

Abstract
The extension of processes of oligodendrocyte (OLG) and their precursor cells (OPCs) are crucial for migration, axonal contact and myelination. Here we show that a non-lethal oxidative stress induced by 3-nitropropionic acid (3-NP) elicited a rapid shortening of processes (~24%) in primary OLGs and in OLN-93 cells (~36%) as compared to vehicle-exposed cells. This was reversible and prevented by antioxidants. Proteomics of OLG lysates with and without 3-NP treatment yielded collapsin response mediator protein 2 (CRMP-2) as a candidate effector molecule. Inhibition of rho kinase (ROCK) was sufficient to prevent process retraction in both OLGs and OLN-93 cells. Oxidative stress increased phosphorylation of CRMP-2 at T555 that was completely prevented by Y27632. Moreover, transfection of OLN-93 cells with the mutant CRMP-2 T555A which cannot be phosphorylated by ROCK, prevented process shortening induced by 3-NP as compared to wild-type CRMP-2. Our results suggest a role for endogenous reactive oxygen species in a pathway that regulates OLG process extension. The vulnerability of late myelinated neurons in the adult brain and the presence of white matter pathology in human dementias warrant the study of this oligodendroglial pathway in the early stages of neurodegenerative conditions characterized by oxidative stress.
Keywords: oligodendrocyte, oxidative stress, CRMP-2, rho kinase, axonal myelination, Alzheimer’s disease
Introduction
In the human brain, axonal myelination starts in late fetal life and has its peak during the first postnatal year (Back et al. 2001, Jakovcevski et al. 2007, Ulfig et al. 1998, Baumann & Pham-Dinh 2001). Yet, certain associative regions continue to increase their myelin content into the 5th–6th decades of life. This protracted myelination may impose a high metabolic burden to oligodendrocyte precursor cells (OPCs) and oligodendrocytes (OLGs) in specific meso-cortical areas of the aging brain (Benes et al. 1994). Neurons with long, unmyelinated axons in this brain region are known to be affected in very early stages of major human dementias such as Alzheimer’s disease (AD) (Bartzokis et al. 2004). The extension and retraction of processes is crucial for 1) migration of OPCs throughout the CNS and 2) for mature OLGs to establish axonal contacts and subsequent myelin ensheathment (Dawson et al. 2003b). Extension of OPC and OLG processes is dependent on actin polymerization-driven protrusion that involves actin filament nucleation-promoting proteins such as N-WASP (Bacon et al. 2007). Molecular motors and cargo proteins that travel along actin filaments such as myosin Va and VAMP2, respectively, also promote process extension (Sloane & Vartanian 2007). The inhibition of myosin II, in turn, promotes branching and lamella formation in vitro (Wang et al. 2008). Secreted proteins known to act as guidance cues are implicated in the regulation of OPC and OLG process dynamics such as netrin-1, via its receptor Deleted in Colorectal Cancer (Dcc) and semaphorin3A (sema3A) via a receptor complex consisting of neuropilins and the Plexin-A subfamily (Spassky et al. 2002, Okada et al. 2007). Netrin-1 promotes the extension of processes in mature OLG. This effect is triggered by netrin-1 upon binding to its receptor Dcc through the phosphorylation of the Src family tyrosine kinase Fyn and inhibition of RhoA activity (Rajasekharan et al. 2010, Rajasekharan et al. 2009) Activation of the sema3A-neuropilin pathway, in turn, results in process retraction in OLGs and OPCs mediated by collapsing response mediator protein-2-(CRMP-2) (Ricard et al. 2001, Spassky et al. 2002). In addition, lysophospholipids including sphingosine-1-phosphate (S1P) and lysophosphatidic acid (LPA) can induce the retraction of pre-OLGs upon interaction with the G-protein-coupled Edg receptor family (Jaillard et al. 2005). The effect of lysophospholipids seems to be mediated by the activation of the small GTP-binding protein RhoA followed by activation of Rho kinase (ROCK) and phosphorylation of CRMP-2 (Dawson et al. 2003a, Novgorodov et al. 2007). Two different pathways have been reported for CRMP-2 phosphorylation, one mediated by ROCK which is crucial for neuronal retraction induced by LPA and ephrin-A5 (Arimura et al. 2000, Arimura et al. 2005) and the other one mediated by cdk5 (as a priming kinase) for further phosphorylation at upstream residues by GSK3β (Cole et al. 2006, Uchida et al. 2005). Both pathways are involved in neuronal process retraction yet, their role in OLG process dynamic is poorly understood.
In neurons, CRMP-2 binds and delivers tubulin dimers to the plus ends of growing microtubules (Gu & Ihara 2000, Fukata et al. 2002), co-polymerizes with tubulin heterodimers and promotes tubulin polymerization in vitro (Yoshimura et al. 2005, Chae et al. 2009). Upon phosphorylation, CRMP-2 decreases its affinity for tubulin and favors microtubule disassembly (Uchida et al. 2005, Arimura et al. 2005).
The mammalian brain is characterized by a high rate of reactive oxygen species (ROS) production. Together with its limited antioxidant capacity as compared to other organs, this results in an increasing age-dependent oxidative stress. (Perez-Campo et al. 1998, Smith et al. 1999, Sastre et al. 2000). OLGs and OPCs are particularly vulnerable to oxidative stress due to their high metabolic rate and low levels of glutathione (GSH) and glutathione peroxidase (Back et al. 1998, Juurlink et al. 1998). In addition, oligodendroglial cells have the highest content of iron (Fe) in the brain (Connor & Menzies 1995). The effects of diverse oxidative stimuli upon OPCs and OLGs have been focused mainly on the mechanisms of apoptosis involving the release of cytochrome C from mitochondria and activation of caspases 3 and 9 (Hollensworth et al. 2000, Mronga et al. 2004). Mature OLGs under oxidative stress also show chromatin segmentation related to the activation of the transcription factors c-fos and c-jun (Richter-Landsberg & Vollgraf 1998). More recently, it has been shown that ROS can induce apoptosis of human primary OLGs through the activation of neutral sphingomyelinase and the production of ceramide (Lee et al. 2004, Jana & Pahan 2007).
Such increased susceptibility to oxidative stress may account for the role of OPC and OLG dysfunction and death in a broad range of sporadic diseases across the human life span including periventricular leukomalacia due to perinatal hypoxia, inflammation-related multiple sclerosis, ischemia-associated leukoaraiosis and AD (Takashima et al. 2009, Haider et al. 2011, Szolnoki 2007, Sjobeck & Englund 2003).
In this paper we focused on the effect of a non-lethal oxidative stress upon the dynamic changes of OLGs processes including branching and length, in mature primary rat OLGs and in the cell line OLN-93. We found that mitochondrial inhibition induced a rapid and reversible shortening of OLG processes that was prevented by antioxidants. Our findings using proteomics, kinase inhibitors, phospho-specific antibodies and transfection with CRMP-2 mutants, suggest a major role of phosphorylated CRMP-2 by ROCK as a mechanism involved in process retraction induced by ROS.
Materials and Methods
Antibodies
All the antibodies, their specificities and sources used in this study are described in Appendix S1
Reagents
The reagents used in this work are listed in Appendix S1
Cell cultures and treatments
Primary rat OLGs were isolated as described previously (McCarthy & de Vellis 1980). Newborn (P0-P2) Wistar rats were obtained from the Animal Facility of FIL. The protocol for the care, handling and use of animals followed the ARRIVE guidelines (Kilkenny et al 2010) and was approved by the Institutional Animal Care and Use Committee of FIL. Details are presented in Appendix S1. OLN-93 rat oligodendroglial cell line (Richter-Landsberg & Heinrich 1996) was cultured in Dulbeco’s Modified Eagle’s Medium (DMEM) containing 10% FCS, 50 U/ml penicillin, and 50 μg/ml streptomycin at 37°C in 10% CO2. After 48 h, cells were rinsed with PBS and subjected to treatments. Details are presented in Appendix S1.
Viability assay
The proportion of viable OLGs was measured by the uptake of neutral red as described (Richter-Landsberg & Vollgraf 1998). Details are presented in Appendix S1.
Assessment of ROS accumulation
OLGs were incubated with PBS containing 1 mM 3-NP or PBS alone for 30 min after which 100 μM of 6CDFDA was added to the culture medium and incubated at 37°C under 5% CO2 in the dark for additional 30 min. Details are presented in Appendix S1.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde, permeabilized with PBS containing 0.1% Triton X-100, and blocked in 5% normal goat serum (NGS) in PBS at room temperature. Details are presented in Appendix S1.
Morphometric analysis
OLG diameter and branching were estimated by a modification of the method described by Sholl (Sholl 1953). Details are presented in Appendix S1 and S2 Fig. S1.
2-D difference gel electrophoresis (DIGE)
Mature OLGs with at least 95% purity were treated with 1 mM 3-NP or PBS for 1 h, harvested and cell pellets containing approximately 6 × 106 cells for each condition were kept at −80°C. This procedure was repeated 8 times. Pellets were pooled and solubilized into a buffer containing 7 M urea; 2 M thiourea and 4% CHAPS detergent. The samples, each containing 100 μg of protein (determined by amino acid analysis), were differentially labeled with Cy2, Cy3 and Cy5 N-hydroxysuccinimidyl ester dyes as described previously (Wu 2006). Details about technical procedures are presented in Appendix S1.
Mass spectrometry analysis of digests
Spots of interest were subjected to robotic tryptic (Promega) digestion on a GE Healthcare Ettan TA Digester and desalted using a C-18 ZipTip. Details about technical procedure for peptides elution, loading onto MALDI target plates and search for protein candidates are presented in Appendix S1.
SDS-PAGE and Western blots
Cells were harvested in PBS and resuspended in cold deoxycholate buffer (10 mM NaH2PO4, 150 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.5% SDS) containing complete protease inhibitor cocktail, 5 mM EDTA, 5 mM EGTA, 50 mM NaF and 5 mM sodium orthovanadate. For de-phosphorylation with calf intestinal alkaline phosphatase, cocktail of proteases, chelators and kinase inhibitors were omitted. Details about homogenates preparation, determination of protein concentration, SDS-PAGE and immunoblottings are presented in Appendix S1.
Plasmids, mutagenesis and cell transfection
The plasmid pRK5 containing the cDNA of human wild type (wt) CRMP-2 with a Flag sequence at its N-terminus was kindly provided by Dr. Calum Sutherland, University of Dundee, Scotland (Bacsi et al. 2006, Cole et al. 2004). A mutant of Rho-kinase phosphorylation site replacing threonine for alanine at position 555 (T555A) CRMP-2 cDNA was generated by PCR-based directed mutagenesis. See details in Appendix S1.
Statistics
Each experiment was repeated at least three times unless otherwise stated. Significant differences between multiple groups were analyzed using one way ANOVA with Tukey’s pot-hoc test and differences between two groups were determined by Student’s t test. A p value < 0.05 was considered significant. The software Graph Pad Prism version 4.0 was used.
Results
Oxidative stress leads to a rapid shortening of OLG processes
To investigate the effect of acute oxidative stress on mature OLGs, these were exposed to 3-NP, an irreversible inhibitor of mitochondrial complex II known to induce the accumulation of superoxide (Bacsi et al. 2006). Treatment of OLG with 3-NP at 1 mM for 1 h did not result in loss of cell viability (Fig. 1A) however induces the intracellular accumulation of ROS as confirmed by staining with 6CDFDA (Fig. 1B, upper right panel). In addition, after immunofluorescence with anti-MBP (Fig. 1B, upper left panel) morphometric analysis showed that the average diameters were significant different in controls as compared to 3-NP-treated cells (81 ± 2.8 μm vs. 61.3± 4 μm; p<0.001) (Fig. 1B, right panel). To rule out a rapid redistribution of MBP from peripheral membranes, measurements were also done after immunofluorescence with anti-α tubulin (Fig. 1B, lower panel). A significant reduction of OLG diameter was seen in 3-NP treated as compared to controls (65.4 ± 3.24 vs. 56.75 ± 2.32; p< 0.05) (Fig. 1B, right panel). Moreover, α-tubulin-positive intersections measured with a concentric grid (as described in Appendix S1 and S2 Fig. S1) were significantly fewer starting at 15 μm from the center toward the periphery in 3-NP-treated as compared to controls (26.35 ± 0.45 vs. 20.8 ± 0.1; p< 0.005) (Fig. 1C). Although unlikely, it was possible that 3-NP halted process extension accounting for the observed differences. Thus, processes’ lengths were calculated in OLGs grown with a 1 h difference in regular medium in the absence of 3-NP. No significant differences were observed (not shown).
Figure 1.

A) Quantitative estimation of 3-NP toxicity at increasing concentrations assessed by the uptake of neutral red. Viability is expressed as the percentage of control (mean ± SEM). NS, non-significant, one-way ANOVA. *p<0.05 ; **p<0.001 Student’s t test as compared to control (CNT). B) Left panels, microphotographs of immunofluorescence with anti-MBP (red, upper) and anti-α-tubulin (green, lower) of rat primary oligodendrocytes (OLGs) incubated in the absence or the presence of 1 mM 3-NP for 1 h. Arrow indicates ROS accumulation in the merge of immunofluorescence with anti-MBP (red) and the fluorescence of 6CDFDA (green). Scale bar=10μm. Right panel, quantification of OLGs diameter after immunofluorescence as describe above. Bars represent the mean ± SEM of at least 25 cells for each condition. Three independent experiments were done. **p<0.001 Student’s t test as compared to control (CNT). C) Plot of the number of α-tubulin-positive processes intersections with concentric circles at increasing distance from the centre of the cell body. *p<0.05, Student’s t test of controls (○) vs 3-NP (▲) at each distance point. D) Reversibility of OLG process retraction induced by 3-NP after replacement with fresh medium (FM) without 3-NP (3-NP-FM). Effect of preincubation with E) Trolox and F) NAC in the process retraction mediated by 3-NP. Bars represent the mean ± SEM of OLG diameters. *p<0.05; **p<0.001, one-way ANOVA. Veh, PBS.
Process retraction is reversible and prevented by antioxidants
To assess reversibility, OLGs were treated for 1 h with 3-NP that was then washed out by replacement with fresh medium. After 1 h, no differences were found in the diameter between 3-NP-treated and control cells (65.9 ± 4.22 vs. 65.1 ± 2.76) (Fig. 1D). To test the effect of antioxidants, OLGs were exposed to 3-NP with or without pre-incubation with antioxidants, Trolox or NAC. A reversible retraction of processes was evident after 3-NP treatment that was fully prevented by Trolox (56 ± 2.82 vs. 71.24 ± 2.21) (Fig. 1E), and NAC (51 ± 1.82 vs. 57.51 ± 2.91) (Fig. 1F), indicating that the accumulation of ROS was necessary for the observed process of shortening upon mitochondrial inhibition. Representative microphotographs of cells in each condition are shown in Appendix S2 Fig. S2.
Proteomics of mature OLGs reveals an increase in CRMP-2 after 3-NP treatment
In an unbiased attempt to relate the dynamic changes induced by oxidative stress with changes in the pattern of protein expression, primary rat OLGs were treated with 3-NP at 1 mM for 1h and total lysates analyzed by 2D-DIGE using vehicle-treated cells as control. A representative 2D-DIGE analysis is shown in Fig. 2A. Eight spots with Cy5/Cy3 or Cy3/Cy5 ratios greater than 1.5 were identified by tryptic digestion and MS/MS (See Table I in Appendix S3). Four spots were identified as vimentin: one of these spots had a molecular mass of 53 kDa, a pI ~6 and was increased two-fold while the other 3 spots were of slightly lower molecular mass, in the ~4–5 pI range and showed a 1.7 to 1.9 reduction in 3-NP-treated samples. One spot of ~33 kDa and pI ~4.8 was identified as ribosomal protein SA (rpSA) with a 2-fold reduction after treatment. At ~67 kDa and ~pI 5.8, a spot with an increased intensity of 1.76-fold induced by 3-NP exposure was identified as CRMP-2. To confirm that these proteins with quantitative changes were expressed in mature OLGs, double immunofluorescence with anti-MBP and anti-rpSA or CRMP-2 was used. While rpSA was below levels of detection (not shown) CRMP-2 staining was strong in cell bodies and main branches and showed no co-localization with MBP, consistent with its known cytosolic location (Fig. 2B). The assessment of branching by using anti-CRMP-2 immunofluorescence yielded similar results as those found with anti-α-tubulin, consistent with process retraction after 3-NP treatment (11.2 ± 0.81 vs. 8.35 ± 0.65; NS) (Fig. 2C). To confirm the changes observed after 2-D-DIGE, the levels of CRMP-2 were assessed by Western blots of total OLG lysates normalized with 2′,3′-cyclic-nucleotide-3′ phosphodiesterase (CNP), an early marker of OLG differentiation, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a housekeeping protein (Fig. 2D and E). Since CNP interacts with tubulin heterodimers (Lee et al. 2005), we confirmed that the levels of CNP relative to GAPDH did not change after 3-NP exposure (Appendix S2 Fig. S3). An antibody against the N-terminus of CRMP-2 showed a minor band at ~60 and a major one at ~70 kDa, the latter was increased ~2-fold after treatment with 3-NP (101 ± 0.57 vs. 131.5 ± 3.65; p< 0.005) (Fig. 2D lower panel). Since the expected molecular mass for the only splicing isoform of CRMP-2 detected in OLG is 62 kDa (Bretin et al. 2005), we tested whether the upper band could be a phosphorylated isoform. Treatment of lysates with alkaline phosphatase showed a significant reduction (101 ± 0.57 vs. 68.33 ± 6; p< 0.005) in the intensity of the 70 kDa band while the 62 kDa was increased, compatible with a partial de-phosphorylation of CRMP-2 (Fig. 2Elower panel).
Figure 2.
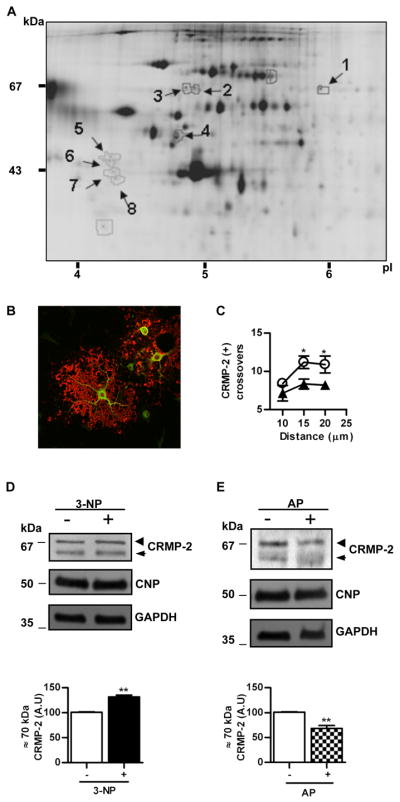
A) Representative 2D-DIGE of proteins from mature OLG homogenates after incubation with 3-NP for 1h. First dimension, isoelectric point (pI); second dimension, molecular mass (kDa). Arrows point spots with a Cy5/Cy3 fluorescent signal change higher or lower than 1.5 selected for identification by tryptic digestion and mass spectrometry. Numbers refer to those proteins identified and listed in Table I (Appendix S3). B) Merge of confocal immunofluorescence of primary OLGs with anti-MBP (red) and anti-CRMP-2 (green). Note the lack of co-localization, as expected from the cytosolic and membrane location of CRMP-2 and MBP, respectively. Scale bar=10μm. C) Plot of the number of CRMP-2 immunoreactive processes that intersect concentric circles at increasing distance from the centre of the cell body in controls (○) or after treatment with 3-NP (▲). *p<0.05, Student’s t test at each distance point. D) Upper panel, representative Western blot of OLG homogenates probed with anti-CRMP-2 in control cells (−) or after treatment with 3-NP (+). Lower panel, bars represent the mean ± SEM of the immunoreactivity of the phosphorylated CRMP-2 isoform (~70 kDa) expressed in arbitrary units (A.U.). E) Upper panel, representative Western blot probed with anti-CRMP-2 of proteins from OLGs after exposure to 3-NP and treated (+) or not (−) with alkaline phosphatase (AP) before SDS-PAGE. Lower panel, bars represent the mean ± SEM of the immunoreactivity of phosphorylated CRMP-2 isoform (~70 kDa) expressed in arbitrary units (A.U.). Membranes were probed with anti-CNP and anti-GAPDH for protein loading control. Arrowheads, phosphorylated CRMP-2 isoform.(~70 kDa); Arrows, unphosphorylated CRMP-2 isoform (~62 kDa). **p<0.001 Student’s t test.
Oxidative stress induces a reversible process retraction and increases CRMP-2 in cell line OLN-93
The cell line OLN-93 was derived from primary glial cultures of neonatal rat cerebral hemispheres. They express markers of mature primary OLGs such as galactocerebroside, MBP, proteolipid protein, myelin-associated glycoprotein and CNP (Richter-Landsberg & Heinrich 1996). Yet, morphologically, OLN-93 at low density are mainly bipolar with long cellular extensions that make testing and measuring of retraction easier and faster than with primary OLGs. Exposure to 1 mM 3-NP for 1 h had no effect upon cell viability (100.5 ± 0.5 vs. 100.05 ± 18.75), yet, OLN-93 showed a significant shortening of the main processes (16.30 ± 2.03 μm vs. 9.11 ± 0.95 μm in control and 3-NP, respectively, p< 0.001). This retraction was fully reversible after 3-NP removal and Trolox (not shown) or by treatment with NAC (Fig. 3A), reproducing the effects found in primary OLGs (Fig. 1D, E and F). Representative microphotographs of treated cells are shown in Appendix S2 Fig. S4. Moreover, as determined by Western blot of total cell lysates (Fig. 3B, left panel) there was a 1.5-fold increase in the levels of a single ~70 kDa CRMP-2 band after exposure to 3-NP (Fig. 3B, right panel). Therefore, the dynamic response of process retraction after oxidative stress is shared by primary mature OLGs and OLN-93 cell line and the increase in CRMP-2 induced by 3-NP suggests that these similarities may extend to basic molecular mechanisms.
Figure 3.

A) Quantitative assessment of the preventive effect of NAC upon process shortening induced by 3-NP. Bars represent the mean ± SEM of process length for each condition. Veh, PBS. B) Left panel, Western blot probed with anti-CRMP-2 of homogenates from OLN-93 cells incubated with PBS in the absence (−) or presence (+) of 3-NP. Anti-CNP in the same membrane was used as a control of protein loading. Right panel, bars represent the mean ± SEM of the immunoreactivity of CRMP-2 expressed in arbitrary units (A.U.). C) Assessment of the lack of effect of roscovitine (Rosc) upon the process retraction induced by 3-NP in primary OLGs. Bars represent the mean ± SEM of OLG diameter in each condition. Vehicle (Veh), DMSO. D) Quantitative estimation of the effect of the ROCK inhibitor, Y27632, upon the shortening of process of primary OLGs elicited by 3-NP. Bars represent the mean ± SEM of OLG diameter. Note the complete prevention of process shortening by Y27632. Vehicle (Veh), PBS. E) Prevention of 3-NP-induced process retraction in OLN-93 cells by Y27632. Bars represent the mean ± SEM of process length. **p<0.001, one-way ANOVA.
3-NP treatment does not modify Netrin-1-Dcc signaling
Netrin-1-Dcc signaling promotes OLG process extension and branching through Fyn phosphorylation and inactivation of RhoA. (Rajasekharan et al. 2009, Rajasekharan et al. 2010). In addition, mitochondrial redox state may modulate PKA activity, which in turn regulates the recruitment of Dcc to the cell surface (Ryu et al. 2005, Bouchard et al. 2004). To test if the effect of 3-NP treatment could be mediated by down-regulation of this pathway, OLN-93 cells treated with 3-NP or vehicle were analyzed by quantitative immunocytochemistry of the Dcc extracellular domain. No significant differences in the levels of cell surface Dcc were observed between control and 3-NP treated cells (Appendix S2 Fig. S5A). Moreover, Western blots showed that the levels of p-Fyn at Tyr416, a key down-stream effector of Dcc did not change in response to 3-NP exposure (Appendix S2 Fig. S5B).
The effect of kinase inhibitors on OLGs process retraction induced by ROS
Roscovitine (an inhibitor of cdc2, cdk2 and cdk5) and Y27632 (an inhibitor of ROCK) were used in the absence or presence of 3-NP. No effect was seen with roscovitine (Fig. 3C) yet, a complete prevention of process retraction after 3-NP exposure was found after pre-incubation with Y27632, suggesting a role for ROCK activation in response to oxidative stress in OLGs (Fig. 3D). Likewise, OLN-93 cells showed that inhibition with Y27632 totally prevented process retraction (Fig. 3E). Representative microphotographs of treated OLN-93 cells are shown in Appendix S2 Fig. S4C.
CRMP-2 phosphorylation at ROCK site in response to ROS
To further assess CRMP-2 phosphorylation induced by 3-NP treatment, OLGs or OLN-93 total cell lysates were analyzed by Western blot with antibodies that recognize specifically CRMP-2 T555 phospho-epitope (ROCK site) and normalized by CNP. In primary OLGs, despite an increase of a ~70 kDa CRMP-2 isoform that was likely phosphorylated, as described above, the signals obtained with anti-pT555 were very low, even after loading 100 μg of protein (not shown). Yet, a mouse brain lysate used as positive control showed a very strong labeling at the same amount of protein loading (not shown). Considering the similar behavior of OLN-93 in terms of process shortening induced by 3-NP, its reversibility, and its prevention by antioxidants and the ROCK inhibitor Y27632, these cells were tested by Western blots. A single major CRMP-2 positive band of ~70 kDa showed a ~1.5 fold increase in 3-NP treated cells as compared to control (Fig. 4A, upper left panel). When probed with anti-CRMP-2 specific for phosphorylated T555 (Fig. 4A, lower left panel), a significant 40% increase was found (Fig. 4A, right panel). As expected from previous studies, phosphorylation at T555 did not induce a shift in electrophoretic mobility (Patrakitkomjorn et al. 2008), precluding the normalization with anti-total CRMP-2 to estimate the phosphorylation ratio. The increase in the CRMP-2 T555 phospho-isoform induced by 3-NP was fully prevented in the presence of Y27632, further confirming the involvement of ROCK in this response.
Figure 4.
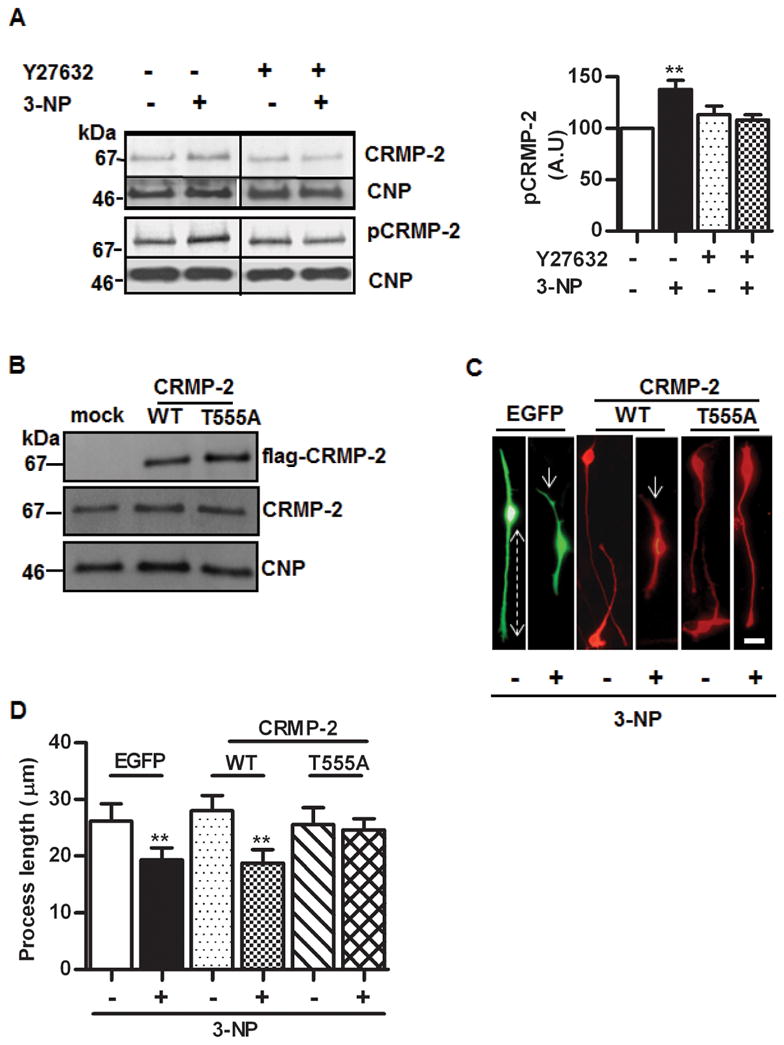
A) Left upper panel, representative Western blots of OLN-93 proteins with anti-CRMP-2 showing the effect of 3-NP and its prevention by ROCK inhibitor Y27632. Lower panel, Western blots with anti-CRMP-2 specific for the phosphorylated epitope T555 (the ROCK site). In both cases, membranes were probed with anti-CNP for control of protein loading. Right panel, quantitative assessment of the increase of CRMP-2 phosphorylated at T555 induced by 3-NP and its prevention by Y27632. Bars represent the mean ± SEM of immunoreactivity, in arbitrary units (A.U.), expressed as percentage of control. B) Western blot of OLN-93 cells transiently transfected with empty vector (mock), wild type CRMP-2 (WT) or the mutant CRMP-2 T555A (T555A). Anti-flag was used to detect transfected proteins (upper panel); anti-CRMP2 was used to assess levels of total CRMP-2 (middle panel). In both experiments, the same membranes were probed with anti-CNP (lower panel) for protein loading control. C) Representative microphotographs showing OLN-93 cells transfected with EGFP, or with wild type CRMP-2 (WT) or CRMP-2 T555A mutant (T555A), treated (+) or not (−) with 3-NP and probed with anti-flag. Dotted line shows the distance from the centre of the cell to the end of the process, used to quantify the retraction. Arrows depict process shortening induced by 3-NP. Note the prevention of the effect in T555A cells. Scale bar=10μm. D) Quantitative assessment of the process length in EGFP, CRMP-2 WT or CRMP-2 T555A transfected cells in the presence (+) or absence (−) of 3-NP. Bars represent the mean ± SEM for each experimental situation. **p<0.001, one-way ANOVA. RE: JNC-E-2011-1028.R1
Expression of the mutant CRMP-2 T555A prevents ROS-dependent process retraction
Based on our results using kinase inhibitors and phospho-specific Western blots, phosphorylation at T555 by ROCK was a major candidate to mediate the OLG process retraction induced by ROS. Therefore, OLN-93 cells were transiently transfected with flag-tagged wild type CRMP-2 (wt) or CRMP-2 T555A and analyzed by Western blot (Fig. 4B) and immunofluorescence (Fig. 4C). To include an additional control, OLN-93 cells were transfected with EGFP as an unrelated, cytosolic protein. Remarkably, the levels of total CRMP-2 normalized by CNP, were similar between non-transfected and transfected cells, indicating that the over-expression of total CRMP-2 was compensated within the 48 h period after transfection. In mock-transfected cells and in cells transfected with EGFP or wt CRMP-2, 3-NP treatment induced a significant process retraction as compared to untreated cells. In contrast, transfection with the CRMP-2 T555A mutant (resistant to phosphorylation by ROCK) fully prevented the effect of 3-NP (Fig. 4D).
Discussion
Our results indicate that in rat primary OLGs and in the rat cell line OLN-93, sub-lethal oxidative stress induced by mitochondrial inhibition results in a rapid and reversible retraction of processes. The complete prevention of this effect by two different antioxidants indicates that accumulation of ROS is necessary for such retraction to take place, ruling out a major role of other acute effects that may be induced by 3-NP such as the reduction of ATP intracellular levels (Liot et al. 2009). Proteomics of the primary OLGs in the presence or absence of 3-NP yielded a few differential proteins including vimentin, rpSA and CRMP-2. The differential spots included fragments of vimentin without a definite quantitative tendency and rpSA, which was undetectable by immunocytochemistry, precluding its further study. CRMP-2 was increased ~1.8 fold, clearly detected in the cytoplasm of mature OLGs and its labeling reflected the same tendency to peripheral retraction as shown with MBP and α-tubulin. Western blots of OLGs homogenates showed an increase of a likely phosphorylated isoform of ~70 kDa thus confirming the 2D-DIGE results. In OLN-93, a single band of 70 kDa had a ~2-fold increase as compared to control. Thus, in both OLGs and OLN-93 cells, CRMP-2 seemed to increase its phosphorylation level after 3-NP exposure. The pyridine-derived smooth muscle relaxant Y27632 is a well known ROCK inhibitor although it can also inhibit other kinases including LRRK2, PRK2 and MNK1 (Davies et al. 2000, Nichols et al. 2009). Of these, only ROCK is known to phosphorylate CRMP-2. CaMK II and PKA have been recently proposed to phosphorylate CRMP-2 in neurons. Yet Y27632 does not inhibit these kinases (Hou et al. 2009, Boudreau et al. 2009). This point is of interest because CaMK II phosphorylates CRMP-2 at T555, just like ROCK (Hou et al. 2009). In addition, Fyn-dependent phosphorylation of CRMP-2 at Tyr32 is involved in Sema3A signaling in neurons and phosphorylation at Y479 by the Src-family kinase Yes mediates T-cell polarization induced by chemokines (Uchida et al. 2009, Varrin-Doyer et al. 2009). The lack of Fyn phosphorylation at Tyr 416 after 3-NP exposure in OLN-93 cells suggests that involvement of netrin-1-Dcc pathway in process shortening was very unlikely. Process retraction elicited by 3-NP was completely inhibited by Y27632 in primary OLGs and OLN-93 cells. In addition, the increase of CRMP-2 phosphorylated at T555 by 3-NP was completely prevented by Y27632 in OLN-93. The functional significance of these correlations was tested by transfecting OLN-93 cells with a mutant CRMP-2, T555A, which cannot be phosphorylated by ROCK. Two observations indicated that CRMP-2 T555A may function as a “dominant negative” for the tubulin-unbound and microtubule disassembly-promoting isoform of CRMP-2. Firstly, it is known that CRMPs assemble into homo-and hetero-tetramers (Wang & Strittmatter 1997, Stenmark et al. 2007, Majava et al. 2008), secondly, the total levels of CRMP-2 after 48 h of transfection in our experiments were similar to the untransfected cells, while the anti-flag Western blot and immunofluorescence labeling showed high levels of transfection efficiency. This would imply a high stoichiometry of mutant CRMP-2 over endogenous CRMP-2 and a higher occupancy at the microtubule of less phosphorylated tetrameric forms. This is a likely explanation for the inability of 3-NP to induce process retraction in OLN-93 expressing CRMP-2 T555A. A large body of evidence has involved CRMP-2 phosphorylation in the cytoskeletal dynamic changes of axonal growth cones and neurites regulated by external stimuli (Petratos et al. 2008). In OPCs in culture, S1P can activate Edg8/S1P5 receptor and induce process retraction through a ROCK pathway that involves CRMP-2 phosphorylation (Jaillard et al. 2005). In mature rat OLGs, exposure to sema3A is capable of inducing a rather slow (24 h) and reversible process retraction mediated by neuropilin-1 that is prevented with incubation with anti-CRMP-2 (Ricard et al. 2001). Apart from these two pathways, little is known about CRMP-2 phosphorylation and its impact on process extension in oligodendroglia. Here we propose that accumulation of ROS in viable, mature OLGs may modulate CRMP-2 phosphorylation and process growth by inducing ROCK activation in a fast and reversible way. Two major features suggest that 3-NP is not acting through activation of Edg8 or neuropilin-1. Firstly, the activation of Edg8 by S1P induces retraction only in OPCs and not in mature OLGs (Jaillard et al. 2005), secondly, the effect upon neuropilin-1 stimulation with sema3A in mature OLGs is very slow as compared to what we found with 3-NP (1h vs 24 h for retraction and 1h vs 72 h for reversibility). The major kinase of sema3A-neuropilin pathway that phosphorylates CRMP-2 in neurons seems to be CDK5 rather than ROCK (Uchida et al 2005). Yet, since very little is known in OLG and despite the lack of effect of roscovitine in our study, a possible contribution of CRMP-2 phosphorylation by CDK5-GSK3β in OLG process retraction deserves further exploration. The mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway promotes OLG process elongation and branching (Younes-Rapozo et al. 2009). Although oxidative stress is known to stimulate this pathway leading to apoptosis (Fragoso et al. 2004), we cannot rule out a rapid and transient inhibition of MAPK/ERK contributing to process shortening in our experiments. ROS are known to regulate the activity of GTPases including the Rho family members RhoA, Rac1, and Cdc42 which contain a distinct redox-active motif located in their phosphoryl-binding loop (Heo & Campbell 2005). RhoA can be activated by ROS in rat aorta, alveolar epithelial cells and in fibroblast cell lines (Jin et al. 2004, Dada et al. 2007). Such RhoA activation seems to be mediated by two highly conserved, redox sensitive cysteines at positions 16 and 20 (Aghajanian et al. 2009). Very recently, a role for hydrogen peroxide has been proposed for a thioredoxin-mediated phosphoprylation of CRMP-2 by GSK3 in axonal growth cone (Morinaka et al. 2011). In a pathological context, CRMP-2 is overexpressed in the rat adult brain in response to focal ischemia (Chen et al. 2007) and phospho-CRMP-2 increases in axons near the inflammatory lesions of multiple sclerosis patients (Menon et al. 2011). In AD, CRMP-2 localizes with neurofibrillary tangles in human cortex and its accumulation is considered an early event in the pathogenesis of the disease. Interestingly, CRMP-2 is hyperphosphorylated in brain tissue from human AD subjects and in some mouse models of AD (Gu et al. 2000, Takata et al. 2009, Cole et al. 2007). CRMP-2 is also an oxidatively modified protein in the AD brain (Castegna et al. 2002, Di Domenico et al. 2011). Most of the studies in AD have been focused on neuronal CRMP-2. The vulnerability of late myelinated neurons in the brain (Braak & Braak 1996, Morrison et al. 1998), the age-associated oxidative stress and the presence of white matter pathology in many AD cases (Roher et al. 2002, Roher et al. 2003) together with our present results warrant further studies of the role of CRMP-2 phosphorylation in OLGs and OPCs in disease pathogenesis. The recent description of defects in myelination preceding the appearance of amyloid and tau pathology in triple-transgenic AD (3xTg-AD) mice may provide a model to study the CRMP-2 phosphorylation pathway in OLGs (Desai et al. 2009). In summary, our results suggest a role for endogenous ROS signaling in the regulation of OLG process dynamics that may be deranged in early stages of diseases with rampant oxidative stress in the CNS, including AD.
Supplementary Material
Acknowledgments
This work was supported by the John Simon Guggenheim Memorial Foundation (EMC), a CONICET grant PIP693 (LM) and by the National Institute on Aging (NIA) grant RO1AG619795 (AER). The NIA had no role in the study, design, data collection and analysis, decision to publish or preparation of the manuscript. Our special thanks to Dr. Calum Sutherland, University of Dundee, for the cDNA of human wt CRMP-2 and to Prof. Juana Pasquini, University of Buenos Aires, for anti-Fyn antibodies. The authors declare that they have no conflict of interests. Authorship credit::EMC, LM, AER: Conceived and designed the experiments;: AFG, MCL, CLM: Performed the experiments;: AFG, MCL, EMC, TW, CRL, AER: Analyzed the data; AFG, LM, AER, CLM, TW, EMC: Wrote the paper:
Abbreviations
- ACN
acetonitrile
- AD
Alzheimer’s disease
- AP
alkaline phosphatase
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- 6CDFDA
2,7-dichlorodihydrofluorescein diacetate
- Cdc2
cell division control protein 2 homolog
- Cdc42
cell division control protein 42 homolog
- CDK2
cyclin dependent kinase 2
- CDK5
cyclin dependent kinase 5
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- CNP
-2′,3′-cyclic nucleotide 3′-phosphodiesterase
- CNT
control
- CRMP-2
collapsin response mediator protein-2
- DAPI
4′, 6′ diamidine-2′ phenylindole dihydrochloride
- Dcc
deleted in colorectal cancer
- 2D-DIGE
bidimensional difference gel electrophoresis
- DMEM
Dulbeco’s Modified Eagle’s Medium
- Edg8
endothelial differentiation gene 8
- EGFP
enhanced green fluorescent protein
- ERK
extracellular-signal-regulated kinase
- FCS
fetal calf serum
- Fyn
tyrosine protein kinase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFAP
glial fibrillary acidic protein
- GSK3β
glycogen synthase kinase 3β
- IPG
immobilized pH gradient
- LPA
lysophosphatidic acid
- LRRK2
Leucine Rich Repeat Protein Kinase-2
- MAPK
Mitogen-activated protein kinase
- MBP
myelin basic protein
- MNK1
MAP kinase-interacting serine/threonine-protein kinase 1
- MS/MS
tandem mass spectrometry
- NAC
N-acetyl-L-cysteine
- 3-NP
3-nitropropionic acid
- N-WASP
neuronal Wiskott–Aldrich Syndrome protein
- OLGs
oligodendrocytes
- OLN-93
oligodendroglial cell line
- OPCs
oligodendrocyte precursor cells
- PBS
phosphate buffered saline
- PKA
protein kinase A
- PRK2
protein kinase C-related kinase
- Rac1
ras-related C3 botulinum toxin substrate 1
- RhoA
ras homolog gene family, member A
- ROCK
rho kinase
- rpSA
ribosomal protein SA
- ROS
reactive oxygen species
- ROSC
roscovitine
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- Sema3A
semaphorin3A
- S1P
sphingosine-1-phosphate
- TFA
trifluoroacetic acid
- VAMP2
vesicle-associated membrane protein 2
- Veh
vehicle
- WT
wild type
References
- Aghajanian A, Wittchen ES, Campbell SL, Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One. 2009;4:e8045. doi: 10.1371/journal.pone.0008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura N, Inagaki N, Chihara K, Menager C, Nakamura N, Amano M, Iwamatsu A, Goshima Y, Kaibuchi K. Phosphorylation of collapsin response mediator protein-2 by Rho-kinase. Evidence for two separate signaling pathways for growth cone collapse. J Biol Chem. 2000;275:23973–23980. doi: 10.1074/jbc.M001032200. [DOI] [PubMed] [Google Scholar]
- Arimura N, Menager C, Kawano Y, et al. Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol Cell Biol. 2005;25:9973–9984. doi: 10.1128/MCB.25.22.9973-9984.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon C, Lakics V, Machesky L, Rumsby M. N-WASP regulates extension of filopodia and processes by oligodendrocyte progenitors, oligodendrocytes, and Schwann cells-implications for axon ensheathment at myelination. Glia. 2007;55:844–858. doi: 10.1002/glia.20505. [DOI] [PubMed] [Google Scholar]
- Bacsi A, Woodberry M, Widger W, Papaconstantinou J, Mitra S, Peterson JW, Boldogh I. Localization of superoxide anion production to mitochondrial electron transport chain in 3-NPA-treated cells. Mitochondrion. 2006;6:235–244. doi: 10.1016/j.mito.2006.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartzokis G, Sultzer D, Lu PH, Nuechterlein KH, Mintz J, Cummings JL. Heterogeneous age-related breakdown of white matter structural integrity: implications for cortical “disconnection” in aging and Alzheimer’s disease. Neurobiol Aging. 2004;25:843–851. doi: 10.1016/j.neurobiolaging.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Benes FM, Turtle M, Khan Y, Farol P. Myelination of a key relay zone in the hippocampal formation occurs in the human brain during childhood, adolescence, and adulthood. Arch Gen Psychiatry. 1994;51:477–484. doi: 10.1001/archpsyc.1994.03950060041004. [DOI] [PubMed] [Google Scholar]
- Bouchard JF, Moore SW, Tritsch NX, Roux PP, Shekarabi M, Barker PA, Kennedy TE. Protein kinase A activation promotes plasma membrane insertion of DCC from an intracellular pool: A novel mechanism regulating commissural axon extension. J Neurosci. 2004;24:3040–3050. doi: 10.1523/JNEUROSCI.4934-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Ferrario CR, Glucksman MJ, Wolf ME. Signaling pathway adaptations and novel protein kinase A substrates related to behavioral sensitization to cocaine. J Neurochem. 2009;110:363–377. doi: 10.1111/j.1471-4159.2009.06140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol. 1996;92:197–201. doi: 10.1007/s004010050508. [DOI] [PubMed] [Google Scholar]
- Bretin S, Reibel S, Charrier E, et al. Differential expression of CRMP1, CRMP2A, CRMP2B, and CRMP5 in axons or dendrites of distinct neurons in the mouse brain. J Comp Neurol. 2005;486:1–17. doi: 10.1002/cne.20465. [DOI] [PubMed] [Google Scholar]
- Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem. 2002;82:1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- Cole AR, Causeret F, Yadirgi G, et al. Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. J Biol Chem. 2006;281:16591–16598. doi: 10.1074/jbc.M513344200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN, Sutherland C. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J Biol Chem. 2004;279:50176–50180. doi: 10.1074/jbc.C400412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole AR, Noble W, van Aalten L, et al. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J Neurochem. 2007;103:1132–1144. doi: 10.1111/j.1471-4159.2007.04829.x. [DOI] [PubMed] [Google Scholar]
- Connor JR, Menzies SL. Cellular management of iron in the brain. J Neurol Sci. 1995;134(Suppl):33–44. doi: 10.1016/0022-510x(95)00206-h. [DOI] [PubMed] [Google Scholar]
- Chae YC, Lee S, Heo K, Ha SH, Jung Y, Kim JH, Ihara Y, Suh PG, Ryu SH. Collapsin response mediator protein-2 regulates neurite formation by modulating tubulin GTPase activity. Cell Signal. 2009;21:1818–1826. doi: 10.1016/j.cellsig.2009.07.017. [DOI] [PubMed] [Google Scholar]
- Chen A, Liao WP, Lu Q, Wong WS, Wong PT. Upregulation of dihydropyrimidinase-related protein 2, spectrin alpha II chain, heat shock cognate protein 70 pseudogene 1 and tropomodulin 2 after focal cerebral ischemia in rats--a proteomics approach. Neurochem Int. 2007;50:1078–1086. doi: 10.1016/j.neuint.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Dada LA, Novoa E, Lecuona E, Sun H, Sznajder JI. Role of the small GTPase RhoA in the hypoxia-induced decrease of plasma membrane Na, K-ATPase in A549 cells. J Cell Sci. 2007;120:2214–2222. doi: 10.1242/jcs.003038. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson J, Hotchin N, Lax S, Rumsby M. Lysophosphatidic acid induces process retraction in CG-4 line oligodendrocytes and oligodendrocyte precursor cells but not in differentiated oligodendrocytes. J Neurochem. 2003a;87:947–957. doi: 10.1046/j.1471-4159.2003.02056.x. [DOI] [PubMed] [Google Scholar]
- Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003b;24:476–488. doi: 10.1016/s1044-7431(03)00210-0. [DOI] [PubMed] [Google Scholar]
- Desai MK, Sudol KL, Janelsins MC, Mastrangelo MA, Frazer ME, Bowers WJ. Triple-transgenic Alzheimer’s disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia. 2009;57:54–65. doi: 10.1002/glia.20734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico F, Sultana R, Barone E, Perluigi M, Cini C, Mancuso C, Cai J, Pierce WM, Butterfield DA. Quantitative proteomics analysis of phosphorylated proteins in the hippocampus of Alzheimer’s disease subjects. J Proteomics. 2011;74:1091–1103. doi: 10.1016/j.jprot.2011.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragoso G, Martinez-Bermudez AK, Liu HN, Khorchid A, Chemtob S, Mushynski WE, Almazan G. Developmental differences in HO-induced oligodendrocyte cell death: role of glutathione, mitogen-activated protein kinases and caspase 3. J Neurochem. 2004;90:392–404. doi: 10.1111/j.1471-4159.2004.02488.x. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Itoh TJ, Kimura T, et al. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol. 2002;4:583–591. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- Gu Y, Hamajima N, Ihara Y. Neurofibrillary tangle-associated collapsin response mediator protein-2 (CRMP-2) is highly phosphorylated on Thr-509, Ser-518, and Ser-522. Biochemistry. 2000;39:4267–4275. doi: 10.1021/bi992323h. [DOI] [PubMed] [Google Scholar]
- Gu Y, Ihara Y. Evidence that collapsin response mediator protein-2 is involved in the dynamics of microtubules. J Biol Chem. 2000;275:17917–17920. doi: 10.1074/jbc.C000179200. [DOI] [PubMed] [Google Scholar]
- Haider L, Fischer MT, Frischer JM, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134:1914–1924. doi: 10.1093/brain/awr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J, Campbell SL. Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J Biol Chem. 2005;280:31003–31010. doi: 10.1074/jbc.M504768200. [DOI] [PubMed] [Google Scholar]
- Hollensworth SB, Shen C, Sim JE, Spitz DR, Wilson GL, LeDoux SP. Glial cell type-specific responses to menadione-induced oxidative stress. Free Radic Biol Med. 2000;28:1161–1174. doi: 10.1016/s0891-5849(00)00214-8. [DOI] [PubMed] [Google Scholar]
- Hou ST, Jiang SX, Aylsworth A, Ferguson G, Slinn J, Hu H, Leung T, Kappler J, Kaibuchi K. CaMKII phosphorylates collapsin response mediator protein 2 and modulates axonal damage during glutamate excitotoxicity. J Neurochem. 2009;111:870–881. doi: 10.1111/j.1471-4159.2009.06375.x. [DOI] [PubMed] [Google Scholar]
- Jaillard C, Harrison S, Stankoff B, et al. Edg8/S1P5: an oligodendroglial receptor with dual function on process retraction and cell survival. J Neurosci. 2005;25:1459–1469. doi: 10.1523/JNEUROSCI.4645-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakovcevski I, Mo Z, Zecevic N. Down-regulation of the axonal polysialic acid-neural cell adhesion molecule expression coincides with the onset of myelination in the human fetal forebrain. Neuroscience. 2007;149:328–337. doi: 10.1016/j.neuroscience.2007.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana A, Pahan K. Oxidative stress kills human primary oligodendrocytes via neutral sphingomyelinase: implications for multiple sclerosis. J Neuroimmune Pharmacol. 2007;2:184–193. doi: 10.1007/s11481-007-9066-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Ying Z, Webb RC. Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am J Physiol Heart Circ Physiol. 2004;287:H1495–1500. doi: 10.1152/ajpheart.01006.2003. [DOI] [PubMed] [Google Scholar]
- Juurlink BH, Thorburne SK, Hertz L. Peroxide-scavenging deficit underlies oligodendrocyte susceptibility to oxidative stress. Glia. 1998;22:371–378. doi: 10.1002/(sici)1098-1136(199804)22:4<371::aid-glia6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: The ARRIVE Guidelines for Reporting Animal Research. PLoS Biol. 8(6):e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Gravel M, Zhang R, Thibault P, Braun PE. Process outgrowth in oligodendrocytes is mediated by CNP, a novel microtubule assembly myelin protein. J Cell Biol. 2005;170:661–673. doi: 10.1083/jcb.200411047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT, Xu J, Lee JM, Ku G, Han X, Yang DI, Chen S, Hsu CY. Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol. 2004;164:123–131. doi: 10.1083/jcb.200307017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liot G, Bossy B, Lubitz S, Kushnareva Y, Sejbuk N, Bossy-Wetzel E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA-and ROS-dependent pathway. Cell Death Differ. 2009;16:899–909. doi: 10.1038/cdd.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majava V, Loytynoja N, Chen WQ, Lubec G, Kursula P. Crystal and solution structure, stability and post-translational modifications of collapsin response mediator protein 2. FEBS J. 2008;275:4583–4596. doi: 10.1111/j.1742-4658.2008.06601.x. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon KN, Steer DL, Short M, Petratos S, Smith I, Bernard CC. A novel unbiased proteomic approach to detect the reactivity of cerebrospinal fluid in neurological diseases. Mol Cell Proteomics. 2011;10:M110 000042. doi: 10.1074/mcp.M110.000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinaka A, Yamada M, Itofusa R, et al. Thioredoxin mediates oxidation-dependent phosphorylation of CRMP2 and growth cone collapse. Sci Signal. 2011;4:ra26. doi: 10.1126/scisignal.2001127. [DOI] [PubMed] [Google Scholar]
- Morrison BM, Hof PR, Morrison JH. Determinants of neuronal vulnerability in neurodegenerative diseases. Ann Neurol. 1998;44:S32–44. doi: 10.1002/ana.410440706. [DOI] [PubMed] [Google Scholar]
- Mronga T, Stahnke T, Goldbaum O, Richter-Landsberg C. Mitochondrial pathway is involved in hydrogen-peroxide-induced apoptotic cell death of oligodendrocytes. Glia. 2004;46:446–455. doi: 10.1002/glia.20022. [DOI] [PubMed] [Google Scholar]
- Nichols RJ, Dzamko N, Hutti JE, Cantley LC, Deak M, Moran J, Bamborough P, Reith AD, Alessi DR. Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson’s disease. Biochem J. 2009;424:47–60. doi: 10.1042/BJ20091035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novgorodov AS, El-Alwani M, Bielawski J, Obeid LM, Gudz TI. Activation of sphingosine-1-phosphate receptor S1P5 inhibits oligodendrocyte progenitor migration. FASEB J. 2007;21:1503–1514. doi: 10.1096/fj.06-7420com. [DOI] [PubMed] [Google Scholar]
- Okada A, Tominaga M, Horiuchi M, Tomooka Y. Plexin-A4 is expressed in oligodendrocyte precursor cells and acts as a mediator of semaphorin signals. Biochem Biophys Res Commun. 2007;352:158–163. doi: 10.1016/j.bbrc.2006.10.176. [DOI] [PubMed] [Google Scholar]
- Patrakitkomjorn S, Kobayashi D, Morikawa T, et al. Neurofibromatosis type 1 (NF1) tumor suppressor, neurofibromin, regulates the neuronal differentiation of PC12 cells via its associating protein, CRMP-2. J Biol Chem. 2008;283:9399–9413. doi: 10.1074/jbc.M708206200. [DOI] [PubMed] [Google Scholar]
- Perez-Campo R, Lopez-Torres M, Cadenas S, Rojas C, Barja G. The rate of free radical production as a determinant of the rate of aging: evidence from the comparative approach. J Comp Physiol B. 1998;168:149–158. doi: 10.1007/s003600050131. [DOI] [PubMed] [Google Scholar]
- Petratos S, Li QX, George AJ, et al. The beta-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain. 2008;131:90–108. doi: 10.1093/brain/awm260. [DOI] [PubMed] [Google Scholar]
- Rajasekharan S, Baker KA, Horn KE, Jarjour AA, Antel JP, Kennedy TE. Netrin 1 and Dcc regulate oligodendrocyte process branching and membrane extension via Fyn and RhoA. Development. 2009;136:415–426. doi: 10.1242/dev.018234. [DOI] [PubMed] [Google Scholar]
- Rajasekharan S, Bin JM, Antel JP, Kennedy TE. A central role for RhoA during oligodendroglial maturation in the switch from netrin-1-mediated chemorepulsion to process elaboration. J Neurochem. 2010;113:1589–1597. doi: 10.1111/j.1471-4159.2010.06717.x. [DOI] [PubMed] [Google Scholar]
- Ricard D, Rogemond V, Charrier E, Aguera M, Bagnard D, Belin MF, Thomasset N, Honnorat J. Isolation and expression pattern of human Unc-33-like phosphoprotein 6/collapsin response mediator protein 5 (Ulip6/CRMP5): coexistence with Ulip2/CRMP2 in Sema3a-sensitive oligodendrocytes. J Neurosci. 2001;21:7203–7214. doi: 10.1523/JNEUROSCI.21-18-07203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter-Landsberg C, Heinrich M. OLN-93: a new permanent oligodendroglia cell line derived from primary rat brain glial cultures. J Neurosci Res. 1996;45:161–173. doi: 10.1002/(SICI)1097-4547(19960715)45:2<161::AID-JNR8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Richter-Landsberg C, Vollgraf U. Mode of cell injury and death after hydrogen peroxide exposure in cultured oligodendroglia cells. Exp Cell Res. 1998;244:218–229. doi: 10.1006/excr.1998.4188. [DOI] [PubMed] [Google Scholar]
- Roher AE, Kuo YM, Esh C, et al. Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer’s disease. Mol Med. 2003;9:112–122. [PMC free article] [PubMed] [Google Scholar]
- Roher AE, Weiss N, Kokjohn TA, et al. Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry. 2002;41:11080–11090. doi: 10.1021/bi026173d. [DOI] [PubMed] [Google Scholar]
- Ryu H, Lee J, Impey S, Ratan RR, Ferrante RJ. Antioxidants modulate mitochondrial PKA and increase CREB binding to D-loop DNA of the mitochondrial genome in neurons. Proc Natl Acad Sci U S A. 2005;102:13915–13920. doi: 10.1073/pnas.0502878102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre J, Pallardo FV, Garcia de la Asuncion J, Vina J. Mitochondria, oxidative stress and aging. Free Radic Res. 2000;32:189–198. doi: 10.1080/10715760000300201. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Sjobeck M, Englund E. Glial levels determine severity of white matter disease in Alzheimer’s disease: a neuropathological study of glial changes. Neuropathol Appl Neurobiol. 2003;29:159–169. doi: 10.1046/j.1365-2990.2003.00456.x. [DOI] [PubMed] [Google Scholar]
- Sloane JA, Vartanian TK. Myosin Va controls oligodendrocyte morphogenesis and myelination. J Neurosci. 2007;27:11366–11375. doi: 10.1523/JNEUROSCI.2326-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, Kapoor R, Felts PA. Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol. 1999;9:69–92. doi: 10.1111/j.1750-3639.1999.tb00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassky N, de Castro F, Le Bras B, Heydon K, Queraud-LeSaux F, Bloch-Gallego E, Chedotal A, Zalc B, Thomas JL. Directional guidance of oligodendroglial migration by class 3 semaphorins and netrin-1. J Neurosci. 2002;22:5992–6004. doi: 10.1523/JNEUROSCI.22-14-05992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark P, Ogg D, Flodin S, Flores A, Kotenyova T, Nyman T, Nordlund P, Kursula P. The structure of human collapsin response mediator protein 2, a regulator of axonal growth. J Neurochem. 2007;101:906–917. doi: 10.1111/j.1471-4159.2006.04401.x. [DOI] [PubMed] [Google Scholar]
- Szolnoki Z. Pathomechanism of leukoaraiosis: a molecular bridge between the genetic, biochemical, and clinical processes (a mitochondrial hypothesis) Neuromolecular Med. 2007;9:21–33. doi: 10.1385/nmm:9:1:21. [DOI] [PubMed] [Google Scholar]
- Takashima S, Itoh M, Oka A. A history of our understanding of cerebral vascular development and pathogenesis of perinatal brain damage over the past 30 years. Semin Pediatr Neurol. 2009;16:226–236. doi: 10.1016/j.spen.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Takata K, Kitamura Y, Nakata Y, Matsuoka Y, Tomimoto H, Taniguchi T, Shimohama S. Involvement of WAVE accumulation in Abeta/APP pathology-dependent tangle modification in Alzheimer’s disease. Am J Pathol. 2009;175:17–24. doi: 10.2353/ajpath.2009.080908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida Y, Ohshima T, Sasaki Y, et al. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells. 2005;10:165–179. doi: 10.1111/j.1365-2443.2005.00827.x. [DOI] [PubMed] [Google Scholar]
- Uchida Y, Ohshima T, Yamashita N, Ogawara M, Sasaki Y, Nakamura F, Goshima Y. Semaphorin3A signaling mediated by Fyn-dependent tyrosine phosphorylation of collapsin response mediator protein 2 at tyrosine 32. J Biol Chem. 2009;284:27393–27401. doi: 10.1074/jbc.M109.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulfig N, Nickel J, Saretzki U. Alterations in myelin formation in fetal brains of twins. Pediatr Neurol. 1998;19:287–293. doi: 10.1016/s0887-8994(98)00068-x. [DOI] [PubMed] [Google Scholar]
- Varrin-Doyer M, Vincent P, Cavagna S, Auvergnon N, Noraz N, Rogemond V, Honnorat J, Moradi-Ameli M, Giraudon P. Phosphorylation of collapsin response mediator protein 2 on Tyr-479 regulates CXCL12-induced T lymphocyte migration. J Biol Chem. 2009;284:13265–13276. doi: 10.1074/jbc.M807664200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Tewari A, Einheber S, Salzer JL, Melendez-Vasquez CV. Myosin II has distinct functions in PNS and CNS myelin sheath formation. J Cell Biol. 2008;182:1171–1184. doi: 10.1083/jcb.200802091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LH, Strittmatter SM. Brain CRMP forms heterotetramers similar to liver dihydropyrimidinase. J Neurochem. 1997;69:2261–2269. doi: 10.1046/j.1471-4159.1997.69062261.x. [DOI] [PubMed] [Google Scholar]
- Wu TL. Two-dimensional difference gel electrophoresis. Methods Mol Biol. 2006;328:71–95. doi: 10.1385/1-59745-026-X:71. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Younes-Rapozo V, Felgueiras LO, Viana NL, Fierro IM, Barja-Fidalgo C, Manhaes AC, Barradas PC. A role for the MAPK/ERK pathway in oligodendroglial differentiation in vitro: stage specific effects on cell branching. Int J Dev Neurosci. 2009;27:757–768. doi: 10.1016/j.ijdevneu.2009.08.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.