flagellar cap and its expression among Clostridium difficile isolates from different serogroups. J Clin Microbiol. 2001;39:1178–83. doi: 10.1128/JCM.39.3.1178-1183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guerry P, Ewing CP, Schirm M, Lorenzo M, Kelly J, Pattarini D, et al. Changes in flagellin glycosylation affect Campylobacter autoagglutination and virulence. Mol Microbiol. 2006;60:299–311. doi: 10.1111/j.1365-2958.2006.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Canals R, Ramirez S, Vilches S, Horsburgh G, Shaw JG, Tomás JM, et al. Polar flagellum biogenesis in Aeromonas hydrophila. J Bacteriol. 2006;188:542–55. doi: 10.1128/JB.188.2.542-555.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dingle TC, Mulvey GL, Armstrong GD. Mutagenic analysis of the Clostridium difficile flagellar proteins, FliC and FliD, and their contribution to virulence in hamsters. Infect Immun. 2011;79:4061–7. doi: 10.1128/IAI.05305-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marsden GL, Davis IJ, Wright VJ, Sebaihia M, Kuijper EJ, Minton NP. Array comparative hybridisation reveals a high degree of similarity between UK and European clinical isolates of hypervirulent Clostridium difficile. BMC Genomics. 2010;11:389. doi: 10.1186/1471-2164-11-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Martin CE, Weishaupt MW, Seeberger PH. Progress toward developing a carbohydrate-conjugate vaccine against Clostridium difficile ribotype 027: synthesis of the cell-surface polysaccharide PS-I repeating unit. Chem Commun (Camb) 2011;47:10260–2. doi: 10.1039/c1cc13614c. [DOI] [PubMed] [Google Scholar]
- 103.Danieli E, Lay L, Proietti D, Berti F, Costantino P, Adamo R. First synthesis of C. difficile PS-II cell wall polysaccharide repeating unit. Org Lett. 2011;13:378–81. doi: 10.1021/ol1026188. [DOI] [PubMed] [Google Scholar]
- 104.Ganeshapillai J, Vinogradov E, Rousseau J, Weese JS, Monteiro MA. Clostridium difficile cell-surface polysaccharides composed of pentaglycosyl and hexaglycosyl phosphate repeating units. Carbohydr Res. 2008;343:703–10. doi: 10.1016/j.carres.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 105.Weidenmaier C, Peschel A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat Rev Microbiol. 2008;6:276–87. doi: 10.1038/nrmicro1861. [DOI] [PubMed] [Google Scholar]
- 106.Weidenmaier C, Kokai-Kun JF, Kulauzovic E, Kohler T, Thumm G, Stoll H, et al. Differential roles of sortase-anchored surface proteins and wall teichoic acid in Staphylococcus aureus nasal colonization. Int J Med Microbiol. 2008;298:505–13. doi: 10.1016/j.ijmm.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 107.Vollmer W. Structural variation in the glycan strands of bacterial peptidoglycan. FEMS Microbiol Rev. 2008;32:287–306. doi: 10.1111/j.1574-6976.2007.00088.x. [DOI] [PubMed] [Google Scholar]
- 108.Ho TD, Ellermeier CD. PrsW is required for colonization, resistance to antimicrobial peptides, and expression of extracytoplasmic function σ factors in Clostridium difficile. Infect Immun. 2011;79:3229–38. doi: 10.1128/IAI.00019-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Peltier J, Courtin P, El Meouche I, Leme´e L, Chapot-Chartier MP, Pons JL. Clostridium difficile has an original peptidoglycan structure with a high level of N-acetylglucosamine deacetylation and mainly 3-3 cross-links. J Biol Chem. 2011;286:29053–62. doi: 10.1074/jbc.M111.259150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Boneca IG, Dussurget O, Cabanes D, Nahori MA, Sousa S, Lecuit M, et al. A critical role for peptidoglycan N-deacetylation in Listeria evasion from the host innate immune system. Proc Natl Acad Sci U S A. 2007;104:997–1002. doi: 10.1073/pnas.0609672104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vollmer W, Tomasz A. Peptidoglycan N-acetylglucosamine deacetylase, a putative virulence factor in Streptococcus pneumoniae. Infect Immun. 2002;70:7176–8. doi: 10.1128/IAI.70.12.7176-7178.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Drudy D, Calabi E, Kyne L, Sougioultzis S, Kelly E, Fairweather N, et al. Human antibody response to surface layer proteins in Clostridium difficile infection. FEMS Immunol Med Microbiol. 2004;41:237–42, 42. doi: 10.1016/j.femsim.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 113.Bianco M, Fedele G, Quattrini A, Spigaglia P, Barbanti F, Mastrantonio P, et al. Immunomodulatory activities of surface-layer proteins obtained from epidemic and hypervirulent Clostridium difficile strains. J Med Microbiol. 2011;60:1162–7. doi: 10.1099/jmm.0.029694-0. [DOI] [PubMed] [Google Scholar]
- 114.Takeuchi O, Akira S. Toll-like receptors; theirphysiological role and signal transduction system. Int Immunopharmacol. 2001;1:625–35. doi: 10.1016/S1567-5769(01)00010-8. [DOI] [PubMed] [Google Scholar]
- 115.Ryan A, Lynch M, Smith SM, Amu S, Nel HJ, McCoy CE, et al. A role for TLR4 in Clostridium difficile infection and the recognition of surface layer proteins. PLoS Pathog. 2011;7:e1002076. doi: 10.1371/journal.ppat.1002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jarchum I, Liu M, Lipuma L, Pamer EG. Toll-like receptor 5 stimulation protects mice from acute Clostridium difficile colitis. Infect Immun. 2011;79:1498–503. doi: 10.1128/IAI.01196-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McBride SM, Sonenshein AL. The dlt operon confers resistance to cationic antimicrobial peptides in Clostridium difficile. Microbiology. 2011;157:1457–65. doi: 10.1099/mic.0.045997-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McBride SM, Sonenshein AL. Identification of a genetic locus responsible for antimicrobial peptide resistance in Clostridium difficile. Infect Immun. 2011;79:167–76. doi: 10.1128/IAI.00731-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sibartie S, O’Hara AM, Ryan J, Fanning A, O’Mahony J, O’Neill S, et al. Modulation of pathogen-induced CCL20 secretion from HT-29 human intestinal epithelial cells by commensal bacteria. BMC Immunol. 2009;10:54. doi: 10.1186/1471-2172-10-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Johnson S, Gerding DN, Janoff EN. Systemic and mucosal antibody responses to toxin A in patients infected with Clostridium difficile. J Infect Dis. 1992;166:1287–94. doi: 10.1093/infdis/166.6.1287. [DOI] [PubMed] [Google Scholar]
- 121.Aronsson B, Granström M, Möllby R, Nord CE. Serum antibody response to Clostridium difficile toxins in patients with Clostridium difficile diarrhoea. Infection. 1985;13:97–101. doi: 10.1007/BF01642866. [DOI] [PubMed] [Google Scholar]
- 122.Savidge TC, Urvil P, Oezguen N, Ali K, Choudhury A, Acharya V, et al. Host S-nitrosylation inhibits clostridial small molecule-activated glucosylating toxins. Nat Med. 2011;17:1136–41. doi: 10.1038/nm.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Qiu B, Pothoulakis C, Castagliuolo I, Nikulasson Z, LaMont JT. Nitric oxide inhibits rat intestinal secretion by Clostridium difficile toxin A but not Vibrio cholerae enterotoxin. Gastroenterology. 1996;111:409–18. doi: 10.1053/gast.1996.v111.pm8690206. [DOI] [PubMed] [Google Scholar]
- 124.Sandolo C, Pe´chine´ S, Le Monnier A, Hoys S, Janoir C, Coviello T, et al. Encapsulation of Cwp84 into pectin beads for oral vaccination against Clostridium difficile. Eur J Pharm Biopharm. 2011;79:566–73. doi: 10.1016/j.ejpb.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 125.Permpoonpattana P, Hong HA, Phetcharaburanin J, Huang JM, Cook J, Fairweather NF, et al. Immunization with Bacillus spores expressing toxin A peptide repeats protects against infection with Clostridium difficile strains producing toxins A and B. Infect Immun. 2011;79:2295–302. doi: 10.1128/IAI.00130-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Leav BA, Blair B, Leney M, Knauber M, Reilly C, Lowy I, et al. Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI) Vaccine. 2010;28:965–9. doi: 10.1016/j.vaccine.2009.10.144. [DOI] [PubMed] [Google Scholar]
- 127.Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, et al. Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med. 2010;362:197–205. doi: 10.1056/NEJMoa0907635. [DOI] [PubMed] [Google Scholar]
- 128.Pe´chine´ S, Denève C, Le Monnier A, Hoys S, Janoir C, Collignon A. Immunization of hamsters against Clostridium difficile infection using the Cwp84 protease as an antigen. FEMS Immunol Med Microbiol. 2011;63:73–81. doi: 10.1111/j.1574-695X.2011.00832.x. [DOI] [PubMed] [Google Scholar]
- 129.Oberli MA, Hecht ML, Bindschädler P, Adibekian A, Adam T, Seeberger PH. A possible oligosaccharide-conjugate vaccine candidate for Clostridium difficile is antigenic and immunogenic. Chem Biol. 2011;18:580–8. doi: 10.1016/j.chembiol.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 130.Bakken JS, Borody T, Brandt LJ, Brill JV, Demarco DC, Franzos MA, et al. Fecal Microbiota Transplantation Workgroup Treating Clostridium difficile infection with fecal microbiota transplantation. Clin Gastroenterol Hepatol. 2011;9:1044–9. doi: 10.1016/j.cgh.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Landy J, Al-Hassi HO, McLaughlin SD, Walker AW, Ciclitira PJ, Nicholls RJ, et al. Review article: faecal transplantation therapy for gastrointestinal disease. Aliment Pharmacol Ther. 2011;34:409–15. doi: 10.1111/j.1365-2036.2011.04737.x. [DOI] [PubMed] [Google Scholar]
- 132.Mullane KM, Gorbach S. Fidaxomicin: first-in-class macrocyclic antibiotic. Expert Rev Anti Infect Ther. 2011;9:767–77. doi: 10.1586/eri.11.53. [DOI] [PubMed] [Google Scholar]
- 133.Louie TJ, Miller MA, Mullane KM, Weiss K, Lentnek A, Golan Y, et al. OPT-80-003 Clinical Study Group Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med. 2011;364:422–31. doi: 10.1056/NEJMoa0910812. [DOI] [PubMed] [Google Scholar]
- 134.Tannock GW, Munro K, Taylor C, Lawley B, Young W, Byrne B, et al. A new macrocyclic antibiotic, fidaxomicin (OPT-80), causes less alteration to the bowel microbiota of Clostridium difficile-infected patients than does vancomycin. Microbiology. 2010;156:3354–9. doi: 10.1099/mic.0.042010-0. [DOI] [PubMed] [Google Scholar]
- 135.Mullane KM, Miller MA, Weiss K, Lentnek A, Golan Y, Sears PS, et al. Efficacy of fidaxomicin versus vancomycin as therapy for Clostridium difficile infection in individuals taking concomitant antibiotics for other concurrent infections. Clin Infect Dis. 2011;53:440–7. doi: 10.1093/cid/cir404. [DOI] [PMC free article] [PubMed] [Google Scholar]