

Abstract
β -Lactamases and penicillin-binding proteins are bacterial enzymes involved in antibiotic resistance to β-lactam antibiotics and biosynthetic assembly of cell wall, respectively. Members of these large families of enzymes all experience acylation by their respective substrates at an active site serine as the first step in their catalytic activities. A Ser-X-X-Lys sequence motif is seen in all these proteins, and crystal structures demonstrate that the side-chain functions of the serine and lysine are in contact with one another. Three independent methods were used in this report to address the question of the protonation state of this important lysine (Lys-73) in the TEM-1 β-lactamase from Escherichia coli. These techniques included perturbation of the pKa of Lys-73 by the study of the γ-thialysine-73 variant and the attendant kinetic analyses, investigation of the protonation state by titration of specifically labeled proteins by nuclear magnetic resonance, and by computational treatment using the thermodynamic integration method. All three methods indicated that the pKa of Lys-73 of this enzyme is attenuated to 8.0–8.5. It is argued herein that the unique ground-state ion pair of Glu-166 and Lys-73 of class A β-lactamases has actually raised the pKa of the active site lysine to 8.0–8.5 from that of the parental penicillin-binding protein. Whereas we cannot rule out that Glu-166 might activate the active site water, which in turn promotes Ser-70 for the acylation event, such as proposed earlier, we would like to propose as a plausible alternative for the acylation step the possibility that the ion pair would reconfigure to the protonated Glu-166 and unprotonated Lys-73. As such, unprotonated Lys-73 could promote serine for acylation, a process that should be shared among all active-site serine β-lactamases and penicillin-binding proteins.
A number of enzymes have evolved a catalytic strategy that depends on a transient acylation of an active site serine. The catalytic serine residue in these enzymes is followed by a lysine three residues toward the carboxyl termini of the proteins (i.e. … Ser-X-X-Lys …). This sequence motif is seen in serine-dependent β-lactamases and penicillin-binding proteins (PBPs1), of which several hundred members are known. The catalytic implication of this Ser-X-X-Lys sequence motif for β-lactamases is debated in the literature, but the role of these residues in catalysis is likely to be general for the large group of proteins that share this sequence.
β-Lactamases are bacterial resistance enzymes to β-lactam antibiotics, which include penicillins and cephalosporins. Members of the class A β-lactamases are the most common among pathogenic bacteria. These enzymes undergo acylation and deacylation at Ser-70 during substrate turnover (1, 2). The process of deacylation of the acyl-enzyme intermediate is best understood. Glu-166 is the active-site general base that promotes a water molecule in the deacylation step (3–5). On the other hand, how the active-site serine experiences acylation is an issue of much debate (3, 4, 6–14).
The contention stems largely from a lack of clear knowledge of the titration states of the active-site residues involved in catalysis. The active-site moieties that could undergo titration are Glu-166, Lys-73, and Lys-234. Two groups have suggested that Lys-73 is not protonated and can serve as the general base in activation of Ser-70 during its acylation (4, 6). Others believe that a positively charged Lys-73 would necessitate other catalytic strategies by these enzymes (8–12, 14). We present herein experimental evidence that indicates that the active site lysine in the class A TEM-1 β-lactamase from Escherichia coli has a lower pKa than a typical lysine. The pKa of Lys-73 was also determined computationally using the molecular dynamics-based thermodynamic integration method. The results were in good agreement with experiments. Further calculations were carried out on the E166A mutant to determine the influence of Glu-166 on the pKa of Lys-73. It was found that the pKa of Lys-73 in the E166A mutant was reduced to 6.0, arguing that the proximity of Glu-166 to Lys-73 has raised its pKa to 8.0–8.5 in class A β-lactamases. These data collectively do not rule out Glu-166 as the potential base that activates an active site water, which in turn would activate the active site serine for acylation. However, based on the reduced pKa for Lys-73 (unique among the 11 lysines), we propose as a plausible alternative mechanism the possibility of a protonated Glu-166 and an unprotonated Lys-73. In this mechanistic scenario, the unprotonated Lys-73 would promote the active site serine for acylation by the substrate. This notion unifies the mechanism(s) for serine acylation by substrates for all active-site serine β-lactamases and PBPs.
EXPERIMENTAL PROCEDURES
Materials
Penicillin G was purchased from Sigma. Mezlocillin was the generous gift of Dr. Robert Bonomo. The growth medium was purchased either from Difco Laboratories (Detroit, MI) or Fisher Scientific. The chromatography media were from Bio-Rad Laboratories. Isolation and purification of all enzymes (wild-type and mutants) were carried out as described earlier (15). The 99% 15 NH4Cl was purchased from Isotec Inc., and [6-13C]L-lysine (99%) was from Cambridge Isotope Laboratories.
Generation of the Mutant TEM-1 β-Lactamases
Mutagenesis of Lys-73 of the TEM-1 β-lactamase to Ala was accomplished by using a QuikChange® site-directed mutagenesis kit (Stratagene), in accordance with the manufacturer’s recommendations. The TEM-1 β-lactamase gene that was previously subcloned into an expression vector pET24a(+) was mutated using two primers: forward, 5′-CAATGCTG-CGCACTTTTGCAGTTCTGCTATGTGGC-3′, and reverse, 5′-GCCAC-ATAGCAGAACTGCAAAAGTGCTCATCATTG-3′ (the codon for alanine is underlined). Mutated DNA was used to transform E. coli JM83 and selection of transformants was performed on agar supplemented with kanamycin A (30 μg/ml). DNA from several transformants were initially screened by digestion with the restriction endonuclease DraI (this site was removed by mutation). Subsequently the nucleotide sequence of the entire gene was verified.
The double mutant C77S/C123A and the triple mutant K73C/C77S/C123A of the TEM β-lactamase were generated using the pTZ19-4 vector essentially as described earlier (16). First, C123A substitution was generated, utilizing primer TEM123 (5′-GGTTATGGCAGCACTGGCTAATTCTCTTACTG-3′; mutated codon underlined). Subsequently, C77S substitution was introduced using primer TEM77 (5′-AATACCGCGCCAGATAGCAGAACTTTAAAAG-3′; mutated codon underlined) to produce the double mutant enzyme. Finally, Lys-73 was mutated to cysteine with primer TEM73 (5′-CGCCAGATAGCAGAACACAAAAAGTGCTCATC-3′; mutated codon underlined). Both DNA strands of the mutant genes were sequenced.
Cloning of Mutant TEM β-Lactamase Genes into Expression Vector and Purification of Enzymes
To facilitate β-lactamase secretion into the growth medium, we fused the PCR-generated genes for the C77S/C123A and K73C/C77S/C123A mutant β-lactamases with the leader sequence of the OmpA protein in the pSV106 vector, as described earlier (15). After resequencing of the entire genes, they were recloned into the NdeI-HindIII sites of the pET24 expression vector. Growth conditions for protein induction and purification were essentially the same as described recently (15).
Labeling (15N and 13C) of Enzymes and Protein Preparation
To prepare the proteins uniformly labeled with 15N, we utilized E. coli BL21DE3 and E. coli BL21DE3 pLysS strains, carrying vector pET24a(+) with the TEM-1 β-lactamase and the K73A mutant β-lactamase genes, respectively. Strains were incubated at 37 °C in a minimal medium containing 15 NH4Cl as the sole nitrogen source (17), 400 mM D-sorbitol, 2.5 mM betaine, and 20 μg/ml kanamycin A. When the optical density at 600 nm for bacterial growth (A600) reached 0.3, expression of proteins was induced by the addition of isopropyl-1-thio-β-D-galactopyranoside (0.4 mM final concentration), and bacteria were grown for another 20 h at room temperature. The proteins were isolated both from the growth medium and from the periplasm by osmotic shock.
For production of proteins selectively labeled with 13C(ε)-lysine, each strain was incubated at 37 °C in minimal medium containing 100 μg/ml of each amino acid (excluding lysine), 150 μg/ml 13C-labeled lysine (18, 19), 400 mM D-sorbitol, 2.5 mM betaine, and 20 μg/ml kanamycin A to13C-labeled lysine (150 μg/ml) was A600 of 0.3. An additional portion of added just before induction. Induction and isolation of the selectively labeled proteins was performed essentially as described above. The desired proteins were purified from the growth medium to homogeneity in one step by DEAE-Sepharose ion-exchange chromatography (2.5 × 20 cm) using a linear gradient of 10 –100 mM Tris (1.0 liter), pH 7.0.
Modification of the K73C/C77S/C123A Mutant Protein
A total of 5 mg of the triple mutant protein was denatured in 8 M urea, 200 mM AMPSO, 15 mM EDTA, pH 8.5, in a final volume of 1.0 ml. The exposure of cysteine to solution was assessed by its titration (10-μl aliquot of the denaturation mixture) with 5,5′-dithiobis-2-nitrobenzoic acid, per the method of Ellman (20). The mixture was kept at 25 °C with gentle mixing under an atmosphere of argon for 15 min. Upon full denaturation, the protein was incubated with a freshly prepared solution of 2-bromoethylamine in the denaturing buffer that provided a final concentration of 40 mM. The mixture was then gently mixed for 20 h at 25°C, at which point the cysteine was fully modified.
Refolding of the enzyme was carried out according to a modified literature method (21). The enzyme was diluted into 100 mM sodium phosphate, 600 mM ammonium sulfate, 20% glycerol, pH 7.0 (refolding buffer; total volume of 50 ml) and was kept gently mixing for 1 h at 4 °C. It was necessary to maintain a protein concentration of ≤ 0.1 mg/ml after the dilution. Removal of urea and ammonium sulfate was accomplished by extensive stepwise dialysis against 100 mM sodium phosphate buffer, pH 7.0, containing 20% glycerol and ammonium sulfate (0.3 M, 0.1 M, and 0 M, in three steps). The protein was concentrated to a maximum of 0.3 mg/ml. In excess of this concentration the protein precipitated.
The pH Dependence of Catalysis by the TEM-1 β-Lactamase and of Its Modified K73C/C77S/C123A Mutant Variant
Mezlocillin was used as the substrate for these experiments. Buffers for the pH range of 5.0 to 9.5 were 50 mM sodium acetate (pH 5–6), sodium phosphate (pH 6 –7.5), and Tris (pH 8 –9), supplemented with 150 mM sodium chloride to keep the ionic strength constant. Analyses of the data were carried out according to the methods of Lineweaver-Burk and Hanes-Wolf. The pH dependence of kcat and kcat/Km were fitted to a double ionization model (Equation 1), and the pH dependence of Km data and the NMR titration findings were fitted to a single-ionization model (Equation 2).
| (Eq. 1) |
| (Eq. 2) |
NMR Spectrometry
Protein samples were dialyzed against 25 mM sodium phosphate buffer, pH 6.0, and 10% D2O. Final concentrations for all samples were 1.0 mM protein, with initial sample volumes of 550 μl. Samples were adjusted for a range of pH values (6.00, 6.50, 7.06, 7.50, 8.06, 8.77, 9.30, 9.33, and 11.06) by adding small (microliter) aliquots of 10 mM HCl or 100 mM NaOH directly into the 5-mm NMR tubes, which contained the protein solution. Samples were then gently mixed, and the final pH was measured in the NMR tube using a Mettler Toledo Analytical NMR tube pH electrode and a Corning pH meter.
All NMR spectra were collected on the Varian Unity Plus 720 MHz NMR spectrometer with a Varian (1H, 13C, and 15N) triple resonance probe equipped with z-axis pulsed-field gradients. The instrument was operating at the unique proton frequency of 719.86 MHz, with carbon and nitrogen frequencies of 181.02 and 72.95 MHz, respectively. NMR spectra were collected at 293 K. Transmitter and decoupler offsets were referenced with sodium 3-(trimethylsilyl)-1-propanesulfonate as an external standard dissolved in 25 mM phosphate buffer and 10% D2O.
Carbon-filtered proton resonances from the 13C(ε)-Lys-labeled samples were observed using the 13C HSQC experiment (22). Transmitter and decoupler offsets and spectral widths for the 1H and 13C dimension were set at 2.59 ppm/2000 Hz and 42.61 ppm/1000 Hz, respectively. Data were collected with 128 transients at 2048 complex points in the 1H and 64 increments in the 13C dimension (linear prediction to 96 points and zero-filling to 128 points were applied during the Fourier transformation process), which resulted in a total data collection time of 4.5 h per pH point. Data were analyzed using Felix 2000 (23). Felix matrices were generated for each spectrum in the identical fashion (i.e. matrix size, linear prediction in the carbon dimension, phasing parameters in both dimensions, and referencing) to ensure continuity of signals.
Quality control spectra to monitor the protein integrity at all pH values were measured on globally 15N-labeled protein using the 15N HSQC experiment (24). Transmitter and decoupler offsets and spectral widths for the 1H and 15N dimension were set at 4.73 ppm/12,001.20 Hz and 83.57 ppm/7,199.42 Hz, respectively. 15N HSQC spectra were measured over the pH range of 6.0 to 11.5. Data were collected with 32 transients at 2048 complex points in the 1H and 256 increments in the 15N dimension (linear predicted to 384 points and zero-filled to 512 applied during the Fourier transformation process) for a total data collection time of 30 min per pH point.
Computational Methods
The thermodynamic integration method was used to determine the free energy change for the protonation of Lys-73 and Lys-146 (a surface lysine with normal pKa) in the wild-type and in the E166A mutant enzymes. The protonation of each residue constitutes one-half of the full thermodynamics cycle used to determine the pKa of Lys-73 (see below). The protein was fully solvated, and the particle mesh Ewald method (PME (25)) was used to treat long range electrostatics as implemented in the Sander module of the AMBER 7 package. The Cornell et al. (26) force field, with the parm99 parameters, was used to carry out the simulations.
Calculation of the pKa of Lys-73, pKa(BH+), was based on the following equation (27),
| (Eq. 3) |
where pKa(AH+) corresponds to that of Lys-146, expected to have a typical pKa value of 10.8, and pKa(BH+) is the pKa of Lys-73. Free energy simulations using the thermodynamic integration technique were used to determine ΔΔGaq (B – BH+) and ΔΔGaq (A –AH+), the free energy changes for the deprotonation of Lys-73 and Lys-146, respectively. Along with the known pKa of Lys-146, the computed values of ΔΔGaq (B – BH+) and ΔΔGaq (A – AH+) were used in Equation 1 to determine the pKa of Lys-73.
The thermodynamic integration method can be used to evaluate the free energy differences between the two states X and Y,
| (Eq. 4) |
where λ is a coupling parameter, GX and GY and the free energies of states X and Y, and V(λ) is the potential energy. The integrand is determined by carrying out molecular dynamics simulations at discrete values of λ ranging from 0 to 1.
The initial coordinates for the wild-type TEM-1 β-lactamase were taken from the Research Collaboratory for Structural Bioinformatics (www.rcsb.org/pdb/index.html) Protein Data Bank (accession number 1BTL). The E166A mutant enzyme was generated by mutating Glu-166 to alanine in silico using the “mutate” option in the Sybyl 6.9 software package. The enzyme was protonated, and counter ions were added to the enzyme using the “protonate” and “addions” programs, respectively, which are parts of the AMBER 7 package (28). Crystallographic water molecules were retained, and the system was fully solvated in a box of TIP3P (29) waters, such that no atom in the enzyme was less than 10 Å from any edge of the box. The total number of atoms in the fully solvated system was 37,012 for the wild-type enzyme and 37,015 for the mutant protein. Before starting the simulations for the free energy calculations, the system was equilibrated based on a combination of energy minimizations, slow heating, and 300 K molecular dynamics simulation, as described in a previous study (30). All bonds involving hydrogen atoms in the system were constrained using the SHAKE algorithm. Particle mesh Ewald (PME) was used to treat long range electrostatics, and a 9-Å van der Waals cutoff was applied to the system. All simulations were carried out in parallel using the Sander program of the AMBER 7 package on eight processors of a Linux cluster.
The integral in Equation 4 was estimated numerically using a 12-point Gaussian quadrature. A total of 40 ps of simulation was carried out at each of the 12 λ values, which consisted of 10 ps of equilibration and 30 ps of data collection.
RESULTS AND DISCUSSION
Determination of the protonation state of Lys-73 would clarify its mechanistic role in the catalytic turnover of substrates by class A β-lactamases, and the elucidation of the mechanism of these enzymes would have implications for those of the related enzymes. We have approached this task for the mechanism of the TEM-1 β-lactamase from E. coli, as a representative member of these families of enzymes, by three complementary methods in this report. First, we have generated a lysine analogue, γ-thialysine, by combining site-directed mutagenesis and chemical modification of the cysteine (31), introduced in place of Lys-73 to perturb its pKa in comparison with the wild-type enzyme. Second, we have evaluated the pKa value of Lys-73 by NMR experiments. Finally, we have evaluated the pKa for Lys-73 by state-of-the-art computational treatment using the molecular dynamics-based thermodynamic integration method.
It has been reported by Hermann and Lemke that the side chain of N-acetyl-γ-thialysine titrates at a pKa value of 9.39 (32). We verified this observation by preparing N-acetyl-S-ethyl[15N]amine-cysteine (i.e. N-acetyl-γ-thialysine with the side chain amine labeled), which gave a pKa of 9.52 ± 0.01 in our hands, in good agreement with the earlier report. The presence of the sulfide moiety in γ-thialysine attenuates the pKa of the side chain by approximately 1 pK unit, compared with N-acetyllysine. This pKa attenuation gives an experimental handle for the evaluation of the influence of residue 73 in the course of enzymic catalysis. γ-Thialysine has been used previously to elucidate the role of lysine residues in the active sites of several proteins such as aspartate aminotransferase (33), ribonuclease A (34), acetoacetate decarboxylase (35), and leader peptidase (36).
We set out to generate a γ-thialysine at position 73 of the TEM-1 β-lactamase. These experiments turned out substantially more complicated than anticipated. For example, to generate γ-thialysine according to earlier methodology (37), one generates a cysteine at the position, and then the cysteine thiolate is modified by bromoethylamine to give γ-thialysine. This seemingly straightforward process did not work in our hands with the TEM β-lactamase. After considerable troubleshooting, we concluded that a disulfide exchange reaction was taking place between the disulfide bond linking residues Cys-77 and Cys-123 and the introduced cysteine at position 73. Hence, the resultant protein was misfolded. We took out the disulfide bond in the enzyme by generating the double mutant C77S/C123A. The choice of the amino acid substitutions was based on earlier precedents (21, 38, 39). This enzyme showed properties very much similar to those of the wild-type enzyme (e.g. a mere 2- to 3-fold effect on the steady-state kinetic parameters for substrate turnover). These results agreed well with previous studies on the role and effect of the disulfide bridge in the TEM-1 β-lactamase (21, 38, 39). Hence, the loss of the disulfide bond was not detrimental from a mechanistic point of view. Having determined this, the triple mutant K73C/C77S/C123A was prepared. We observed that this protein would not express well (2 mg per liter of growth). Despite this challenge, we were able to purify to homogeneity sufficient amounts of this protein for our experiments. The protein was devoid of activity (0.04% activity compared with the wild-type), as the critical Lys-73 was mutated. Subsequently, it became clear that modification of Cys-73 in the triple mutant protein (both by the Ellman reagent and by 2-bromoethylamine) was not taking place in attempts at generating γ-thialysine. According to the x-ray structure, Lys-73 in the wild-type enzyme is solvent-inaccessible, so we reasoned the same may be true for the mutant protein. Therefore, the protein was denatured for modification by 2-bromoethylamine, and then the reconstituted enzyme was refolded. The reaction with 5,5′-dithiobis-2-nitrobenzoic acid with the denatured protein indicated that modification by bromoethylamine was complete. Several different methods for denaturation and folding were tried before we settled on the method that worked best in our hands (see “Experimental Procedures”). The yield of the “modified triple-mutant enzyme” (possessing the γ-thialysine) after renaturation was quantitative. The reconstituted modified triple-mutant protein was an active enzyme, with roughly 50% of the wild-type activity (see below), the same level of activity seen for the C77S/C123A double-mutant enzyme. It has been reported that residues such as histidine and methionine could experience modification by bromoethylamine (40). We used isoelectric focusing gel electrophoresis to look at the extent of the protein modification. The isoelectric focusing indicated that the modified triple mutant was focused at the same point as the wild-type TEM-1 enzyme. In addition, as a control experiment, when the wild-type protein was put through the precise condition for the chemical modification procedure that we have described here, the protein did not show any attenuation of its activity.
One of the goals of preparing the γ -thialysine-73 variant of the TEM-1 β-lactamase was our expectation to study the γ-thialysine moiety suitably labeled for titration by NMR experiments. For example, 15Nε- or 13Cδ-labeled γ-thialysine would be useful to study the protonation state of the side chain of residue 73, because NMR signals are sensitive to the protonation states. In essence, we would be able to determine the pKa of a single residue in the fully constituted enzyme. Unfortunately, the high concentration of the γ-thialysine variant of the TEM-1 β-lactamase that was necessary to attempt the NMR experiments could not be attained, because the protein precipitated at such concentrations, precluding this experiment. This is in contrast to the wild-type enzyme, which is stable at high protein concentrations, but similar to the case of the C77S/C123A double-mutant enzyme.
The pH dependence of kcat/Km of the wild-type enzyme with mezlocillin (a penicillin), an indicator of the state of the free enzyme or the substrate, revealed two titratable residues with pKa values of 5.0 ± 0.2 and 8.0 ±0.1 (Fig. 1). For the γ-thialysine-73 variant, the pKa for the basic limb decreased to 7.5 ± 0.1, and the pKa for the acidic limb was 4.6 ± 0.4, which is virtually unchanged within the error. The errors on each of the kinetic parameters at each pH value were not taken into account in this data-fitting procedure. If these errors in the fitting processes were to be taken into account for the modified triple mutant, the new values become pK1= 5.0 ± 0.2 and pK2 = 7.2 ± 0.2 (no differences were seen for the corresponding wild-type values with this kind of analysis). The optimum pH was also shifted slightly to lower pH values for the γ-thialysine mutant (Fig. 1). The pH dependence of kcat, an indicator of the complex of enzyme and the substrate, for the wild-type TEM-1 enzyme with mezlocillin was bell-shaped with pK1 = 4.5 ± 0.6 and pK2 = 8.5 ± 0.1, and also for γ-thialysine we measured pK1 = 4.9 ± 0.1 and pK2 = 8.4 ± 0.1. The pH dependence profile for Km was essentially the same for the wild-type and the mutant (Fig. 1C).
Fig. 1. The pH dependence of kcat/Km (A), kcat (B), Km (C) of the wild-type (closed circles; left y-axis) and of γ-thialysine (open circles; right y-axis) TEM-1 β-lactamases with mezlocillin.

The error bars in the plots represent the standard deviations for each data point.
These findings indicate that the pKa for the basic limb of the kcat/Km versus pH profile was attenuated in the case of the γ-thialysine-73 variant enzyme, hence, the basic limb is due to titration of Lys-73. Therefore, the implication is that at the free state of the enzyme (uncomplexed by the substrate), Lys-73 is protonated. In light of a lack of observation of any difference for the basic limb of the plots for the two enzymes with reference to the pH dependence for kcat (Fig. 1B), we surmise that the basic limb for kcat is not due to Lys-73. Because the pH dependence for kcat (Fig. 1B) is an indicator of the complex of enzyme and the substrate, we cannot conclude anything about the protonation state of Lys-73 within the complex. Indeed, the titratable residues that define the pH dependence of kcat/Km and of kcat need not be the same.
To follow up on these measurements using an additional technique, we utilized NMR spectrometry to explore the titration states of the 11 lysines in the TEM-1 β-lactamase. Proton and carbon chemical shift changes at the lysine ε position were used to determine a pKa profile of all 11 lysines. Aliphatic H2C (ε) resonances are unaffected by solvent exchange broadening, and their chemical shifts serve as useful markers in characterizing the chemical environment of the N(ζ) group directly attached to them. By selectively labeling only the C(ε) positions in all lysines in the TEM-1 β-lactamase, we were able to remove ambiguity of signal overlap with additional aliphatic resonances from other residues and focus directly on the pKa values of the 11 lysines.
The pH profile for the 11 lysine H2C(ε) resonances was determined by measuring 13C HSQC spectra over the pH range of 6.0 to 11.1. The overlap of signals in a complimentary set of 15N HSQC spectra collected on uniformly 15N-labeled TEM-1 β-lactamase samples between pH 4 and 11.7 were used to monitor the integrity of the protein. Samples above pH 11.1 showed precipitation in the NMR tube and shifts in the amide backbone chemical shifts to the unfolded region in the 15N-filtered HSQC spectra (spectra not shown). The data from the globally 15N-labeled TEM-1 β-lactamase indicated that the enzyme was stable in the pH range of 5–11 (data not shown).
Carbon-filtered HSQC spectra were collected on both 13C(ε)-labeled wild-type and K73A mutant TEM-1 β-lactamases to determine which of the one or more resonances in the wild-type spectra were due to the active site Lys-73. The wild-type and mutant 13C(ε) HSQC spectra are shown in Fig. 2 at two different pH values. Fig. 2A shows the 13C HSQC spectrum of the 11 lysine H2C(ε) resonances in the TEM-1 β-lactamase at pH 6.0. Eleven distinct lysine signals were observed, and these were labeled 1–11 for clarity, with peaks correlated to the same lysine labeled with the a/b label extensions. At pH 6.0, all peaks seen in the wild-type sample (Fig. 2A) appeared to be present also in the K73A mutant spectrum (Fig. 2B). However, when the pH of these samples was raised to 9.3, differences in these spectra were observed (Fig. 2, C and D). Peak 4 in the wild-type protein spectrum is the feature that is absent when compared with the mutant at high pH spectrum. Peak 7 in the mutant spectra still followed the chemical shift versus pH profile seen in the wild-type spectra, whereas peak 1b in the mutant protein resisted the behavior of merging with peak 1a, which was seen in the wild-type spectra. As in the spectrum for the mutant protein, the remaining peaks were unaffected by the elevated pH up to a value of 9.3. Because peak 4 was the absent peak in the K73A mutant spectrum at high pH when compared with the wild-type spectrum, we attribute this peak to the H2C(ε) resonance of Lys-73 in the TEM-1 β-lactamase.
Fig. 2. 13C HSQC spectra for wild-type and K73A mutant TEM-1 β-lactamase, specifically 13C labeled at the C(ε) position, and the corresponding 1H-13C(ε) chemical shifts from all lysines for the wild-type protein, obtained from the pH titration.

Spectral features are labeled in panel A. The wild-type spectra at pH 6.0 (A) and pH 9.3 (C), and those of the K73A mutant protein at pH 6.0 (B) and pH 9.3 (D) are depicted. Red boxed areas designate the position (or lack thereof) for peak 4, attributed to the Lys-73 signal. Chemical shifts of proton (E) and carbon (F) as a function of pH for all lysine H2C(ε) resonances. The profile for peak 4 is shown in red. Peak labels are as follows: Peaks 1a (●), 1b (○), 2 ([+]), 3a (▲), 3b (▼), 4 (■), 5 (×), 6 (□), 7 (◆), 8 (⊙),9 (¦), 10 (+), and 11 (\). G, the NMR chemical shifts of the proton and carbon for peak 4 (E and F) were fitted to a single-ionization model (Equation 2).
Panels E and F of Fig. 2 show the 1H(ε) and 13C(ε) chemical shift profiles of all lysines from the 13C HSQC spectral titration at all pH values for the wild-type protein. At pH 7.5, peak 4 begins to separate from its adjacent peak, with which it overlaps (called 3b, because it is correlated with 3a in higher pH values). Peak 4 repositioning is monitored in the red box (see Fig. 2, A and C) and shows chemical shift changes in the upfield direction in the proton and downfield direction in the carbon dimensions. In addition, peak 1b begins to merge with peak 1a in the wild-type enzyme. By pH 8.77, peak 4 has emerged from the new peak 3b, and peaks 1a and 1b have merged, while peak 7 has begun to undergo a carbon chemical shift change to the downfield position. However, the nine remaining signals (including peaks 1 and 3) are unchanged in their chemical shifts up to pH 8.77. The results of Fig. 2 indicate that all lysine side chains have normal pKa values of 10 –11, with the sole exception of that for Lys-73. Because the enzyme is virtually inactive at pH above 9.0 (consistent with kinetics), we used the NMR titration data for pH values of 6.0–9.3 for the titration of Lys-73. The fitting of these data to a single-ionization model (Equation 2) revealed a pKa of 8.4 ± 0.1 and 8.7 ± 0.2, whether the 13C or 1H chemical shifts were used for analyses, respectively (Fig. 2G). As a cautionary note, the trends for the chemical shifts of the side chain of Lys-73 cannot be attributed to different “conformational” states, rather than the two titration states, because integration of the signal after its separation from peak 3b did not change to the end of the titration. Furthermore, the degree of changes in chemical shifts for Lys-73 resonances is typical for a titration. The chemical shifts for the lysines at pH > 11 would appear more dramatic as the protein unfolds at and beyond this pH value.
The results from the NMR experiments are inconsistent with similar experiments performed by Damblon et al. (12) earlier. 13C HSQC spectra for the wild-type 13C(ε)-labeled TEM-1 have been previously published at similar spectral resolution as our data, only at a lower spectrometer frequency, with fewer 1H transients and at fewer pH points in the low pH range. Despite the differences in data collection conditions, except for the absence of the low intensity peaks 2 and 8 in the data of Damblon et al., our spectra and those of Damblon et al. look very much similar. The fact that we see these less intense peaks (2 and 8) when the previously published work did not, is most likely a reflection of the higher sensitivity for our instrument (a 720-MHz spectrometer, compared with a 600-MHz instrument for the earlier study). The earlier work had not studied a mutant at position 73 to delineate which NMR signal corresponded to that for the desired residue. As a consequence, the NMR signal that was attributed to Lys-73 was inadvertently incorrect. The experiments performed herein unequivocally identified the NMR signal due to Lys-73 and monitored its titration as a function of changing pH. As evidenced by Fig. 2, Lys-73 is indeed unique among the 11 lysines in the TEM-1β-lactamase.
We decided to complement the experimental results with state-of-the-art computational treatment of the TEM-1 β-lactamase. Computational analyses of the pKa values of the active site residues in the TEM-1 β-lactamase have been reported previously. These studies have used the continuum electrostatic methods to compute pKa of ionizable groups in the native TEM-1 β-lactamase (6, 41, 42) or in complex with substrates (6, 9). The first continuum electrostatics calculation of the pKa of Lys-73 was carried out by Swarén et al. (6) who evaluated its pKa at 8.0 in the substrate-free enzyme. This proposal was subsequently explored further with additional similar calculations by another group, but no decrease in the pKa of Lys-73 was found (42). Subsequent studies, also using continuum electrostatics calculations, have estimated the pKa of Lys-73 to be around 10 (41).
It would be desirable to use more sophisticated methods. Free-energy perturbation and thermodynamic integration methods have been shown to determine free energy change within 1 kcal/ mol of the experimental values; hence they afford high accuracy (43). The thermodynamic integration approach takes into consideration several factors that are important for the accurate calculations of the pKa of ionizable groups. These factors include protein flexibility, explicit treatment of water and counter ions, and a robust and extensively used AMBER force field for the treatment of electrostatics and van der Waals interactions surrounding the ionizable residues. For these studies, we have used the fully solvated structure of the TEM-1 β-lactamase with PME for treatment of long range electrostatics, which, needless to say, preserves the unique environment of Lys-73.
The pKa of an ionizable residue is related to the free energy difference between the protonated and the unprotonated states. The following free energy cycle, which forms the basis of Equation 3, was used to calculate the pKa of Lys-73,
Reaction 1.
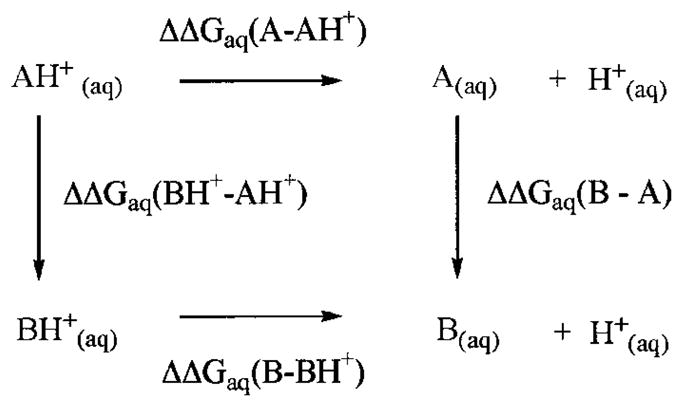
where AH+ is the protonated Lys-73, and BH+ is the protonated Lys-146. Because the pKa of Lys-146 is expected to be that of a typical lysine (as per NMR experiments), Equation 3 can be used to determine the pKa of Lys-73, given that ΔΔGaq (B – BH+) and ΔΔGaq (A – AH+) are known. These free energy terms are computed using the thermodynamic integration free energy method. In general, the free energy difference between two states X and Y can be determined, if an ensemble of structures is available for both states. However, the difference in energy between states X and Y can be large and might lead to inaccuracies in the free energy. This problem is solved with the introduction of a series of non-physical intermediate states joining the two physical states X and Y. In practice, these intermediate states are defined by introducing a coupling parameter λ and the potential energy of these states is defined by Equation 5.
| (Eq. 5) |
Calculation of the free energy difference between the protonated and unprotonated states of Lys-73 and Lys-146 using the thermodynamic integration methods thus consists of collecting an ensemble of structures using molecular dynamics simulations at each λ value and using a 12-point Gaussian quadrature to determine the integral in Equation 4. The resulting difference between ΔΔGaq (B – BH+) and ΔΔGaq (A – AH+) was found to be −3.8 kcal/mol. Using Equation 3, along with the known pKa of 10.8 for a typical lysine assigned to Lys-146, the computed pKa of Lys-73 was found to be 8.0 ± 0.1 (determined in triplicate). Typical calculation of pKa by this method is known to give standard deviations in the range of ± 0.05–0.20 pK unit (44), consistent with that of our determination (i.e. ± 0.1).
As determined by kinetic experiments with the γ-thialysine mutant and with the wild-type β-lactamase, the pKa of Lys-73 is around 8.0 –8.5 for the native enzyme without a ligand in the active site. The NMR experiment and computations also indicated that Lys-73 of the native TEM-1 β-lactamase has a reduced pKa, with a value of the same range. Swarén et al. (6) had computed a pKa value of 8.0 for Lys-73 based on electrostatic considerations of the x-ray structure of the enzyme. The more sophisticated computational analyses reported herein are supportive of this earlier determination (6) and contradict other computations (41, 42).
The protonation state of this lysine residue has been debated in the literature. There are proponents for both a protonated (3, 7–11, 14) and unprotonated (4, 6) Lys-73, each favoring a different mechanism for the turnover chemistry by class A β-lactamases. The proponents of a free-base Lys-73 argue that the side-chain amine of this residue is the basic entity that activates Ser-70 for the acylation event by the substrate. Those favoring a protonated Lys-73 have proposed that Glu-166, via an intervening water molecule, activates Ser-70 for acylation. The fact that Glu-166 functions as the general base for promotion of a water molecule for the deacylation step is undisputed by the two camps.
The sequence analysis of over 140 penicillin-binding proteins and β-lactamases that undergo transient acylation in the active-site serine revealed that the only motif that is conserved in all these proteins is the Ser-X-X-Lys sequence in the active site (45). In essence, this is a minimal requirement shared by all these enzymes that transiently undergo acylation at the active site serine in the course of catalysis. There are several representative x-ray structures for both PBPs and β-lactamases in the literature by now (1). It is revealing that practically in all the cases among members of both families of enzymes, class A β-lactamases being the exception, there is no other basic residue other than the lysine three residues downstream of the active site serine to carry out the activation step. We include the Nε-carboxylated lysine in class D β-lactamases in this reasoning, as the function of the plain lysine or a Nε-carboxylated lysine is the same in the mechanisms of the respective enzymes. Acylation of the active site by the respective substrates clearly happens in these enzymes, so it would appear that there should be no choice other than relying on the active site lysine to facilitate it. Insofar as β-lactamases are widely accepted to have descended from PBPs (2, 45–47), it would appear that the mechanism of acylation is handed down from the parental PBPs to various β-lactamases, as suggested earlier (45). As such, the side chain of an unprotonated lysine interacting with that of the active-site serine in the preacylation complex would be an intuitive mechanism for its activation. It is important to note that there are no natural variants of any of these proteins that have experienced substitutions at either active-site serine or lysine, so both residues are critical for the activities of these enzymes.
The evolution of a nascent β-lactamase from a PBP would require the advent of a catalytic step for the deacylation event (3, 45, 47). It has been conjectured that as class A β-lactamases evolved, Glu-166 was introduced within the active site to activate a water molecule for deacylation of the acyl-enzyme species (45, 47) (Fig. 3). We acknowledge that introduction of a polar amino acid such as Glu-166 within the active site for evolution of the deacylation step could also influence the acylation event. A number of reports on properties of mutant variants of β-lactamases at positions 73 and 166 have appeared (7, 11, 48–50, 52, 53). Mutations at position 166 have been revealed to have effects on both the rates of acylation and deacylation of class A β-lactamases (7, 52). However, the effect is substantially more for the deacylation step (six orders of magnitude), such that a mutant variant at position 166 has been useful in determining an x-ray structure for the acyl-enzyme species with a preferred substrate, namely a penicillin (4). This x-ray structure argues that the glutamate carboxylate at position 166 is dispensable for the acylation event, although one would not argue against it having some influence on the microscopic rate constant for the acylation step (7, 52), because the residue is within the active site. In essence, in the absence of the Glu-166 carboxylate in the mutant variant, acylation of the active site Ser-70 by the β-lactam substrate takes place, although with a somewhat attenuated microscopic rate constant.
Fig. 3.

Stereo view of the active site of the TEM-1 β-lactamase from the x-ray structure depicted as a Connolly surface.
We have documented in this report that clearly Lys-73 is different than a typical lysine in a protein by having its side chain pKa attenuated to 8.0–8.5. Attenuation of the pKa of Lys-73 is due to the electrostatic influence of Lys-234 (4.7 Å away), also an active site residue. This type of electrostatic suppression of the pKa of the side chain of lysine was first reported by Schmidt and Westheimer (54), and precedent has been set in several enzymes (33, 35, 55). Although Lys-73 of class A β-lactamases has an attenuated pKa compared with a typical lysine, the results of Fig. 1 indicate that it is protonated in the native state in the absence of substrate in the active site. This result is a direct consequence of the electrostatic effect of the side chain of Glu-166, which is within 3.5 Å of the Lys-73 side-chain nitrogen. The proximity of the carboxylate of Glu-166 indeed would elevate the pKa of Lys-73.
To explore this possibility, further calculations of the pKa of Lys-73 were carried out with the E166A mutant enzyme to determine the pKa of Lys-73 in the absence of the glutamate carboxylate. The free energy change for protonation of Lys-73 and Lys-146 were determined using the same procedure that was used for the wild-type enzyme. Two distinct molecular dynamics simulation, one for Lys-73 and the other for Lys-146, were carried out at various intermediate states, because the lysine residues were converted from the deprotonated to their protonated forms. The results were intriguing. Mutation of Glu-166 to alanine resulted in a pKa of 6.0 for Lys-73. In essence, the advent of Glu-166 in evolution of class A β-lactamases created a unique electrostatic balance that settled at a pKa of 8.0 – 8.5 for the side chain of Lys-73 in the native state.
Therefore, the question remains how acylation of the active site serine in class A β-lactamases, and in other β-lactamases and PBPs, takes place. As for class A β-lactamases, the presence of Glu-166 might have altered the catalytic processes of these enzymes such that Glu-166 now assumes the role of activating the active site serine, as proposed earlier (7, 8, 52). An alternative might also be possible. We suggest here that introduction of Glu-166 might not necessarily have altered the overall mechanism for acylation of class A β-lactamases, compared with PBPs, their kin enzymes. It is conceivable that an unprotonated lysine is involved in activation of the active site serine. In the native ground state, Glu-166 and Lys-73 of class A β-lactamases form an ion pair (i.e. a protonated Lys-73; so the earlier proposal for ground-state free-base Lys-73 would appear to be incorrect (4)). In the course of catalysis, at the non-covalent Michaelis complex stage Glu-166 becomes protonated, resulting in an unprotonated Lys-73 that activates Ser-70. In this proposal, the mechanism for acylation for all known active-site serine β-lactamases and PBP would appear to be the same, and class A β-lactamases are not exceptions. We underscore that a deprotonated Glu-166 and a free-base Lys-73 would be unsuitable, as the lone electron pairs in the two side chains would repel each other, probably causing local detrimental conformational changes within the active site, consistent with the enzyme losing activity above pH of 9. This is supported by previous molecular dynamics simulations of TEM-1 with deprotonated Glu-166 and free-base Lys-73 that resulted in unstable trajectories (56).
We forthrightly state that the proposed proton disproportionation between Lys-73 and Glu-166 upon formation of the enzyme-substrate complex is presently unsupported. Furthermore, we have not studied the complex in our investigations reported herein. In addition, this assertion is not supported by the pKa calculations of the Michaelis complex carried out by another method in a different laboratory (42).
We acknowledge that the catalytic event that precipitates active site acylation might benefit from protonation of the β-lactam nitrogen in the substrate by Ser-130, facilitated by the carboxylate of the substrate itself, such as proposed by Makinen and colleagues (9). Furthermore, we note that Ishiguro and Imajo (14) were the first to invoke involvement of the substrate carboxylate in proton transfer events. Indeed, Ishiguro and Imago too have proposed that in the native ground-state, Lys-73 of class A β-lactamases should be protonated, but the substrate carboxylate would abstract a proton from Lys-73 through the intervening involvement of Ser-130 to generate the free-base form of Lys-73, which activated Ser-70 for the acylation event. We acknowledge the possibility for this deprotonation event of Lys-73 for the acylation steps, but the crystallographic evidence presented below persuaded us to conclude that it is Glu-166 that abstracts the proton and not the substrate carboxylate.
It is interesting that class D β-lactamases (57, 58) and a penicillin-binding protein from Staphylococcus aureus, the BlaR protein (51), have an Nε-carboxylated version of the active site lysine. The side-chain amines of these enzymes, as free bases, undergo modification by carbon dioxide to give carbamates. The carbamate has been shown to serve as the active site base for serine acylation in both the OXA-10 class D β-lactamase (57) and in BlaR from S. aureus (51). Therefore, despite this post-translational alteration of the structure of the active site lysine in these proteins, the function remains the same, namely as a base in promotion of serine acylation.
Two recent ultrahigh resolution x-ray structures of class A β-lactamases have appeared in the literature that are worthy of comment (10, 13). Because of the resolutions of better than 1 Å in these structures, many of the hydrogens in the two proteins were visualized. Whereas, neither structure could settle the number of hydrogens that reside on the side-chain nitrogen of Lys-73, the protonation state of this residue can be inferred from the immediate environment around it. We hasten to add that both structures are consistent with the possibility that Lys-73 is protonated. One structure, that of the native SHV-2 β-lactamase, shows that the hydrogen on the side-chain hydroxyl of Ser-70 points toward the active site water molecule, and not at Lys-73 (13). This is to be expected as proposed herein, because Lys-73 is protonated in the native state in the absence of any ligand (substrate) bound to the active site. The other is an x-ray structure of the TEM-1 β-lactamase with a boronate inhibitor bound to the active site serine. This structure shows that Glu-166 is protonated (10). Whereas the authors surmised that the protonated nature of Glu-166 argues for it abstracting a proton from the active site water, which in turn would activate Ser-70 for acylation, it is also consistent with our proposal that the ion pair of Glu-166 and Lys-73 might reconfigure to a protonated Glu-166 and free-base Lys-73 for active site acylation. In essence, in our view, what the structure of Minasov et al. might reveal is that the free-base lysine has activated Ser-70 for modification by the boronate inhibitor, as a consequence of which both Glu-166 and Lys-73 would be trapped in their protonated forms; the protonation state of the latter residue could not be visualized in the x-ray structure.
Concluding Remarks
The mechanism(s) of active site acylation of penicillin-binding proteins and β-lactamases by their respective substrates might likely be the same. Certain aspects of the mechanistic features from earlier reports are consistent with our findings. For example, we concur with the earlier reports that the native ground-state Lys-73 of class A β-lactamases is protonated (8 –11, 12, 14), it is so because of the proximity of Glu-166 that elevates the pKa of Lys-73, as per earlier discussion in this report. However, our proposed alternative mechanism is consistent with the role of Ser-70 activation taking place by a free-base form of Lys-73 (4, 6, 14), which would be formed in the preacylation step. In the case of class A β-lactamases, introduction of Glu-166 into the active site has perturbed the pKa of the active site Lys-73, but its role for promotion of serine for acylation would be preserved. The equilibrium for the existence of the ion pair between Glu-166 and Lys-73 and their respective uncharged versions (protonated Glu-166 and free-base Lys-73) should be important in the reaction of the enzyme, consistent with the ultrahigh resolution structures for these enzymes reported recently. However, these data do not reject out of hand the possibility that the mechanism of class A β-lactamases has been altered such that Glu-166 promotes the active site serine for acylation.
Footnotes
The abbreviations used are: PBP, penicillin-binding protein; BEA, (2-bromoethyl)amine; AMPSO, 3-[(1,1-dimethyl-2-hydroxyethyl)-amino]-2-hydroxypropanesulfonic acid; HSQC, heteronuclear single quantum coherence; PME, particle mesh Ewald method.
The work was supported by Grants AI33170 and GM61629 from the National Institutes of Health. NMR studies were carried out at the National High Magnet Field Laboratory in Tallahassee, FL through support of their Solutions NMR User’s Facility.
References
- 1.Kotra LP, Samama JP, Mobashery S. In: Bacterial Resistance to Antimicrobials, Mechanisms, Genetics, Medical Practice and Public Health. Lewis A, Salyers A, Haber H, Wax RG, editors. Marcel Dekker, Inc; New York: 2002. pp. 123–159. [Google Scholar]
- 2.Bush K, Mobashery S. Adv Exp Med Biol. 1998;456:71–98. [PubMed] [Google Scholar]
- 3.Herzberg O, Moult J. Science. 1987;236:694–701. doi: 10.1126/science.3107125. [DOI] [PubMed] [Google Scholar]
- 4.Strynadka NCJ, Adachi H, Jensen SE, Johns K, Sielecki A, Betzel C, Sutoh K, James MNG. Nature. 1992;359:700–705. doi: 10.1038/359700a0. [DOI] [PubMed] [Google Scholar]
- 5.Maveyraud L, Massova I, Birck C, Miyashita K, Samama JP, Mobashery S. J Am Chem Soc. 1996;118:7435–7440. [Google Scholar]
- 6.Swarén P, Maveyraud L, Guillet V, Masson JM, Mourey L, Samama JP. Structure. 1995;3:603–613. doi: 10.1016/s0969-2126(01)00194-0. [DOI] [PubMed] [Google Scholar]
- 7.Gibson RM, Christensen H, Waley SG. Biochem J. 1990;272:613–619. doi: 10.1042/bj2720613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lamotte-Brasseur J, Dive G, Dideberg O, Frère JM, Ghuysen JM. Biochem J. 1991;279:213–221. doi: 10.1042/bj2790213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atanasov BP, Mustafi D, Makinen MW. Proc Natl Acad Sci U S A. 2000;97:3160–3165. doi: 10.1073/pnas.060027897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minasov G, Wang X, Shoichet BK. J Am Chem Soc. 2002;124:5333–5340. doi: 10.1021/ja0259640. [DOI] [PubMed] [Google Scholar]
- 11.Lietz EJ, Truher H, Kahn D, Hokenson MJ, Fink AL. Biochemistry. 2000;39:4971–4981. doi: 10.1021/bi992681k. [DOI] [PubMed] [Google Scholar]
- 12.Damblon C, Raquet X, Lian LY, Lamotte-Brasseur J, Fonze E, Charlier P, Roberts GCK, Frère JM. Proc Natl Acad Sci U S A. 1996;93:1747–1752. doi: 10.1073/pnas.93.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nukaga M, Mayama K, Hujer AM, Bonomo RA, Knox JR. J Mol Biol. 2003;328:289–301. doi: 10.1016/s0022-2836(03)00210-9. [DOI] [PubMed] [Google Scholar]
- 14.Ishiguro M, Imajo S. J Med Chem. 1996;39:2207–2218. doi: 10.1021/jm9506027. [DOI] [PubMed] [Google Scholar]
- 15.Vakulenko S, Golemi D. Antimicrob Agents Chemother. 2002;46:646–653. doi: 10.1128/AAC.46.3.646-653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vakulenko SB, Geryk B, Kotra LP, Mobashery S, Lerner SA. Antimicrob Agents Chemother. 1998;42:1542–1548. doi: 10.1128/aac.42.7.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramesh V, Frederick RO, Syed SEH, Gibson CF, Yang JC, Roberts GCK. Eur J Biochem. 1994;225:601–608. doi: 10.1111/j.1432-1033.1994.00601.x. [DOI] [PubMed] [Google Scholar]
- 18.Yamazaki T, Yoshida M, Kanaya S, Nakamura H, Nagayama K. Biochemistry. 1991;30:6036–6047. doi: 10.1021/bi00238a030. [DOI] [PubMed] [Google Scholar]
- 19.Roth SM, Schneider DM, Strobel LA, Van Berkum MFA, Means AR, Wand AJ. Biochemistry. 1992;31:1443–1451. doi: 10.1021/bi00120a022. [DOI] [PubMed] [Google Scholar]
- 20.Ellman GL. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 21.Laminet AA, Pluckthun A. EMBO J. 1989;8:1469–1477. doi: 10.1002/j.1460-2075.1989.tb03530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kay L, Keifer P, Saarinen T. J Am Chem Soc. 1992;114:10663–10665. [Google Scholar]
- 23.Accelrys Inc. Felix ND Spectral Processing Software, Release 2000. San Diego, CA: 2000. [Google Scholar]
- 24.Mori S, Abeygunawardana C, O’Neil Johnson M, van Zijl PCM. J Magn Reson Ser B. 1995;108:94–98. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
- 25.Darden TA, York D, Pedersen LG. J Chem Phys. 2003;98:10089–10092. [Google Scholar]
- 26.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 27.Merz KM., Jr J Am Chem Soc. 1991;113:3572–3575. [Google Scholar]
- 28.Case DA, Pearlman DA, Caldwell JW, Cheatham TE, III, Wang J, Ross WS, Simmerling CL, Darden TA, Merz KM, Stanton RV, Cheng AL, Vincent JJ, Crowley M, Tsui V, Gohlke H, Radmer RJ, Duan Y, Pitera J, Massova I, Seibel GL, Singh UC, Weiner PK, Kollman PA. AMBER. 7. University of California; San Francisco: 2002. [Google Scholar]
- 29.Jorgensen WL, Chandraseklar J, Maduar JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 30.Maveyraud L, Golemi D, Ishiwata A, Meroueh O, Mobashery S, Samama JP. J Am Chem Soc. 2002;124:2461–2465. doi: 10.1021/ja016736t. [DOI] [PubMed] [Google Scholar]
- 31.Smith HB, Hartman FC. J Biol Chem. 1988;263:4921–4925. [PubMed] [Google Scholar]
- 32.Hermann P, Lemke K. Hoppe-Seyler’s Z Physiol Chem. 1968;349:390–394. [PubMed] [Google Scholar]
- 33.Planas A, Kirsch J. Biochemistry. 1991;30:8268–8276. doi: 10.1021/bi00247a023. [DOI] [PubMed] [Google Scholar]
- 34.Messmore JM, Fuchs DN, Raines RT. J Am Chem Soc. 1995;117:8057–8060. doi: 10.1021/ja00136a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heighbarger LA, Gerlt JA, Kenyon GL. Biochemistry. 1996;35:41–46. doi: 10.1021/bi9518306. [DOI] [PubMed] [Google Scholar]
- 36.Paetzel M, Strynadka NCJ, Tschantz WR, Casareno R, Bullinger PR, Dalbey RE. J Biol Chem. 1997;272:9994–10003. doi: 10.1074/jbc.272.15.9994. [DOI] [PubMed] [Google Scholar]
- 37.Toney MD, Kirsch JF. Science. 1989;243:1485–1488. doi: 10.1126/science.2538921. [DOI] [PubMed] [Google Scholar]
- 38.Schultz SC, Dalbadie-McFarland G, Neitzel JJ, Richards JH. Proteins Struct Funct Genet. 1987;2:290–297. doi: 10.1002/prot.340020405. [DOI] [PubMed] [Google Scholar]
- 39.Vanhove M, Guillaume G, Ledent P, Richards P, Pain R, Frère JM. Biochem J. 1997;321:413–417. doi: 10.1042/bj3210413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hopkins CE, O’Connor PB, Allen KN, Costello CE, Tolan DR. Protein Sci. 2002;11:1591–1599. doi: 10.1110/ps.3900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raquet X, Lounnas V, Lamotte-Brasseur J, Frère JM, Wade RC. Biophys J. 1997;73:2416–2426. doi: 10.1016/S0006-3495(97)78270-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamotte-Brasseur J, Lounnas V, Raquet X, Wade RC. Protein Sci. 1999;8:404–409. doi: 10.1110/ps.8.2.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao BG, Kim EE, Murcko MA. J Comput-Aided Mol Des. 1996;10:23–30. doi: 10.1007/BF00124462. [DOI] [PubMed] [Google Scholar]
- 44.Gouda H, Kuntz ID, Case DA, Kollman PA. Biopolymers. 2003;68:16–34. doi: 10.1002/bip.10270. [DOI] [PubMed] [Google Scholar]
- 45.Massova I, Mobashery S. Antimicrob Agents Chemother. 1998;42:1–17. doi: 10.1128/aac.42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelly JA, Moews PC, Knox JR, Frère JM, Ghuysen JM. Science. 1982;218:479–481. doi: 10.1126/science.7123246. [DOI] [PubMed] [Google Scholar]
- 47.Knox JR, Moews PC, Frère JM. Chem Biol. 1996;3:937–947. doi: 10.1016/s1074-5521(96)90182-9. [DOI] [PubMed] [Google Scholar]
- 48.Escobar WA, Tan AK, Fink AL. Biochemistry. 1991;30:10783–10787. doi: 10.1021/bi00108a025. [DOI] [PubMed] [Google Scholar]
- 49.Chen CC, Smith TJ, Kapadia G, Wäsch S, Zawadzke LE, Coulson A, Herzberg O. Biochemistry. 1996;35:12251–12258. doi: 10.1021/bi961153v. [DOI] [PubMed] [Google Scholar]
- 50.Leung YC, Robinson CV, Aplin RT, Waley SG. Biochem J. 1994;299:671–678. doi: 10.1042/bj2990671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Golemi-Kotra D, Cha JY, Meroueh SO, Vakulenko SB, Mobashery S. J Biol Chem. 2003;278:18419–18425. doi: 10.1074/jbc.M300611200. [DOI] [PubMed] [Google Scholar]
- 52.Guillaume G, Vanhove M, Lamotte-Brasseur J, Ledent P, Jamin M, Joris B, Frère JM. J Biol Chem. 1997;272:5438–5444. doi: 10.1074/jbc.272.9.5438. [DOI] [PubMed] [Google Scholar]
- 53.Madgwick PJ, Waley SG. Biochem J. 1987;248:657–662. doi: 10.1042/bj2480657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidt DE, Jr, Westheimer FH. Biochemistry. 1971;10:1249–1253. doi: 10.1021/bi00783a023. [DOI] [PubMed] [Google Scholar]
- 55.Dao-pin S, Anderson DE, Baase WA, Dahlquist FW, Matthews BW. Biochemistry. 1991;30:11521–11529. doi: 10.1021/bi00113a006. [DOI] [PubMed] [Google Scholar]
- 56.Massova I, Kollman PA. J Comput Chem. 2002;23:1559–1576. doi: 10.1002/jcc.10129. [DOI] [PubMed] [Google Scholar]
- 57.Golemi D, Maveyraud L, Vakulenko S, Samama JP, Mobashery S. Proc Natl Acad Sci U S A. 2001;98:14280–14285. doi: 10.1073/pnas.241442898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun T, Nukaga M, Mayama K, Braswell EH, Knox JR. Protein Sci. 2003;12:82–91. doi: 10.1110/ps.0224303. [DOI] [PMC free article] [PubMed] [Google Scholar]