

Abstract
Monoamine oxidase (MAO) isoenzymes A and B are mitochondrial-bound proteins, catalyzing the oxidative deamination of monoamine neurotransmitters as well as xenobiotic amines. Although they derive from a common ancestral progenitor gene, are located at X-chromosome and display 70% structural identity, their substrate preference, regional distribution, and physiological role are divergent. In fact, while MAO-A has high affinity for serotonin and norepinephrine, MAO-B primarily serves the catabolism of 2-phenylethylamine (PEA) and contributes to the degradation of other trace amines and dopamine. Convergent lines of preclinical and clinical evidence indicate that variations in MAO enzymatic activity—due to either genetic or environmental factors—can exert a profound influence on behavioral regulation and play a role in the pathophysiology of a large spectrum of mental and neurodegenerative disorders, ranging from antisocial personality disorder to Parkinson’s disease. Over the past few years, numerous advances have been made in our understanding of the phenotypical variations associated with genetic polymorphisms and mutations of the genes encoding for both isoenzymes. In particular, novel findings on the phenotypes of MAO-deficient mice are highlighting novel potential implications of both isoenzymes in a broad spectrum of mental disorders, ranging from autism and anxiety to impulse-control disorders and ADHD. These studies will lay the foundation for future research on the neurobiological and neurochemical bases of these pathological conditions, as well as the role of gene × environment interactions in the vulnerability to several mental disorders.
I. General Characteristics of Monoamine Oxidase
A. Catalytic Reaction
Monoamine oxidases [MAOs; amine: oxygen oxidoreductase (deaminating) (flavin-containing); EC 1.4.3.4] are a family of mitochondrial-bound flavoproteins catalyzing the oxidative deamination of monoamine neurotransmitters, neuro-modulators, and hormones to the corresponding aldehydes:
This reaction requires flavin adenine dinucleotide (FAD) as a covalently bound redox cofactor and consists of three main steps (for a detailed analysis of the current knowledge on the catalytic mechanisms of MAO, see Edmondson et al., 2009):
- Following the formation of a FAD-substrate adduct, the cofactor is reduced to its hydroquinone form (FADH2), while the amine is converted into the corresponding imine.
- Once dissociated from the enzyme, the imine is spontaneously hydrolyzed, with production of aldehyde and ammonium:
- FADH2 is reoxidized to FAD, with formation of hydrogen peroxide from molecular oxygen. This reaction is the rate-limiting step of the whole enzymatic process:
As shown in Table I, the endogenous substrates of MAO include key brain neurotransmitters, such as serotonin (5-hydroxytryptamine, 5-HT), dopamine (DA), norepinephrine (NE), and epinephrine (E), as well as a number of trace amines, such as tyramine, tryptamine, 2-phenylethylamine (PEA), octopamine, and 3-iodothyronamine (T1AM). Notably, the oxidative deamination of short-chain primary amines (including PEA, tyramine, and T1AM) is not exclusively mediated by MAO but also contributed by the copper/topaquinone-containing semicarbazide-sensitive amine oxidase (SSAO; encoded by the gene AOC3; Obata, 2002; Saba et al., 2010). The role of MAO in the homeostasis of these compounds is essential to modulate the neuroendocrine regulation of the central nervous system and many peripheral organs.
Table I.
Synoptic View of the Main Substrates and Products of MAO-Mediated Metabolism (Coupled with Aldehyde Dehydrogenase (ALDH) or Aldehyde Reductase (ALR)).
| Substrates | Products | |||
|---|---|---|---|---|
| MAO | ALDH | ALR | ||
| Indolamines |
 Serotonin |
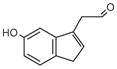 5-HIAAL |
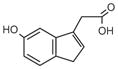 5-HIAA |
 5-HIET |
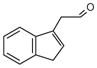
 Tryptamine |
 IAAL |
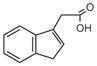 IAA |
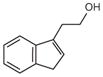 IET |
|
| Cathecolamines |
Dopamine |
DOPAL |
DOPAC |
DOPET |
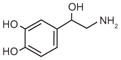 Norepinephrine |
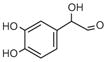 DOPGAL |
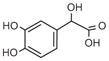 DOMA |
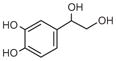 DOPEG |
|
| Other trace amines |
PEA |
PAAL |
PAA |
PET |
|
Tyramine |
HPAL |
HPA |
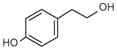 HPET |
|
Epinephrine is not listed, as its metabolites are the same as those indicated for norepinephrine.
Abbreviations: 5-HIAAL, 5-hydroxyindolaldehyde; 5-HIAA, 5-hydroxyindolacetic acid; 5-HIET, 5-hydroxyindolethanol; IAAL, indole-3-acetaldehyde; IAA, indole-3-acetic acid, IET, indole-3-ethanol (tryptophol); DOPAL, 3,4-dihydroxyphenylacetaldehyde; DOPAC, 3,4-dihydroxyphenylacetic acid; DOPET, 3,4-dihydroxyphenylethanol; DOPGAL, 3,4-dihydroxyphenylglycolaldehyde; DOMA, 3,4-dihydroxymandelic acid; DOPEG, 3,4-dihydroxylphenylethyleneglycol; PEA, 2-phenylethylamine; PAAL, 2-phenylacetaldehyde; PAA, 2-phenylacetic acid; PET, 2-phenylethanol; HPAL, 4-hydroxy-phenylaldehyde; HPA, 4-hydroxyphenylacetic acid; HPET, 4-hydroxyphenylethanol.
The aldehydes produced by MAO are toxic species (for a review on the pathogenic potential of aldehydes, see O’Brien et al., 2005) which need to be converted in less harmful metabolites. Thus, this enzyme is functionally coupled with a NAD(P)+-dependent aldehyde dehydrogenase (ALDH), which oxidizes the aldehyde to the corresponding carboxylic acid; alternatively (depending on the location and the intracellular conditions), aldehydes can be reduced to alcohols or glycols by aldehyde reductase (ALR) or alcohol dehydrogenase (ADH) (Table I). The main metabolic pathway of 5-HT consists in the conversion of this mono-amine into 5-hydroxyindolacetic acid (5-HIAA) by joint action of MAO and ALDH. Like other MAO metabolites, 5-HIAA is rapidly eliminated by diffusion into the bloodstream and excreted through the kidneys by glomerular filtration and active tubular excretion (Udenfriend et al., 1956; Despopoulos and Weissbach, 1957). Given the predominance of the MAO-ALDH pathway in 5-HT metabolism, urinary levels of 5-HIAA are used as an index for measurement of plasma 5-HT content (with diagnostic value as a biomarker for carcinoid syndrome, a paraneoplastic disorder caused by gastrointestinal apudomas secreting 5-HT). Small amounts of 5-HT (1–5%) are converted into 5-hydroxyindolethanol (5-HIET, also termed 5-hydroxytryptophol) by either ALR or ADH (Feldstein and Williamson, 1968; Beck et al., 1984; Consalvi et al., 1986; Svensson et al., 1999) (Table I). Interestingly, the amount of 5-HIET can be enhanced by compounds that compete with endogenous 5-HT metabolite for ALDH, such as ethanol (Helander et al., 1993).
The metabolism of catecholamines (DA, NE, E) is served by both MAO (in conjunction with either ALDH or ALR) and catecholamine-O-methyl-transferase (COMT). The combined action of the two enzymes converts DA into either homovanillic acid (HVA; MAO/ALDH + COMT pathway) or, less frequently, into 3-methoxy-4-hydroxyphenylethanol (MHPE; MAO/ALR + COMT pathway). The latter can be processed by ADH into HVA (Fig. 1).
Fig. 1.

Metabolic pathways of dopamine. DOPAL, 3,4-dihydroxyphenylacetaldehyde; 3-MT, 3-methoxytyramine; DOPET, 3,4-dihydroxyphenylethanol; DOPAC, 3,4-dihydroxyphenylacetic acid; MOPAL, 3-methoxy-4-hydroxyphenylacetaldehyde; MHPE, 3-methoxy-4-hydroxyphenylethanol; HVA, homovanillic acid.
NE and E undergo similar degradation pathways (Fig. 2). Specifically, MAO converts both monoamines into 3,4-dihydroxyphenylglycol aldehyde (DOPGAL), which is further processed by ALR into 3,4-dihydroxylphenylethylene glycol (DOPEG). A much smaller aliquote of DOPGAL is oxidized to 3,4-dihydroxymandelic acid (DOMA). COMT converts DOPEG into 3-methoxy-4-hydroxyphenylethylene glycol (MHPG) and DOMA into vanillyl mandelic acid (VMA). Alternatively, NE and E can be methylated by COMT to normetanephrine and metanephrine, respectively. These metabolites can be conjugated with sulfate groups by sulfatransferase 1A3 (SULT1A3) or processed by either MAO/ALR or MAO/ALDH into MHPG and VMA.
Fig. 2.

Metabolic pathways of norepinephrine. DOPGAL, 3,4-dihydroxyphenylglycol aldehyde; DOPEG, 3,4-dihydroxylphenylethylene glycol; DOMA, 3,4-dihydroxymandelic acid; MOPGAL, 3-methoxy-4-hydroxyphenylglycol aldehyde; MHPG, 3-methoxy-4-hydroxyphenylethylene glycol; VMA, vanillyl mandelic acid.
MAO function is highly critical for the regulation the intracellular redox state in neurons and other cells; indeed, one of the byproducts of MAO-mediated reaction, hydrogen peroxide, is a potent oxidizer which can trigger the formation of superoxide radicals and other reactive oxygen species, which can in turn induce mitochondrial and cytoplasmic damage. Under physiological conditions, the overall redox potential is kept in equilibrium by antioxidant enzymes, such as superoxide dismutase, catalase, and glutathione peroxidase; nevertheless, high concentrations of ammonia (the other by-product of the reaction) have been shown to decrease the activity of these enzymes and lead to the formation of superoxide radicals (Kosenko et al., 1997). The excess of oxidizing species in the central nervous system leads to permanent damages through death of neurons and glia. These mechanisms lay the theoretical foundations for the implication of MAO in the pathophysiology of certain neurodegenerative disorders, such as Parkinson’s disease (PD) and dementias (Danielczyk et al., 1988). In line with this concept, an increase in the activity of the isoenzyme MAO-B in platelets has been found in Alzheimer’s disease patients, leading to the proposal that this parameter may be an early biomarker for diagnosis of this condition (Grünblatt et al., 2005).
Finally, MAO serves a primary role in the degradation of primary, secondary, and some tertiary xenobiotic amines, which is particularly important to preventing their cardio- and neurotoxicity. A well-characterized example of these detrimental effects is the “cheese reaction,” a vasoconstrictive crisis (often lethal) caused by the absorption of sympathomimetic amines in fermented food (such as cheese, wine, etc.) following administration of irreversible MAO inhibitors (Anderson et al., 1993).
B. MAO Isoenzymes
In higher vertebrates, the MAO family comprises two isoenzymes, termed A and B, which, despite a substantial structural overlap, are remarkably different for substrate preference, inhibitor selectivity, anatomical distribution, and functional role in behavioral regulation.
The existence of multiple MAO isoenzymes was initially postulated to account for a number of experimental data indicating divergent neurochemical effects of different inhibitors. Indeed, MAO-A is selectively blocked by low doses of clorgyline (Johnston, 1968), while MAO-B is inhibited by nanomolar concentrations of (R)-deprenyl (Knoll and Magyar, 1972). The two isoenzymes can be separated electrophoretically (Shih and Eiduson, 1969; Youdim et al., 1969).
Subsequent studies showed that MAO-A had very high affinity for 5-HT (120 folds higher than MAO-B) and, to a lower degree, NE; by contrast, MAO-B preferred PEA and benzylamine as its substrates. The degradation of DA, tryptamine, and tyramine is mediated by both MAOs, but the relative contribution of each isoenzyme appears to vary greatly in relation to the species and the tissue under consideration. For example, DA metabolism is prevalently served by MAO-A in rodents and by MAO-B in humans and other primates (Garrick and Murphy, 1980; Fornai et al., 1999). The dichotomy between MAO-A and -B in terms of substrate preference is not absolute; in fact, in the absence of one isoenzyme, the other can deaminate a certain amount of its nonpreferred mono-amine substrates (Chen et al., 2004). This mechanism of partial compensation mechanism is fully revealed by the neurochemical outcomes of MAO deficiency in murine models (see below) and indicates the physiological significance of the presence of two isoenzymes in vivo. The unequivocal demonstration of the existence of two isoenzymes came in 1988, with the cloning of human MAO-A and MAO-B genes (Bach et al., 1988). Subsequent studies elucidated the structural configuration of both genes, revealing that they are located on the locus Xp11.23, in a tail-to-tail arrangement, with the 3′-coding sequences separated by about 50 kb (Ozelius et al., 1988; Lan et al., 1989a,b; Levy et al., 1989; Chen et al., 1992). MAO-A and MAO-B encode for two proteins of 527 and 520 amino acids, with molecular weights of 59.7 and 58.8 kDa, respectively. Interestingly, the two genes share ~70% sequence identity and an identical intron–exon organization, with 15 exons and 14 introns; these findings provided one of the first lines of evidence to define the structure and evolution of the isoenzymes, by suggesting that both genes derive from duplication of a common ancestor gene (Grimsby et al., 1991). In agreement with this interpretation, numerous phylogenetic studies have revealed the existence of only one MAO in early eukaryotes (Schilling and Lerch, 1995), invertebrates (Boutet et al., 2004), and teleost fish (Chen et al., 1994; Setini et al., 2005; Anichtchik et al., 2006). Conversely, the presence of two MAO isoenzymes can be traced back to anuran amphibians (Kobayashi et al., 1981); in frogs, MAO-A is predominant in the tadpole stage, while MAO-B expression increases through metamorphosis (Nicotra and Senatori, 1988), pointing at the possibility that the duplication of the gene may have been selected as an advantageous trait to maintain redox homeostasis in response to the development of lung-based respiration and the consequent hyperoxic shift. Interestingly, MAO-B has a much higher Km (lower affinity) for O2 (~ 250 μM) than MAO-A ~ (6 μM) (Edmondson et al., 2004). Given the role of monoamines in the regulation of cardiovascular function, the development of substrate-selective isoenzymes may have also been instrumental to withstand the new challenges posed by terrestrial life to blood circulation.
A more detailed insight in the structural characteristics of MAOs was afforded by several mutagenesis and chimerization studies (Bach et al., 1988; Gottowik et al., 1993; Wu et al., 1993; Chen et al., 1996; Geha et al., 2000), as well as by the crystallization of the two isoenzymes (Binda et al., 2002, 2003; Ma et al., 2004; De Colibus et al., 2005).
Nascent MAO polypeptidic chains undergo a number of posttranslational modifications; the best-characterized processes are the removal of the initiator methionine in MAO-B (but not in MAO-A) and the acetylation of the N-terminus in both molecules (methionine for MAO-A and serine for MAO-B) (Newton-Vinson et al., 2000; Li et al., 2002). Another critical modification is the covalent attachment of FAD to cysteinyl residues 406 in MAO-A and 397 in MAO-B; both amino acids are encoded by exon 12 of the respective gene, and their mutation ablates enzymatic activity (Wu et al., 1993). The coenzyme is attached by a thioether bond with the 8α-methylene group of its isoalloxazine ring (Kearney et al., 1971), and maintained in a position opposite to the entrance of the mono-amine-binding cavity within the active site (Edmondson et al., 2007). The substrate preference and inhibitor specificity appear to be conferred by a number of internal residues, such as Ile 335 in MAO-A and Tyr 326 in MAO-B (Geha et al., 2001).
Both proteins are anchored to the mitochondrial outer membrane through a transmembrane helix located within the carboxyl-terminal domain. In their membrane-bound conformations, human MAO-A and MAO-B are both dimeric (Ma et al., 2004; De Colibus et al., 2005; Binda et al., 2007; Upadhyay et al., 2008; Edmondson et al., 2009).
C. Anatomical Localization of MAO-A and -B
Although both isoenzymes are expressed in most tissues, only MAO-A is characteristically abundant in fibroblasts and placenta; in contrast, MAO-B is the only isoenzyme expressed in platelets and lymphocytes (Bond and Cundall, 1977; Donnelly and Murphy, 1977). MAO-A and -B are present in most brain regions; however, certain areas display only one isoenzyme. MAO-A is found mainly in DAergic and NEergic neurons; conversely, MAO-B is the only isoenzyme expressed in the cell bodies of 5-HTergic neurons (as well as in histaminergic neurons and astrocytes) (Konradi et al., 1989); the significance of this localization remains partially unclear, since 5-HTappears to be mainly metabolized by MAO-A in vivo. To account for this apparent mismatch, we have hypothesized that MAO-A protein may be translated in the cell body and segregated to the axon terminals (Bortolato et al., 2010); this hypothesis is supported by the discovery that MAO-B is absent from the mitochondria of the axon terminals (Arai et al., 2002), as well as by the documentation of MAO-A mRNA in the 5-HTergic cells (Luque et al., 1995, 1996; Jahng et al., 1997; Filipenko et al., 2002; Wylie et al., 2010). The proposed compartmentalization may facilitate the specific degradation of 5-HT in the synaptic terminal; further, the expression of MAO-B in the somata of 5-HTergic neurons may serve protective functions for 5-HT.
II. Phenotypical Outcomes of MAO-A Deficit
A. Clinical Findings
The serendipitous discovery of the mood-enhancing effects elicited by MAO pharmacological blockade (Fox and Gibas, 1953) was a historical breakthrough in the pharmacotherapy of mental disorders and gave impetus to the first investigations on the role of MAO in behavioral regulation. It was subsequently discovered that the antidepressant properties of MAO inhibitors were mainly due to the inactivation of MAO-A, which resulted in increased synaptic 5-HT concentrations (Sharp et al., 1997) and modifications of the firing rate of 5-HTergic neurons (Blier and de Montigny, 1985).
The significant side effects of MAO inhibitors, however, led to a progressive decline in the employment of these agents in the therapy of depression, in favor of other categories of antidepressants. The discussion of the antidepressant properties of MAO-A inhibitors and their therapeutic usage is beyond the scope of the present chapter; the interested reader is referred to Amrein et al. (1993) and Kennedy (1997).
Interest in the clinical implications of MAO was rekindled by a number of reports on the implications of its deficiency in atypical Norrie disease (ND) patients. This recessive X-linked disease is caused by loss-of-function mutations of NDP (Norrie disease pseudoglioma) gene, which encodes for norrin, a protein involved in the development and vascularization of the retina and inner ear. In affected males, total norrin deficiency results in congenital blindness, cataracts, and progressive deterioration of the iris; additionally, several patients experience progressive hearing loss and other abnormalities of the cardiovascular, respiratory, and digestive systems.
As a result of the close proximity of NDP (located in Xp11.4) and the two MAO genes, a relatively sizable contingent of ND patients are reported to harbor deletion of these genes. In particular, the deletion of MAO-A and MAO-B in ND patients is conducive to severe mental retardation, growth failure, alterations of sleep pattern, and autistic-like symptoms (Lan et al., 1989a,b; Sims et al., 1989a.b; Murphy et al., 1990; Collins et al., 1992).
Further insight into the phenotypical outcomes of selective MAO-A deficiency was gained with the discovery of a behavioral syndrome in eight males of a large Dutch kindred (Brunner et al., 1993a,b), characterized by borderline mental retardation and maladaptive regulation of impulsive aggression. The genetic defect was a point mutation in exon 8 of the MAO-A gene, resulting in the substitution of a glutamine codon (CAG) with a stop codon (TAG) at position 296 of the amino acid sequence. The main nosographic feature of the disorder was a high proclivity to engage in violent and antisocial behavior (including arson, attempted rape and murder, exhibitionism and voyeurism), often in response to minor stressors. The affected individuals also exhibited stereotyped hand movements and sleep disturbances. These alterations were paralleled by a set of abnormalities in the urinary concentrations of monoamine metabolites, including decreased content of 5-HIAA, HVA, and VMA and increased levels of 5-HT (fivefolds higher than the normal range) and normetanephrine (from COMT metabolism of NE). To the best of our knowledge, no other case of Brunner syndrome has been described in the medical literature to date, even despite specific attempts to identify the disorder in cohorts of aggressive individuals (Mejia et al., 2001).
The sequencing of MAO-A gene allowed the characterization of its variants (for a review, see Shih and Thompson, 1999) and their different influence in behavioral regulation; among the numerous allelic variants identified to date, four polymorphisms have been particularly studied as potential biomarkers/risk factors for psychiatric disorders:
MAO-A (CA)n, a dinucleotide repeat polymorphism in intron 2 (Black et al., 1991);
a 23 bp variable-number tandem repeat (VNTR) near exon 1 (Hinds et al., 1992);
Fnu4HI and EcoRV, two restriction fragment length polymorphisms (Lim et al., 1994);
MAO-A-uVNTR, a 30 bp VNTR polymorphism located 1.2 kb upstream of MAO-A transcription initiation site (Sabol et al., 1998).
Variants of the first three polymorphisms (localized in MAO-A gene) have been associated to higher susceptibility to several mental conditions; in particular, a robust association was found between bipolar disorder and MAO-A (CA)n and 23 bp-VNTR polymorphisms (Lim et al., 1994, 1995; Kawada et al., 1995; Rubinsztein et al., 1996; Preisig et al., 2000). This association, albeit not confirmed by few studies (Craddock et al., 1995; Muramatsu et al., 1997), was also supported by a meta-analysis study (Furlong et al., 1999).
The MAO-A-uVNTR promoter polymorphism has been extensively studied, in consideration of its well-characterized functional nature. Six MAO-A-uVNTR variants have been characterized based on a different number of repeats (2, 3, 3.5, 4, 5, and 6) (Huang et al., 2004); in particular, the 3-repeat (3R) and 4-repeat (4R) alleles are the most common in the population (Sabol et al., 1998; Deckert et al., 1999; Jonsson et al., 2000); of these, the 4R variant has been associated to higher transcriptional efficiency and enzymatic activity (Sabol et al., 1998; Deckert et al., 1999; Denney et al., 1999). In line with this finding, a number of studies have shown that 4R carriers have higher levels of 5-HIAA in the cerebrospinal fluid (Williams et al., 2003), as well as a higher prevalence of panic disorder and major depression (in females), with poor response to chronic fluoxetine treatment (Deckert et al., 1999; Yu et al., 2005).
The 3R variant, which has been found to result in lower MAO-A catalytic activity in fibroblasts, has been linked to a higher risk for behavioral traits related to Brunner syndrome symptoms, namely impulsive aggressiveness and antisocial personality (Samochowiec et al., 1999; Contini et al., 2006; Oreland et al., 2007; Buckholtz and Meyer-Lindenberg, 2008; Williams et al., 2009), as well as impaired stress response (Brummett et al., 2008), lower cognitive functioning (Cohen et al., 2003), and maladaptive emotional processing of affect (Lee and Ham, 2008). Interestingly, this variant has also been shown to influence the clinical course and severity of mental disorders; for example, 3R autistic children exhibit higher severity of the pathological manifestations (Cohen et al., 2003), lower levels of anxiety and attentional deficits (Roohi et al., 2009), and larger cortical volumes (Davis et al., 2008).
The behavioral changes associated to MAO-A-uVNTR polymorphic variants have been documented to be related to a number of morphological and functional differences between the brains of 3R and 4R carriers. In particular, several studies have shown that male individuals with the 3R haplotype exhibit morphological alterations of the orbitofrontal cortex (Meyer-Lindenberg et al., 2006; Cerasa et al., 2008, 2010), as well as functional abnormalities in several cortical and limbic regions, including prefrontal cortex, amygdala, and hippocampus (Meyer-Lindenberg et al., 2006; Passamonti et al., 2006).
Recent findings have challenged the association between MAO-A-uVNTR variants and MAO-A brain activity. For example, postmortem studies showed that, while the average MAO-A catalytic activity in brain samples from 4R carriers was higher than 3R, this difference was not significant (Balciuniene et al., 2002). Similarly, investigations conducted on populations of 3R and 4R male carriers with positron emission tomography [PET] for [11C]clorgyline revealed no significant difference in brain MAO-A activity between the two groups (Fowler et al., 2007; Alia-Klein et al., 2008). However, irrespective of the genetic components, MAO-A activity in cortical and subcortical brain regions was shown to be inversely correlated with the degree of self-reported aggression in men (Alia-Klein et al., 2008).
These data strongly suggest that polymorphic variants, rather than dictating MAO-A activity, may only confer a predisposition to a higher or lower baseline level of this index. Indeed, a large number of environmental factors have been shown to modify MAO-A expression and activity, including stress (Maura and Vaccari, 1975), diet changes (Jahng et al., 1998), tobacco smoking (Fowler et al., 1996), physical exercise (Morishima et al., 2006), social environment (Filipenko et al., 2002), and aging itself (Saura et al., 1994). Thus, the interaction between a genetic predisposition and specific environmental determinants could induce variations of MAO-A activity and increase the vulnerability to develop aggressive conduct and antisocial personality.
In line with this possibility, Caspi and coworkers reported that male 3R-carriers with a history of abuse during childhood had a significantly higher prevalence of aggressive behavior in adulthood than 3R-carriers with no history of early maltreatment or 4R-carriers with history of abuse (Caspi et al., 2002). This important finding has been confirmed by subsequent studies (Foley et al., 2004; Huang et al., 2004; Kim-Cohen et al., 2006; Edwards et al., 2010). Further, recent evidence shows that the effects of early stress on impulsivity are reduced by high levels of perceived parental care in individuals with the 3R-, but not the 4R, allelic variant (Kinnally et al., 2009). These results highlight the importance of gene × environment interactions in the pathophysiology of the conditions associated with low MAO-A activity.
B. Preclinical Findings
The analysis of the psychopathological implications of MAO-A deficiency and their neurobiological underpinnings is greatly limited by the rarity of Brunner syndrome and its elusive nosographic description (Hebebrand and Klug, 1995; Schuback et al., 1999). A useful experimental tool to partially obviate this limitation has been afforded by MAO-A knockout (KO) mice. The first line of these mutants was generated in the C3H/HeJ strain by the insertion of an interferon-β minigene into exon 2 of Maoa gene (Cases et al., 1995). More recently, another murine line has been developed in 129S6 background, carrying a spontaneous nonsense point mutation of the exon 8 (in a position close to that documented in Brunner syndrome patients) (Scott et al., 2008). Although strain differences may play a modulatory role in phenotypical manifestations, our results have shown that the neurochemical and behavioral phenotypes of both lines bear striking resemblances (Scott et al., 2008).
The selective loss of MAO-A enzymatic function leads to high levels of brain 5-HT and NE, as well as a broad spectrum of phenotypical aberrances, highly reminiscent of the symptoms described in Brunner syndrome. The most evident behavioral alteration in MAO-A KO mice consists in their elevated aggressiveness, toward both foreign mice and cage mates (Cases et al., 1995; Scott et al., 2008). Further, MAO-A KO mice display marked reduction in exploratory activity (Godar et al., 2011), low levels of depression-like behavior in the forced swim test (Cases et al., 1995) and high level of mnemonic retention of aversive events (Kim et al., 1997; Dubrovina et al., 2006).
Recent studies in our laboratory have begun to elucidate the potential psychopathological bases of the emotional alterations in MAO-A KO mice. In particular, we have observed that these mutants exhibit maladaptive defensive reactivity to different contextual cues. In particular, MAO-A KO mice display high levels of neophobia and antagonistic behavior in presence of novel neutral objects (particularly if introduced within a familiar context); conversely, they increase their level of exploration (with no aggressive or defensive postures) in the presence of cues associated with high danger, such as objects impregnated with predator urine or an anesthetized rat.
In line with this background, MAO-A deficient mice display exaggerated freezing reactions to relatively minor stressors (such as a mild footshock) (Kim et al., 1997), but reduced endocrine responses to major environmental stress, such as physical restraint, water deprivation, cold temperature, and chronic variable stress (Popova et al., 2006).
In conflict-based models of anxiety (which are based on the contrast of exploratory drive and neophobia), MAO-A KO mice did not display major behavioral alterations associated with anxiety-like behavior, but did show a reduction in risk-assessment postures (Popova et al., 2001; Godar et al., 2011). This phenomenon may be partially explained by the simultaneous decrease in avoidance/fear-related behaviors as well as approach/exploration in most contextual settings (Godar et al., 2011).
Taken together, these studies suggest that most behavioral alterations featured in MAO-A KO mice may depend on their inability to attune their responses to environmental inputs. In particular, their maladaptive responses are similar to the deficits in facial affect processing in schizophrenia and autism patients (Phillips et al., 1999; Bolte and Poustka, 2003; Dawson et al., 2004; Surguladze et al., 2006; Gur et al., 2007; Hall et al., 2008).
Although the assessment of the morphological characteristics in the brain of MAO-A KO mice is still incomplete, the most remarkable feature identified to date is the dysmorphogenesis of the barrel fields in layer IV of the somatosensory cortex (Cases et al., 1995). Barrel fields are the cortical representations of the mystacial vibrissae in the rodent snout, and their formation depends on the thalamocortical projections arising from ventrobasal thalamic nuclei (Erzurumlu and Jhaveri, 1990).
The impairment of barrel fields has been associated with alterations of perceptual processing, exploratory activity, and sensory integration (Hurwitz et al., 1990; Sanders et al., 2001; Dowman and Ben-Avraham, 2008; Straube et al., 2009), suggesting similar deficits in MAO-A KO mice. Nevertheless, we showed that these animals were able to recognize familiar objects both in the presence or absence of environmental light in a fashion comparable with their wild-type (WT) littermates (Godar et al., 2011). This finding challenges the possibility that the behavioral alterations observed in MAO-A KO mice may be strictly reflective of their deficits in vibrissal function.
The analysis of sensory modalities in both MAO-A KO lines has revealed impairments in acoustic reactivity (Cases et al., 1995), in line with defects in the auditory pathways (Thompson, 2008; Thompson and Thompson, 2009). Although MAO-A deficiency may also result in subtle developmental alterations of retinal projections (Upton et al., 1999), these deficits do not seem to affect the visual acuity of the mice, as assessed with the visual cliff paradigm (Godar et al., 2011). Similarly, MAO-A KO mice do not exhibit any overt change in olfactory discrimination (Godar et al., 2011).
Previous studies have shown that long-term treatment with MAO-A inhibitors in adult rodents induces a decrease in defensive behavior against predators (Griebel et al., 1998), and an enhancement in exploratory activity (Steckler et al., 2001). These alterations are distinctly different from those observed in MAO-A KO mice, highlighting the likely contribution of early developmental mechanisms in the alterations associated with MAO-A deficiency. Accordingly, early treatment with clorgyline and other MAO inhibitors was found to induce behavioral alterations and impairments in thalamocortical development similar to those observed in MAO-A KO mice (Whitaker-Azmitia et al., 1994; Boylan et al., 2000; Mejia et al., 2002). Moreover, the neurobehavioral alterations of MAO-A KO mice begin at very early stages, with intense head bobbing, prolonged righting, and trembling and delayed maturation of motor skills (Cases et al., 1995; Cazalets et al., 2000). In addition, MAO-A KO mice feature abnormalities of respiratory activity (Bou-Flores et al., 2000). Between postnatal days 11 and 16, MAO-A KO mice display hyperlocomotion, jumping, abnormal postures, and hyperreactivity to stimuli (Cases et al., 1995).
Several studies have shown that the sensorimotor cortex deficits in these animals are due to neurodevelopmental alterations based on the excessive 5-HT levels and 5-HT1B receptor hyperactivation in the first days of postnatal life (Cases et al., 1995; Vitalis et al., 1998; Salichon et al., 2001). In contrast, the morphological alterations of the respiratory centers in the medulla and the cervical phrenic motoneurons in MAO-A KO are reversed by 5-HT2A receptor inhibition (Bou-Flores et al., 2000).
III. Phenotypical Outcomes of MAO-B Deficit
A. Clinical Findings
Although the functional role of MAO-B in brain and behavioral regulation is more elusive than MAO-A, numerous lines of evidence point to its role in emotional regulation. The main MAO-B substrate, PEA, is widely regarded as an endogenous amphetamine, in view of its similar chemical structure and effects in vivo, which include increased alertness, euphoria, insomnia, and tremor (Baud et al., 1985; Zucchi et al., 2006). In line with this concept, numerous studies have highlighted a key role of this trace amine in the pathophysiology of schizophrenia and other neuropsychiatric disorders (Beckmann et al., 1983; Szymanski et al., 1987; O’Reilly et al., 1991; Berry, 2007).
Alterations of MAO-B activity and expression have been associated with a broad constellation of neuropsychiatric manifestations, including psychotic disorders, depression, alcoholism, impulsivity, and neurodegenerative diseases (Mann and Chiu, 1978; Adolfsson et al., 1980; Sandler et al., 1993). In addition to its well-characterized neuroprotective role in PD therapy, the prototypical MAO-B inhibitor selegiline has been shown to exert mood-enhancing and anxiolytic effects in depression (Mendlewicz and Youdim, 1983; Quitkin et al., 1984; Robinson et al., 2007) and other pathological conditions (Tariot et al., 1987; Tolbert and Fuller, 1996).
Most clinical studies on MAO-B function have focused on the activity of this enzyme in platelets, as these bodies can be easily collected and display exclusively this isoenzyme. Other advantages of this index lie in its high heritability (Oxenstierna et al., 1986; Pedersen et al., 1993), as well as in its potential association to brain MAO-B catalytic activity (af. Klintenberg et al., 2004). Rich evidence shows a robust correlation between low MAO-B platelet activity and a spectrum of psychological traits related to behavioral disinhibition, such as sensation-seeking and novelty-seeking personality, extraversion, poor impulse control, and proclivity to engage in risky behaviors (Buchsbaum et al., 1976; Fowler et al., 1980; von Knorring et al., 1984; Reist et al., 1990; for a review, see Oreland and Hallman, 1995). While the discovery that smoking can reduce MAO-B activity (Simpson et al., 1999; Hauptmann and Shih, 2001) partially tempered this notion, in view of the high prevalence of this habit among sensation-seekers, further studies confirmed that the association between low MAO-B activity and novelty-seeking remain even after controlling for this environmental factor (Ruchkin et al., 2005).
To the best of our knowledge, there have been no reports of clinical conditions characterized by selective MAO-B deficiency. However, in few cases of atypical ND with MAO-B deletion, the latter deficit was reported to result in increased urinary excretion of PEA, but no overt behavioral abnormalities or cognitive deficits (Berger et al., 1992; Lenders et al., 1996).
Several MAO-B polymorphic variants have been significantly associated with a number of psychological traits, such as negative emotionality (Dlugos et al., 2009), and neuropsychiatric conditions, such as attention-deficit hyperactivity disorder (Li et al., 2008; Ribasés et al., 2009). This association is particularly intriguing, as other authors have documented associations between low platelet MAO-B activity with attention-deficit hyperactivity (Shekim et al., 1986; Coccini et al., 2009; Nedic et al., 2010). Notably, sensation-seeking personality and poor impulse control are key features of ADHD psychopathology.
One of the best-characterized functional MAO-B polymorphisms consists in a single-base variation (A or G) in the intron 13 (Kurth et al., 1993); the A allele has been associated with lower MAO-B catalytic activity in platelets (Garpenstrand et al., 2000) and higher activity in the brain (Balciuniene et al., 2002). However, other studies have failed to detect any connection between this polymorphism and the enzymatic activity (Girmen et al., 1992).
An increase in MAO-B activity and/or expression has been associated with PD; accordingly, few studies have revealed associations between MAO-B polymorphisms and specific clusters of this disease (Kang et al., 2006; Bialecka et al., 2007). Nevertheless, the results on the potential connection between the polymorphism of intron 13 and PD remain controversial (Kurth et al., 1993; Ho et al., 1995; Costa et al., 1997).
B. Preclinical Findings
A complementary line of evidence on the phenotypical implications of MAO-B deficits has been provided by the generation of MAO-B KO mice (Grimsby et al., 1997), achieved by targeted insertion of a foreign neomycin resistance cassette in exon 6 of Maob gene. In this line of mice, the lack of MAO-B catalytic activity resulted in significantly higher levels of PEA in brain. However, levels of major monoamine neurotransmitters (5-HT, NE, and DA) were comparable with those in WT mice.
The initial studies aimed at the behavioral characterization of MAO-B KO mice did not highlight many overt behavioral and cognitive changes. For example, unlike MAO-A-deficient animals, MAO-B KO mice display no significant alteration of aggressive behavior. The most remarkable alterations were a reduced level of immobility in the forced swim test (Grimsby et al., 1997). This phenomenon was originally interpreted as an enhancement in stress responsiveness; however, recent studies have shown that MAO-B KO mice display low levels of restraint-induced hyperthermia, a typical parameter of stress reactivity (Bortolato et al., 2009). Taken together, these data suggest that the behavioral performance in MAO-B KO mice was likely reflective of their increased ability to counteract the stress induced by hazardous situations.
We recently documented that MAO-B KO mice display responses reminiscent of behavioral disinhibition, increased novelty-seeking, and reduced anxiety in a number of complementary behavioral paradigms aimed at capturing different aspects of emotional reactivity. For example, MAO-B KO mice displayed significant reduction in anxiety-like behaviors in an elevated plus maze, as well as in the defensive withdrawal, marble burying, hole-board. In addition, MAO-B KO mice displayed a high inclination to explore unfamiliar objects and displayed low novelty-induced grooming, suggesting an enhancement of novelty-seeking behavior (or reduction of neophobia) in these animals. These results are in agreement with the numerous findings on the correlation between low MAO-B platelet activity and novelty-seeking personality, and point to a causal link between the two phenomena.
Interestingly, the abnormal characteristics of MAO-B KO mice were best observed in the presence of several environmental adjustments, such as the reduction of environmental light, a strong anxiogenic factor in mice. This consideration suggests that the ablation of MAO-B may result in variations of personality, rather than actual pathological outcomes.
However, it is extremely likely that low MAO-B activity may be a key vulnerability factor for several conditions associated with sensation-seeking conduct. Future studies will have to evaluate what factors (both genetic and environmental) can interact with low MAO-B activity to induce pathological outcomes.
PEA has been implicated in the regulation of emotional responses, including exploratory activity, arousal, and behavioral reinforcement (Sabelli and Javaid, 1995). MAO-B KO mice display high levels of this monoamine particularly in striatum. Given the implication of PEA in the regulation of DA functions (Kuroki et al., 1990; Sotnikova et al., 2004) and the relevance of DA in behavioral disinhibition (Megens et al., 1992; Black et al., 2002; van Gaalen et al., 2006) and anxiolysis (Shabanov et al., 2005; Picazo et al., 2009), it is possible that DA may play an important role in the behavioral features of MAO-B KO mice. In line with this possibility, MAO-B KO mice have been shown to feature hypersensitivity of D1 receptor (Chen et al., 1999), which have been implicated in the motivational aspects of novelty-seeking behavior (Peters et al., 2007; Olsen and Winder, 2009).
IV. Phenotypical Outcomes of Combined MAO-A and MAO-B Deficit
A. Clinical Findings
As outlined above, the phenotypical outcomes of joint MAO-A and MAO-B deficiency could be initially surmised by comparisons between atypical ND patients with deletion of both genes and their counterparts with mutations restricted to NDP gene. These studies suggested that total congenital MAO deficit resulted in a spectrum of severe developmental deficits, with profound mental retardation and autistic-like behavior (Sims et al., 1989a,b; Murphy et al., 1990; Collins et al., 1992). The ultimate description of the consequences of total MAO deficiency, however, was recently provided by Whibley and colleagues, who reported the case of two male siblings carrying a 240 kb deletion in Xp11.3, which encompassed exons 2–15 of MAO-A and all exons of MAO-B, but no mutations of NDP or other adjacent genes (Whibley et al., 2010).
The two affected children were born to healthy, nonconsanguineous Caucasian parents and presented with a spectrum of severe abnormalities, including major developmental delay (with height and weight on very low percentiles), mental retardation, and stereotypical movements (hand-flapping and lip-smacking) akin to those featured in Rett syndrome and other pervasive developmental disorders (Whibley et al., 2010). Both brothers had exhibited several episodes of profound hypotonia since perinatal stages, which was typically resistant to phenobarbital, sodium valproate, and lamotrigine. One patient, who displayed minor dysmorphic features (inner canthal folds and an extra incisor) and EEG alterations, died at 5 years of age. His autopsy revealed small foci of perivascular calcification with loss of Purkinje cells in the cerebellum and neurons in the cortex (Whibley et al., 2010).
B. Preclinical Findings
Following the fortuitous discovery of a mouse with a spontaneous mutation in Maoa gene within the MAO-B KO colony (Chen et al., 2004), a colony of MAO-A/B KO mice was established, and their phenotypes were characterized. The lack of catalytic activity for either MAO isoenzymes was confirmed by a high increase in brain levels of all monoamines (5-HT: 850%; NE: 220%; DA: 170%; PEA: 1570%) in comparison to WT littermates. Interestingly, the magnitude of these enhancements is significantly greater than those observed in either MAO-A or MAO-B KO mice, suggesting that, in the absence of one isoenzyme, the other can partially overtake its catalytic role. This concept underscores that the alterations induced by joint MAO-A and MAO-B deficiency do not simply result from the summation of the aberrant phenotypes related to each mutation. In fact, it is likely that the abnormalities exhibited by MAO-A/B KO mice may be largely mediated by the exposure to extremely high concentrations of monoamines (in particular 5-HT) in early developmental stages. Preliminary phenotypical analyses of MAO-A/B KO mice revealed that these animals display major developmental alterations (not observed in either MAO-A or MAO-B KO mice), with weight and size at birth significantly lower than WT littermates. These animals also exhibit a complex array of behavioral abnormalities, including aberrant emotional response to novelty, high levels of anxiety-like behaviors in select tasks and low latency to attack in the resident-intruder paradigm (Chen et al., 2004). Recent studies conducted in our laboratory suggest that MAO-A/B KO mice may display alterations in emotional reactivity and informational processing similar (but more severe) to those observed in MAO-A KO mice (unpublished observations). In contrast, these mutants show no patent analogies with the behavioral changes of MAO-B KO mice, likely due to the phenotypical predominance of the defects associated with MAO-A deficiency.
V. Conclusions
Since the cloning of MAO-A and MAO-B genes in 1988, the employment of complementary approaches has paved the way for the elucidation of the structural and functional characteristics of these two genes and their products. Over the past few years, the advances in the analysis of the phenotypical variations associated with the polymorphisms of both genes has revealed a number of promising leads for the understanding of their role in mental illness and behavioral regulation. Novel findings in the phenotypes of MAO-deficient mice are highlighting novel potential implications of both isoenzymes in a broad spectrum of mental disorders, ranging from autism and anxiety to impulse-control disorders and ADHD. These studies will lay the foundation for future research on the neurobiological and neurochemical bases of these pathological conditions, as well as the role of gene × environment interactions in the vulnerability to several mental disorders.
Acknowledgments
The present study was supported by National Institute of Health grants R01MH39085 (to JCS) and R21HD070611 (to MB), as well as the Boyd and Elsie Welin Professorship (to JS), and the USC Zumberge Research Individual Grant (to MB). We are grateful to Kevin Chen for his unique contributions in MAO research, and particularly in generating various lines of MAO-deficient mice.
References
- Adolfsson R, Gottfries CG, Oreland L, Wiberg A, Winblad B. Increased activity of brain and platelet monoamine oxidase in dementia of Alzheimer type. Life Sci. 1980;27:1029–1034. doi: 10.1016/0024-3205(80)90025-9. [DOI] [PubMed] [Google Scholar]
- Alia-Klein N, Goldstein RZ, Kriplani A, Logan J, Tomasi D, Williams B, Telang F, Shumay E, Biegon A, Craig IW, Henn F, Wang GJ, et al. Brain monoamine oxidase A activity predicts trait aggression. J Neurosci. 2008;28:5099–5104. doi: 10.1523/JNEUROSCI.0925-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrein R, Hetzel W, Stabl M, Schmid-Burgk W. RIMA—a new concept in the treatment of depression with moclobemide. Int Clin Psychopharmacol. 1993;7(3–4):123–132. doi: 10.1097/00004850-199300730-00001. [DOI] [PubMed] [Google Scholar]
- Anderson MC, Hasan F, McCrodden JM, Tipton KF. Monoamine oxidase inhibitors and the cheese effect. Neurochem Res. 1993;18:1145–1149. doi: 10.1007/BF00978365. [DOI] [PubMed] [Google Scholar]
- Anichtchik O, Sallinen V, Peitsaro N, Panula P. Distinct structure and activity of monoamine oxidase in the brain of zebrafish (Danio rerio) J Comp Neurol. 2006;498:593–610. doi: 10.1002/cne.21057. [DOI] [PubMed] [Google Scholar]
- Arai R, Karasawa N, Kurokawa K, Kanai H, Horiike K, Ito A. Differential subcellular location of mitochondria in rat serotonergic neurons depends on the presence and the absence of monoamine oxidase type B. Neuroscience. 2002;114:825–835. doi: 10.1016/s0306-4522(02)00351-2. [DOI] [PubMed] [Google Scholar]
- Bach AW, Lan NC, Johnson DL, Abell CW, Bembenek ME, Kwan SW, Seeburg PH, Shih JC. cDNA cloning of human liver monoamine oxidase A and B: molecular basis of differences in enzymatic properties. Proc Natl Acad Sci USA. 1988;85:4934–4938. doi: 10.1073/pnas.85.13.4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balciuniene J, Emilsson L, Oreland L, Petterson U, Jazin EE. Investigation of the functional effect of monoamine oxidase polymorphisms in human brain. Hum Genet. 2002;110:1–7. doi: 10.1007/s00439-001-0652-8. [DOI] [PubMed] [Google Scholar]
- Baud P, Arbilla S, Cantrill RC, Scatton B, Langer SZ. Trace amines inhibit the electrically evoked release of [3H]acetylcholine from slices of rat striatum in the presence of pargyline: similarities between beta-phenylethylamine and amphetamine. J Pharmacol Exp Ther. 1985;235:220–229. [PubMed] [Google Scholar]
- Beck O, Borg S, Edman G, Fyro B, Oxenstierna G, Sedvall G. 5-hydroxytryptophol in human cerebrospinal fluid: conjugation, concentration gradient, relationship to 5-hydroxyindo-leacetic acid, and influence of hereditary factors. J Neurochem. 1984;43:58–61. doi: 10.1111/j.1471-4159.1984.tb06678.x. [DOI] [PubMed] [Google Scholar]
- Beckmann H, Waldmeier P, Lauber J, Gattaz WF. Phenylethylamine and monoamine metabolites in CSF of schizophrenics: effects of neuroleptic treatment. J Neural Transm. 1983;57:103–110. doi: 10.1007/BF01250052. [DOI] [PubMed] [Google Scholar]
- Berger W, Meindl A, van de Pol TJ, Cremers FP, Ropers HH, Döerner C, Monaco A, Bergen AA, Lebo R, Warburg M, et al. Isolation of a candidate gene for Norrie disease by positional cloning. Nat Genet. 1992;1(3):199–203. doi: 10.1038/ng0692-199. [DOI] [PubMed] [Google Scholar]
- Berry MD. The potential of trace amines and their receptors for treating neurological and psychiatric diseases. Rev Recent Clin Trials. 2007;2:3–19. doi: 10.2174/157488707779318107. [DOI] [PubMed] [Google Scholar]
- Bialecka M, Klodowska-Duda G, Honczarenko K, Gawrońska-Szklarz B, Opala G, Safranow K, DroŸdzik M. Polymorphisms of catechol-0-methyltransferase (COMT), monoamine oxidase B (MAOB), N-acetyltransferase 2 (NAT2) and cytochrome P450 2D6 (CYP2D6) gene in patients with early onset of Parkinson’s disease. Parkinsonism Relat Disord. 2007;13:224–229. doi: 10.1016/j.parkreldis.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Binda C, Newton-Vinson P, Hubálek F, Edmondson DE, Mattevi A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat Struct Biol. 2002;9:22–26. doi: 10.1038/nsb732. [DOI] [PubMed] [Google Scholar]
- Binda C, Li M, Hubalek F, Restelli N, Edmondson DE, Mattevi A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc Natl Acad Sci USA. 2003;100:9750–9755. doi: 10.1073/pnas.1633804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda C, Wang J, Pisani L, Caccia C, Carotti A, Salvati P, Edmondson DE, Mattevi A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: safinamide and coumarin analogs. J Med Chem. 2007;50:5848–5852. doi: 10.1021/jm070677y. [DOI] [PubMed] [Google Scholar]
- Black GC, Chen ZY, Craig IW, Powell JF. Dinucleotide repeat polymorphism at the MAOA locus. Nucleic Acids Res. 1991;19:689. doi: 10.1093/nar/19.3.689-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black KJ, Hershey T, Koller JM, Videen TO, Mintun MA, Price JL, Perlmutter JS. A possible substrate for dopamine-related changes in mood and behavior: prefrontal and limbic effects of a D3-preferring dopamine agonist. Proc Natl Acad Sci USA. 2002;99(26):17113–17118. doi: 10.1073/pnas.012260599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blier P, de Montigny C. Serotoninergic but not noradrenergic neurons in rat central nervous system adapt to long-term treatment with monoamine oxidase inhibitors. Neuroscience. 1985;16:949–955. doi: 10.1016/0306-4522(85)90107-1. [DOI] [PubMed] [Google Scholar]
- Bolte S, Poustka F. The recognition of facial affect in autistic and schizophrenic subjects and their first-degree relatives. Psychol Med. 2003;33(5):907–915. doi: 10.1017/s0033291703007438. [DOI] [PubMed] [Google Scholar]
- Bond PA, Cundall RL. Properties of monoamine oxidase (MAO) in human blood platelets, plasma. Clin Chim Acta. 1977;80:317–326. doi: 10.1016/0009-8981(77)90039-0. [DOI] [PubMed] [Google Scholar]
- Bortolato M, Godar SC, Davarian S, Chen K, Shih JC. Behavioral disinhibition and reduced anxiety-like behaviors in monoamine oxidase B-deficient mice. Neuropsychopharmacology. 2009;34(13):2746–2757. doi: 10.1038/npp.2009.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolato M, Chen K, Shih JC. The degradation of serotonin: role of MAO. In: Müller CP, Jacobs BL, editors. Handbook of the Behavioral Neurobiology of Serotonin. Elsevier; Amsterdam: 2010. pp. 203–218. [Google Scholar]
- Bou-Flores C, Lajard AM, Monteau R, De Maeyer E, Seif I, Lanoir J, Hilaire G. Abnormal phrenic motoneuron activity and morphology in neonatal monoamine oxidase A-deficient transgenic mice: possible role of a serotonin excess. J Neurosci. 2000;20:4646–4656. doi: 10.1523/JNEUROSCI.20-12-04646.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet I, Tanguy A, Moraga D. Molecular identification and expression of two non-P450 enzymes, monoamine oxidase A and flavin-containing monooxygenase 2, involved in phase I of xenobiotic biotransformation in the Pacific oyster, Crassostrea gigas. Biochim Biophys Acta. 2004;1679:29–36. doi: 10.1016/j.bbaexp.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Boylan CB, Bennett-Clarke CA, Crissman RS, Mooney RD, Rhoades RW. Clorgyline treatment elevates cortical serotonin and temporarily disrupts the vibrissae-related pattern in rat somatosensory cortex. J Comp Neurol. 2000;427:139–149. doi: 10.1002/1096-9861(20001106)427:1<139::aid-cne9>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Brummett BH, Boyle SH, Siegler IC, Kuhn CM, et al. HPA axis function in male caregivers: effect of the monoamine oxidase-A gene promoter (MAOA-uVNTR) Biol Psychol. 2008;79(2):250–255. doi: 10.1016/j.biopsycho.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner HG, Nelen M, Breakefield XO, Ropers HH, van Oost BA. Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science. 1993a;262:578–580. doi: 10.1126/science.8211186. [DOI] [PubMed] [Google Scholar]
- Brunner HG, Nelen MR, van Zandvoort P, Abeling NG, van Gennip AH, Wolters EC, Kuiper MA, Ropers HH, van Oost BA. X-linked borderline mental retardation with prominent behavioral disturbance: phenotype, genetic localization, and evidence for disturbed monoamine metabolism. Am J Hum Genet. 1993b;52:1032–1039. [PMC free article] [PubMed] [Google Scholar]
- Buchsbaum MS, Coursey RD, Murphy DL. The biochemical high-risk paradigm: behavioral and familial correlates of low platelet monoamine oxidase activity. Science. 1976;194:339–341. doi: 10.1126/science.968488. [DOI] [PubMed] [Google Scholar]
- Buckholtz JW, Meyer-Lindenberg A. MAO A and the neurogenetic architecture of human aggression. Trends Neurosci. 2008;31:120–129. doi: 10.1016/j.tins.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Cases O, Seif I, Grimsby J, Gaspar P, Chen K, Pournin S, Muller U, Aguet M, Babinet C, Shih JC. Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science. 1995;268:1763–1766. doi: 10.1126/science.7792602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, Taylor A, Poulton R. Role of genotype in the cycle of violence in maltreated children. Science. 2002;297:851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- Cazalets JR, Gardette M, Hilaire G. Locomotor network maturation is transiently delayed in the MAOA-deficient mouse. J Neurophysiol. 2000;83:2468–2470. doi: 10.1152/jn.2000.83.4.2468. [DOI] [PubMed] [Google Scholar]
- Cerasa A, Gioia MC, Labate A, Lanza P, Magariello A, Muglia M, Quattrone A. MAO A VNTR polymorphism and variation in human morphology: a VBM study. Neuroreport. 2008;19:1107–1110. doi: 10.1097/WNR.0b013e3283060ab6. [DOI] [PubMed] [Google Scholar]
- Cerasa A, Cherubini A, Quattrone A, Gioia MC, Magariello A, Muglia M, Manna I, Assogna F, Caltagirone C, Spalletta G. Morphological correlates of MAO A VNTR polymorphism: new evidence from cortical thickness measurement. Behav Brain Res. 2010;211:118–124. doi: 10.1016/j.bbr.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Chen K, Wu HF, Grimsby J, Shih JC. Cloning of a novel monoamine oxidase cDNA from trout liver. Mol Pharmacol. 1994;46:1226–1233. [PubMed] [Google Scholar]
- Chen K, Wu HF, Shih JC. Influence of C terminus on monoamine oxidase A and B catalytic activity. J Neurochem. 1996;66:797–803. doi: 10.1046/j.1471-4159.1996.66020797.x. [DOI] [PubMed] [Google Scholar]
- Chen L, He M, Sibille E, Thompson A, Sarnyai Z, Baker H, Shippenberg T, Toth M. Adaptive changes in postsynaptic dopamine receptors despite unaltered dopamine dynamics in mice lacking monoamine oxidase B. J Neurochem. 1999;73:647–655. doi: 10.1046/j.1471-4159.1999.0730647.x. [DOI] [PubMed] [Google Scholar]
- Chen K, Holschneider DP, Wu W, Rebrin I, Shih JC. A spontaneous point mutation produces monoamine oxidase A/B knock-out mice with greatly elevated monoamines and anxiety-like behavior. J Biol Chem. 2004;279:39645–39652. doi: 10.1074/jbc.M405550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Powell JF, Hsu YP, Breakefield XO, Craig IW. Organization of the human monoamine oxidase genes and long-range physical mapping around them. Genomics. 1992;14(1):75–82. doi: 10.1016/s0888-7543(05)80286-1. [DOI] [PubMed] [Google Scholar]
- Coccini T, Crevani A, Rossi G, Assandri F, Balottin U, Nardo RD, Manzo L. Reduced platelet monoamine oxidase type B activity and lymphocyte muscarinic receptor binding in unmedicated children with attention deficit hyperactivity disorder. Biomarkers. 2009;14:513–522. doi: 10.3109/13547500903144436. [DOI] [PubMed] [Google Scholar]
- Cohen IL, Liu X, Schutz C, White BN, et al. Association of autism severity with a monoamine oxidase A functional polymorphism. Clin Genet. 2003;64(3):190–197. doi: 10.1034/j.1399-0004.2003.00115.x. [DOI] [PubMed] [Google Scholar]
- Collins FA, Murphy DL, Reiss AL, Sims KB, Lewis JG, Freund L, Karoum F, Zhu D, Maumenee IH, Antonarakis SE. Clinical, biochemical, and neuropsychiatric evaluation of a patient with a contiguous gene syndrome due to a microdeletion Xp11.3 including the Norrie disease locus and monoamine oxidase (MAOA and MAOB) genes. Am J Med Genet. 1992;42:127–134. doi: 10.1002/ajmg.1320420126. [DOI] [PubMed] [Google Scholar]
- Consalvi V, Mardh G, Vallee BL. Human alcohol dehydrogenases and serotonin metabolism. Biochem Biophys Res Commun. 1986;139:1009–1016. doi: 10.1016/s0006-291x(86)80278-9. [DOI] [PubMed] [Google Scholar]
- Contini V, Marques FZ, Garcia CE, Hutz MH, Bau CH. MAOA-uVNTR polymorphism in a Brazilian sample: further support for the association with impulsive behaviors and alcohol dependence. Am J Med Genet B Neuropsychiatr Genet. 2006;141B(3):305–308. doi: 10.1002/ajmg.b.30290. [DOI] [PubMed] [Google Scholar]
- Costa P, Checkoway H, Levy D, Smith-Weller T, Franklin GM, Swanson PD, Costa LG. Association of a polymorphism in intron 13 of the monoamine oxidase B gene with Parkinson disease. Am J Med Genet. 1997;74:154–156. doi: 10.1002/(sici)1096-8628(19970418)74:2<154::aid-ajmg7>3.3.co;2-a. [DOI] [PubMed] [Google Scholar]
- Craddock N, Daniels J, Roberts E, Rees M, McGuffin P, Owen MJ. No evidence for allelic association between bipolar disorder and monoamine oxidase A gene polymorphisms. Am J Med Genet. 1995;60:322–324. doi: 10.1002/ajmg.1320600412. [DOI] [PubMed] [Google Scholar]
- Danielczyk W, Streifler M, Konradi C, Riederer P, Moll G. Platelet MAO-B activity and the psychopathology of Parkinson’s disease, senile dementia and multi-infarct dementia. Acta Psychiatr Scand. 1988;78:730–736. doi: 10.1111/j.1600-0447.1988.tb06412.x. [DOI] [PubMed] [Google Scholar]
- Davis LK, Hazlett HC, Librant AL, Nopoulos P, Sheffield VC, Piven J, Wassink TH. Cortical enlargement in autism is associated with a functional VNTR in the monoamine oxidase A gene. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1145–1151. doi: 10.1002/ajmg.b.30738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson G, Webb SJ, Carver L, Panagiotides H, et al. Young children with autism show atypical brain responses to fearful versus neutral facial expressions of emotion. Dev Sci. 2004;7(3):340–359. doi: 10.1111/j.1467-7687.2004.00352.x. [DOI] [PubMed] [Google Scholar]
- De Colibus L, Li M, Binda C, Lustig A, Edmondson DE, Mattevi A. Three-dimensional structure of human monoamine oxidase A (MAO A): relation to the structures of rat MAO A and human MAO B. Proc Natl Acad Sci USA. 2005;102:12684–12689. doi: 10.1073/pnas.0505975102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckert J, Catalano M, Syagailo YV, Bosi M, Okladnova O, Di Bella D, Nöthen MM, Maffei P, Franke P, Fritze J, Maier W, Propping P, et al. Excess of high activity monoamine oxidase A gene promoter alleles in female patients with panic disorder. Hum Mol Genet. 1999;8:621–624. doi: 10.1093/hmg/8.4.621. [DOI] [PubMed] [Google Scholar]
- Denney RM, Koch H, Craig IW. Association between monoamine oxidase A activity in human male skin fibroblasts and genotype of the MAO-A promoter-associated variable number tandem repeat. Hum Genet. 1999;105:542–551. doi: 10.1007/s004399900183. [DOI] [PubMed] [Google Scholar]
- Despopoulos A, Weissbach H. Renal metabolism of 5-hydroxyindolacetic acid. Am J Physiol. 1957;189:548–550. doi: 10.1152/ajplegacy.1957.189.3.548. [DOI] [PubMed] [Google Scholar]
- Dlugos AM, Palmer AA, de Wit H. Negative emotionality: monoamine oxidase B gene variants modulate personality traits in healthy humans. J Neural Transm. 2009;116:1323–1334. doi: 10.1007/s00702-009-0281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CH, Murphy DL. Substrate- and inhibitor-related characteristics of human platelet. Biochem Pharmacol. 1977;26:853–858. doi: 10.1016/0006-2952(77)90398-7. [DOI] [PubMed] [Google Scholar]
- Dowman R, Ben-Avraham D. An artificial neural network model of orienting attention toward threatening somatosensory stimuli. Psychophysiology. 2008;45(2):229–239. doi: 10.1111/j.1469-8986.2007.00614.x. [DOI] [PubMed] [Google Scholar]
- Dubrovina NI, Popova NK, Gilinskii MA, Tomilenko RA, Seif I. Acquisition and extinction of a conditioned passive avoidance reflex in mice with genetic knockout of monoamine oxidase A. Neurosci Behav Physiol. 2006;36:335–339. doi: 10.1007/s11055-006-0022-z. [DOI] [PubMed] [Google Scholar]
- Edmondson DE, Mattevi A, Binda C, Li M, Hubálek F. Structure and mechanism of monoamine oxidase. Curr Med Chem. 2004;11:1983–1993. doi: 10.2174/0929867043364784. [DOI] [PubMed] [Google Scholar]
- Edmondson DE, Binda C, Mattevi A. Structural insights into the mechanism of amine oxidation by monoamine oxidases A and B. Arch Biochem Biophys. 2007;464:269–276. doi: 10.1016/j.abb.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson DE, Binda C, Wang J, Upadhyay AK, Mattevi A. Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry. 2009;48:4220–4230. doi: 10.1021/bi900413g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AC, Dodge KA, Latendresse SJ, Lansford JE, Bates JE, Pettit GS, Budde JP, Goate AM, Dick DM. MAOA-uVNTR and early physical discipline interact to influence delinquent behavior. J Child Psychol Psychiatry. 2010;51(6):679–687. doi: 10.1111/j.1469-7610.2009.02196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erzurumlu RS, Jhaveri S. Thalamic axons confer a blueprint of the sensory periphery onto the developing rat somatosensory cortex. Brain Res Dev Brain Res. 1990;56:229–234. doi: 10.1016/0165-3806(90)90087-f. [DOI] [PubMed] [Google Scholar]
- Feldstein A, Williamson O. 5-Hydroxytryptamine metabolism in rat brain and liver homogenates. Br J Pharmacol. 1968;34:38–42. doi: 10.1111/j.1476-5381.1968.tb07948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipenko ML, Beilina AG, Alekseyenko OV, Dolgov VV, Kudryavtseva NN. Repeated experience of social defeats increases serotonin transporter and monoamine oxidase A mRNA levels in raphe nuclei of male mice. Neurosci Lett. 2002;321:25–28. doi: 10.1016/s0304-3940(01)02495-8. [DOI] [PubMed] [Google Scholar]
- Foley DL, Eaves LJ, Wormley B, Silberg JL, et al. Childhood adversity, monoamine oxidase a genotype, and risk for conduct disorder. Arch Gen Psychiatry. 2004;61(7):738–744. doi: 10.1001/archpsyc.61.7.738. [DOI] [PubMed] [Google Scholar]
- Fornai F, Chen K, Giorgi FS, Gesi M, Alessandri MG, Shih JC. Striatal dopamine metabolism in monoamine oxidase B-deficient mice: a brain dialysis study. J Neurochem. 1999;73:2434–2440. doi: 10.1046/j.1471-4159.1999.0732434.x. [DOI] [PubMed] [Google Scholar]
- Fowler CJ, von Knorring L, Oreland L. Platelet monoamine oxidase activity in sensation seekers. Psychiatry Res. 1980;3(3):273–279. doi: 10.1016/0165-1781(80)90057-8. [DOI] [PubMed] [Google Scholar]
- Fowler JS, Volkow ND, Wang GJ, Pappas N, Logan J, Shea C, Alexoff D, MacGregor RR, Schlyer DJ, Zezulkova I, Wolf AP. Brain monoamine oxidase A inhibition in cigarette smokers. Proc Natl Acad Sci USA. 1996;93:14065–14069. doi: 10.1073/pnas.93.24.14065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler JS, Alia-Klein N, Kriplani A, Logan J, Williams B, Zhu W, Craig IW, Telang F, Goldstein R, Volkow ND, Vaska P, Wang GJ. Evidence that brain MAO A activity does not correspond to MAO A genotype in healthy male subjects. Biol Psychiatry. 2007;62:355–358. doi: 10.1016/j.biopsych.2006.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox HH, Gibas JT. Synthetic tuberculostats. V Alkylidene derivatives of isonicotinylhydrazine. J Org Chem. 1953;18:983–989. [Google Scholar]
- Furlong RA, Ho L, Rubinsztein JS, Walsh C, Paykel ES, Rubinsztein DC. Analysis of the monoamine oxidase A (MAOA) gene in bipolar affective disorder by association studies, meta-analyses, and sequencing of the promoter. Am J Med Genet. 1999;88:398–406. [PubMed] [Google Scholar]
- Garpenstrand H, Ekblom J, Forslund K, Rylander G, Oreland L. Platelet monoamine oxidase activity is related to MAOB intron 13 genotype. J Neural Transm. 2000;107:523–530. doi: 10.1007/s007020070075. [DOI] [PubMed] [Google Scholar]
- Garrick NA, Murphy DL. Species differences in the deamination of dopamine and other substrates for monoamine oxidase in brain. Psychopharmacology. 1980;72:27–33. doi: 10.1007/BF00433804. [DOI] [PubMed] [Google Scholar]
- Geha RM, Chen K, Shih JC. Phe(208) and Ile(199) in human monoamine oxidase A and B do not determine substrate and inhibitor specificities as in rat. J Neurochem. 2000;75:1304–1309. doi: 10.1046/j.1471-4159.2000.751304.x. [DOI] [PubMed] [Google Scholar]
- Geha RM, Rebrin I, Chen K, Shih JC. Substrate and inhibitor specificities for human monoamine oxidase A and B are influenced by a single amino acid. J Biol Chem. 2001;276:9877–9882. doi: 10.1074/jbc.M006972200. [DOI] [PubMed] [Google Scholar]
- Girmen AS, Baenziger J, Hotamisligil GS, Konradi C, Shalish C, Sullivan JL, Breakefield XO. Relationship between platelet monoamine oxidase B activity and alleles at the MAOB locus. J Neurochem. 1992;59:2063–2066. doi: 10.1111/j.1471-4159.1992.tb10095.x. [DOI] [PubMed] [Google Scholar]
- Godar SC, Bortolato M, Frau R, Dousti M, Chen K, Shih JC. Maladaptive defensive behaviours in monoamine oxidase A-deficient mice. Int J Neuropsychopharmacol. 2011 doi: 10.1017/S1461145710001483. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottowik J, Cesura AM, Malherbe P, Lang G, Da Prada M. Characterisation of wild-type and mutant forms of human monoamine oxidase A and B expressed in a mammalian cell line. FEBS Lett. 1993;317:152–156. doi: 10.1016/0014-5793(93)81512-x. [DOI] [PubMed] [Google Scholar]
- Griebel G, Curet O, Perrault G, Sanger DJ. Behavioral effects of phenelzine in an experimental model for screening anxiolytic and anti-panic drugs: correlation with changes in monoamine-oxidase activity and monoamine levels. Neuropharmacology. 1998;37(7):927–935. doi: 10.1016/s0028-3908(98)00077-x. [DOI] [PubMed] [Google Scholar]
- Grimsby J, Chen K, Wang LJ, Lan NC, Shih JC. Human monoamine oxidase A and B genes exhibit identical exon-intron organization. Proc Natl Acad Sci USA. 1991;88:3637–3641. doi: 10.1073/pnas.88.9.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsby J, Toth M, Chen K, Kumazawa T, Klaidman L, Adams JD, Karoum F, Gal J, Shih JC. Increased stress response and b-phenylethylamine in MAOB-deficient mice. Nat Genet. 1997;17:206–210. doi: 10.1038/ng1097-206. [DOI] [PubMed] [Google Scholar]
- Grünblatt E, Schlösser R, Fischer P, Fischer MO, Li J, Koutsilieri E, Wichart I, Sterba N, Rujescu D, Möller HJ, Adamcyk W, Dittrich B, et al. Oxidative stress related markers in the “VITA” and the centenarian projects. Neurobiol Aging. 2005;26:429–438. doi: 10.1016/j.neurobiolaging.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Gur RE, Loughead J, Kohler CG, Elliott MA, et al. Limbic activation associated with misidentification of fearful faces and flat affect in schizophrenia. Arch Gen Psychiatry. 2007;64(12):1356–1366. doi: 10.1001/archpsyc.64.12.1356. [DOI] [PubMed] [Google Scholar]
- Hall J, Whalley HC, McKirdy JW, Romaniuk L, et al. Overactivation of fear systems to neutral faces in schizophrenia. Biol Psychiatry. 2008;64(1):70–73. doi: 10.1016/j.biopsych.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Hauptmann N, Shih JC. 2-Naphthylamine, a compound found in cigarette smoke, decreases both monoamine oxidase A and B catalytic activity. Life Sci. 2001;68:1231–1241. doi: 10.1016/s0024-3205(00)01022-5. [DOI] [PubMed] [Google Scholar]
- Hebebrand J, Klug B. Specification of the phenotype required for men with monoamine oxidase type A deficiency. Hum Genet. 1995;96(3):372–376. doi: 10.1007/BF00210430. [DOI] [PubMed] [Google Scholar]
- Helander A, Beck O, Jacobsson G, Lowenmo C, Wikstrom T. Time course of ethanol-induced changes in serotonin metabolism. Life Sci. 1993;53:847–855. doi: 10.1016/0024-3205(93)90507-y. [DOI] [PubMed] [Google Scholar]
- Hinds HL, Hendriks RW, Craig IW, Chen ZY. Characterization of a highly polymorphic region near the first exon of the human MAOA gene containing a GT dinucleotide and a novel VNTR motif. Genomics. 1992;13(3):896–897. doi: 10.1016/0888-7543(92)90181-q. [DOI] [PubMed] [Google Scholar]
- Ho SL, Kapadi AL, Ramsden DB, Williams AC. An allelic association study of monoamine oxidase B in Parkinson’s disease. Ann Neurol. 1995;37:403–405. doi: 10.1002/ana.410370318. [DOI] [PubMed] [Google Scholar]
- Huang Y-y, Cate SP, Battistuzzi C, Oquendo MA, Brent D, Mann JJ. An association between a functional polymorphism in the monoamine oxidase A gene promoter, impulsive traits and early abuse experiences. Neuropsychopharmacology. 2004;29:1498–1505. doi: 10.1038/sj.npp.1300455. [DOI] [PubMed] [Google Scholar]
- Hurwitz BE, Dietrich WD, McCabe PM, Watson BD, et al. Sensory-motor deficit and recovery from thrombotic infarction of the vibrissal barrel-field cortex. Brain Res. 1990;512(2):210–220. doi: 10.1016/0006-8993(90)90628-o. [DOI] [PubMed] [Google Scholar]
- Jahng JW, Houpt TA, Wessel TC, Chen K, Shih JC, Joh TH. Localization of monoamine oxidase A and B mRNA in the rat brain by in situ hybridization. Synapse. 1997;25:30–36. doi: 10.1002/(SICI)1098-2396(199701)25:1<30::AID-SYN4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Jahng JW, Houpt TA, Joh TH, Son JH. Differential expression of monoamine oxidase A, serotonin transporter, tyrosine hydroxylase and norepinephrine transporter mRNA by anorexia mutation and food deprivation. Dev Brain Res. 1998;107:241–246. doi: 10.1016/s0165-3806(98)00013-3. [DOI] [PubMed] [Google Scholar]
- Johnston JP. Some observations upon a new inhibitor of monoamine oxidase in brain tissue. Biochem Pharmacol. 1968;17:1285–1297. doi: 10.1016/0006-2952(68)90066-x. [DOI] [PubMed] [Google Scholar]
- Jonsson EG, Norton N, Gustavsson JP, Oreland L, Owen MJ, Sedvall GC. A promoter polymorphism in the monoamine oxidase A gene and its relationships to monoamine metabolite concentrations in CSF of healthy volunteers. J Psychiatr Res. 2000;34:239–244. doi: 10.1016/s0022-3956(00)00013-3. [DOI] [PubMed] [Google Scholar]
- Kang SJ, Scott WK, Li YJ, Hauser MA, van der Walt JM, Fujiwara K, Mayhew GM, West SG, Vance JM, Martin ER. Family-based case-control study of MAOA and MAOB polymorphisms in Parkinson disease. Mov Disord. 2006;21:2175–2180. doi: 10.1002/mds.21151. [DOI] [PubMed] [Google Scholar]
- Kawada Y, Hattori M, Dai XY, Nanko S. Possible association between monoamine oxidase A gene and bipolar affective disorder. Am J Hum Genet. 1995;56:335–336. [PMC free article] [PubMed] [Google Scholar]
- Kearney EB, Salach JI, Walker WH, Seng RL, Kenney W, Zeszotek E, Singer TP. The covalently-bound flavin of hepatic monoamine oxidase. 1 Isolation and sequence of a flavin peptide and evidence for binding at the 8alpha position. Eur J Biochem. 1971;24:321–327. doi: 10.1111/j.1432-1033.1971.tb19689.x. [DOI] [PubMed] [Google Scholar]
- Kennedy SH. Continuation and maintenance treatments in major depression: the neglected role of monoamine oxidase inhibitors. J Psychiatry Neurosci. 1997;22:127–131. [PMC free article] [PubMed] [Google Scholar]
- Kinnally EL, Huang YY, Haverly R, Burke AK, Galfalvy H, Brent DP, Oquendo MA, Mann JJ. Parental care moderates the influence of MAOA-uVNTR genotype and childhood stressors on trait impulsivity and aggression in adult women. Psychiatr Genet. 2009;19(3):126–133. doi: 10.1097/YPG.0b013e32832a50a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Shih JC, Chen K, Chen L, Bao S, Maren S, Anagnostaras SG, Fanselow MS, De Maeyer E, Seif I, Thompson RF. Selective enhancement of emotional, but not motor, learning in monoamine oxidase A-deficient mice. Proc Natl Acad Sci USA. 1997;94(11):5929–5933. doi: 10.1073/pnas.94.11.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Cohen J, Caspi A, Taylor A, Williams B, Newcombe R, Craig IW, Moffitt TE. MAOA, maltreatment, and gene-environment interaction predicting children’s mental health: new evidence and a meta-analysis. Mol Psychiatry. 2006;11(10):903–913. doi: 10.1038/sj.mp.4001851. [DOI] [PubMed] [Google Scholar]
- Klinteberg B, von Knorring L, Oreland L. On the psychobiology of impulsivity. In: Stelmack RM, editor. On the psychobiology of personality: essays in honour of Marvin Zuckerman. Elsevier; Amsterdam: 2004. pp. 455–478. [Google Scholar]
- Knoll J, Magyar K. Some puzzling pharmacological effects of monoamine oxidase inhibitors. Adv Biochem Psychopharmacol. 1972;5:393–408. [PubMed] [Google Scholar]
- Kobayashi S, Takahara K, Kamijo K. Monoamine oxidase in frog liver and brain. Comp Biochem Physiol C. 1981;69:179–183. doi: 10.1016/0306-4492(81)90126-x. [DOI] [PubMed] [Google Scholar]
- Konradi C, Kornhuber J, Froelich L, Fritze J, Heinsen H, Beckmann H, Schulz E, Riederer P. Demonstration of monoamine oxidase-A and -B in the human brainstem by a histochemical technique. Neuroscience. 1989;33:383–400. doi: 10.1016/0306-4522(89)90218-2. [DOI] [PubMed] [Google Scholar]
- Kosenko E, Kaminsky Y, Kaminsky A, Valencia M, Lee L, Hermenegildo C, Felipo V. Superoxide production and antioxidant enzymes in ammonia intoxication in rats. Free Radic Res. 1997;27:637–644. doi: 10.3109/10715769709097867. [DOI] [PubMed] [Google Scholar]
- Kurth JH, Kurth MC, Poduslo SE, Schwankhaus JD. Association of a monoamine oxidase B allele with Parkinson’s disease. Ann Neurol. 1993;33:368–372. doi: 10.1002/ana.410330406. [DOI] [PubMed] [Google Scholar]
- Kuroki T, Tsutsumi T, Hirano M, Matsumoto T, Tatebayashi Y, Nishiyama K, Uchimura H, Shiraishi A, Nakahara T, Nakamura K. Behavioral sensitization to beta-phenylethylamine (PEA): enduring modifications of specific dopaminergic neuron systems in the rat. Psychopharmacology (Berl) 1990;102(1):5–10. doi: 10.1007/BF02245736. [DOI] [PubMed] [Google Scholar]
- Lan NC, Chen CH, Shih JC. Expression of functional human monoamine oxidase A and B cDNAs in mammalian cells. J Neurochem. 1989a;52:1652–1654. doi: 10.1111/j.1471-4159.1989.tb09223.x. [DOI] [PubMed] [Google Scholar]
- Lan NC, Heinzmann C, Gal A, Klisak I, Orth U, Lai E, Grimsby J, Sparkes RS, Mohandas T, Shih JC. Human monoamine oxidase A and B genes map to Xp 11.23 and are deleted in a patient with Norrie disease. Genomics. 1989b;4:552–559. doi: 10.1016/0888-7543(89)90279-6. [DOI] [PubMed] [Google Scholar]
- Lee BT, Ham BJ. Monoamine oxidase A-uVNTR genotype affects limbic brain activity in response to affective facial stimuli. Neuroreport. 2008;19(5):515–519. doi: 10.1097/WNR.0b013e3282f94294. [DOI] [PubMed] [Google Scholar]
- Lenders JW, Eisenhofer G, Abeling NG, Berger W, Murphy DL, Konings CH, Wagemakers LM, Kopin IJ, Karoum F, van Gennip AH, Brunner HG. Specific genetic deficiencies of the A and B isoenzymes of monoamine oxidase are characterized by distinct neurochemical and clinical phenotypes. J Clin Invest. 1996;97:1010–1019. doi: 10.1172/JCI118492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy ER, Powell JF, Buckle VJ, Hsu YP, Breakefield XO, Craig IW. Localization of human monoamine oxidase-A gene to Xp11.23–11.4 by in situ hybridization: implications for Norrie disease. Genomics. 1989;5(2):368–370. doi: 10.1016/0888-7543(89)90072-4. [DOI] [PubMed] [Google Scholar]
- Li M, Hubálek F, Newton-Vinson P, Edmondson DE. High-level expression of human liver monoamine oxidase A in Pichia pastoris: comparison with the enzyme expressed in Saccharomyces cerevisiae. Protein Expr Purif. 2002;24:152–162. doi: 10.1006/prep.2001.1546. [DOI] [PubMed] [Google Scholar]
- Li J, Wang Y, Hu S, Zhou R, Yu X, Wang B, Guan L, Yang L, Zhang F, Faraone SV. The monoamine oxidase B gene exhibits significant association to ADHD. Am J Med Genet B Neuropsychiatr Genet. 2008;147:370–374. doi: 10.1002/ajmg.b.30606. [DOI] [PubMed] [Google Scholar]
- Lim LC, Powell JF, Murray R, Gill M. Monoamine oxidase A gene and bipolar affective disorder. Am J Hum Genet. 1994;54:1122–1124. [PMC free article] [PubMed] [Google Scholar]
- Lim LC, Powell J, Sham P, Castle D, Hunt N, Murray R, Gill M. Evidence for genetic association between alleles of monoamine oxidase A gene and bipolar affective disorder. Am J Med Genet. 1995;1995(60):325–331. doi: 10.1002/ajmg.1320600413. [DOI] [PubMed] [Google Scholar]
- Luque JM, Kwan SW, Abell CW, Da Prada M, Richards JG. Cellular expression of mRNAs encoding monoamine oxidases A and B in the rat central nervous system. J Comp Neurol. 1995;363:665–680. doi: 10.1002/cne.903630410. [DOI] [PubMed] [Google Scholar]
- Luque JM, Bleuel Z, Hendrickson A, Richards JG. Detection of MAO-A and MAO-B mRNAs in monkey brainstem by cross-hybridization with human oligonucleotide probes. Brain Res Mol Brain Res. 1996;36:357–360. doi: 10.1016/0169-328x(96)88407-5. [DOI] [PubMed] [Google Scholar]
- Ma J, Yoshimura M, Yamashita E, Nakagawa A, Ito A, Tsukihara T. Structure of rat monoamine oxidase A and its specific recognitions for substrates and inhibitors. J Mol Biol. 2004;338:103–114. doi: 10.1016/j.jmb.2004.02.032. [DOI] [PubMed] [Google Scholar]
- Mann J, Chiu E. Platelet monoamine oxidase activity in Huntington’s chorea. J Neurol Neurosurg Psychiatry. 1978;41:809–812. doi: 10.1136/jnnp.41.9.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maura G, Vaccari A. Relationships between age of submission to environmental stress, and monoamine oxidase activity in rats. Experientia. 1975;31:191–193. doi: 10.1007/BF01990699. [DOI] [PubMed] [Google Scholar]
- Megens AA, Niemegeers CJ, Awouters FH. Behavioral disinhibition and depression in amphetaminized rats: a comparison of risperidone, ocaperidone and haloperidol. J Pharmacol Exp Ther. 1992;260(1):160–167. [PubMed] [Google Scholar]
- Mejia JM, Ervin FR, Palmour RM, Tremblay RE. Aggressive behavior and Brunner syndrome: no evidence for the C936T mutation in a population sample. Am J Med Genet. 2001;105:396–397. doi: 10.1002/ajmg.1356. [DOI] [PubMed] [Google Scholar]
- Mejia JM, Ervin FR, Baker GB, Palmour RM. Monoamine oxidase inhibition during brain development induces pathological aggressive behavior in mice. Biol Psychiatry. 2002;52:811–821. doi: 10.1016/s0006-3223(02)01418-x. [DOI] [PubMed] [Google Scholar]
- Mendlewicz J, Youdim MB. L-Deprenil, a selective monoamine oxidase type B inhibitor, in the treatment of depression: a double blind evaluation. Br J Psychiatry. 1983;142:508–511. doi: 10.1192/bjp.142.5.508. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Buckholtz JW, Kolachana BR, Hariri AR, Pezawas L, Blasi G, Wabnitz A, Honea R, Verchinski B, Callicott JH, Egan M, Matty V, et al. Neural mechanisms of genetic risk for impulsivity and violence in humans. Proc Natl Acad Sci USA. 2006;103:6269–6274. doi: 10.1073/pnas.0511311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima M, Harada N, Hara S, Sano A, Seno H, Takahashi A, Morita Y, Nakaya Y. Monoamine oxidase A activity and norepinephrine level in hippocampus determine hyperwheel running in SPORTS rats. Neuropsychopharmacology. 2006;31:2627–2638. doi: 10.1038/sj.npp.1301028. [DOI] [PubMed] [Google Scholar]
- Muramatsu T, Matsushita S, Kanba S, Higuchi S, Manki H, Suzuki E, Asai M. Monoamine oxidase genes polymorphisms and mood disorder. Am J Med Genet. 1997;74:494–496. [PubMed] [Google Scholar]
- Murphy DL, Sims KB, Karoum F, de la Chapelle A, Norio R, Sankila EM, Breakefield XO. Marked amine and amine metabolite changes in Norrie disease patients with an X-chromosomal deletion affecting monoamine oxidase. J Neurochem. 1990;54:242–247. doi: 10.1111/j.1471-4159.1990.tb13307.x. [DOI] [PubMed] [Google Scholar]
- Nedic G, Pivac N, Hercigonja DK, Jovancevic M, Curkovic KD, Muck-Seler D. Platelet monoamine oxidase activity in children with attention-deficit/hyperactivity disorder. Psychiatry Res. 2010;175:252–255. doi: 10.1016/j.psychres.2009.08.013. [DOI] [PubMed] [Google Scholar]
- Newton-Vinson P, Hubalek F, Edmondson DE. High-level expression of human liver monoamine oxidase B in Pichia pastoris. Protein Expr Purif. 2000;20:334–345. doi: 10.1006/prep.2000.1309. [DOI] [PubMed] [Google Scholar]
- Nicotra A, Senatori O. Changes in monoamine oxidase activity by mitochondria isolated from late embryos of Bufo bufo. Comp Biochem Physiol C. 1988;89:5–9. doi: 10.1016/0742-8413(88)90138-7. [DOI] [PubMed] [Google Scholar]
- Obata T. Semicarbazide-sensitive amine oxidase (SSAO) in the brain. Neurochem Res. 2002;27:263–268. doi: 10.1023/a:1014911209925. [DOI] [PubMed] [Google Scholar]
- O’Brien PJ, Siraki AG, Shangari N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit Rev Toxicol. 2005;35:609–662. doi: 10.1080/10408440591002183. [DOI] [PubMed] [Google Scholar]
- Olsen CM, Winder DG. Operant sensation seeking engages similar neural substrates to operant drug seeking in C57 mice. Neuropsychopharmacology. 2009;34:1685–1694. doi: 10.1038/npp.2008.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly R, Davis BA, Durden DA, Thorpe L, Machnee H, Boulton AA. Plasma phenylethylamine in schizophrenic patients. Biol Psychiatry. 1991;30:145–150. doi: 10.1016/0006-3223(91)90168-l. [DOI] [PubMed] [Google Scholar]
- Oreland L, Hallman J. The correlation between platelet MAO activity and personality: short review of findings and a discussion on possible mechanisms. Prog Brain Res. 1995;106:77–84. doi: 10.1016/s0079-6123(08)61204-2. [DOI] [PubMed] [Google Scholar]
- Oreland L, Nilsson K, Damberg M, Hallman J. Monoamine oxidases: activities, genotypes and the shaping of behaviour. J Neural Transm. 2007;114:817–822. doi: 10.1007/s00702-007-0694-8. [DOI] [PubMed] [Google Scholar]
- Oxenstierna G, Edman G, Iselius L, Oreland L, Ross SB, Sedvall G. Concentrations of monoamine metabolites in the cerebrospinal fluid of twins and unrelated individuals—a genetic study. J Psychiatr Res. 1986;20:19–29. doi: 10.1016/0022-3956(86)90020-8. [DOI] [PubMed] [Google Scholar]
- Ozelius L, Hsu YP, Bruns G, Powell JF, Chen S, Weyler W, Utterback M, Zucker D, Haines J, Trofatter JA, et al. Human monoamine oxidase gene (MAOA): chromosome position (Xp21-p11) and DNA polymorphism. Genomics. 1988;3:53–58. doi: 10.1016/0888-7543(88)90159-0. [DOI] [PubMed] [Google Scholar]
- Passamonti L, Fera F, Magariello A, Cerasa A, Gioia MC, Muglia M, Nicoletti G, Gallo O, Provinciali L, Quattrone A. Monoamine oxidase—a genetic variations influence brain activity associated with inhibitory control: new insight into the neural correlates of impulsivity. Biol Psychiatry. 2006;59:334–340. doi: 10.1016/j.biopsych.2005.07.027. [DOI] [PubMed] [Google Scholar]
- Pedersen NL, Oreland L, Reynolds C, McClearn GE. Importance of genetic effects for monoamine oxidase activity in thrombocytes in twins reared apart and twins reared together. Psychiatry Res. 1993;46:239–251. doi: 10.1016/0165-1781(93)90092-u. [DOI] [PubMed] [Google Scholar]
- Peters JR, Vallie B, Difronzo M, Donaldson ST. Role of dopamine D1 receptors in novelty seeking in adult female Long-Evans rats. Brain Res Bull. 2007;74:232–236. doi: 10.1016/j.brainresbull.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Phillips ML, Williams L, Senior C, Bullmore ET, et al. A differential neural response to threatening and non-threatening negative facial expressions in paranoid and non-paranoid schizophrenics. Psychiatry Res. 1999;92(1):11–31. doi: 10.1016/s0925-4927(99)00031-1. [DOI] [PubMed] [Google Scholar]
- Picazo O, Chuc-Meza E, Anaya-Martinez V, Jimenez I, Aceves J, Garcia-Ramirez M. 6-Hydroxydopamine lesion in thalamic reticular nucleus reduces anxiety behaviour in the rat. Behav Brain Res. 2009;197(2):317–322. doi: 10.1016/j.bbr.2008.08.047. [DOI] [PubMed] [Google Scholar]
- Popova NK, Skrinskaya YA, Amstislavskaya TG, Vishnivetskaya GB, Seif I, de Meier E. Behavioral characteristics of mice with genetic knockout of monoamine oxidase type A. Neurosci Behav Physiol. 2001;31:597–602. doi: 10.1023/a:1012364910091. [DOI] [PubMed] [Google Scholar]
- Popova NK, Maslova LN, Morosova EA, Bulygina VV, Seif I. MAO A knockout attenuates adrenocortical response to various kinds of stress. Psychoneuroendocrinology. 2006;31:179–186. doi: 10.1016/j.psyneuen.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Preisig M, Bellivier F, Fenton BT, Baud P, Berney A, Courtet P, Hardy P, Golaz J, Leboyer M, Mallet J, Matthey ML, Mouthon D, et al. Association between bipolar disorder and monoamine oxidase A gene polymorphisms: results of a multicenter study. Am J Psychiatry. 2000;157:948–955. doi: 10.1176/appi.ajp.157.6.948. [DOI] [PubMed] [Google Scholar]
- Quitkin FM, Liebowitz MR, Stewart JW, McGrath PJ, Harrison W, Rabkin JG, Markowitz J, Davies SO. L-Deprenyl in atypical depressives. Arch Gen Psychiatry. 1984;41:777–781. doi: 10.1001/archpsyc.1984.01790190051006. [DOI] [PubMed] [Google Scholar]
- Reist C, Haier RJ, DeMet E, Chicz-DeMet A. Platelet MAO activity in personality disorders and normal controls. Psychiatry Res. 1990;33:221–227. doi: 10.1016/0165-1781(90)90039-8. [DOI] [PubMed] [Google Scholar]
- Ribasés M, Ramos-Quiroga JA, Hervás A, Bosch R, Bielsa A, Gastaminza X, Artigas J, Rodriguez-Ben S, Estivill X, Casas M, Cormand B, Bayés M. Exploration of 19 serotoninergic candidate genes in adults and children with attention-deficit/hyperactivity disorder identifies association for 5HT2A, DDC and MAOB. Mol Psychiatry. 2009;14:71–85. doi: 10.1038/sj.mp.4002100. [DOI] [PubMed] [Google Scholar]
- Robinson DS, Gilmor ML, Yang Y, Moonsammy G, Azzaro AJ, Oren DA, Campbell BJ. Treatment effects of selegiline transdermal system on symptoms of major depressive disorder: a meta-analysis of short-term, placebo-controlled, efficacy trials. Psychopharmacol Bull. 2007;40:15–28. [PubMed] [Google Scholar]
- Roohi J, DeVincent CJ, Hatchwell E, Gadow KD. Association of a monoamine oxidase-a gene promoter polymorphism with ADHD and anxiety in boys with autism spectrum disorder. J Autism Dev Disord. 2009;39(1):67–74. doi: 10.1007/s10803-008-0600-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Leggo J, Goodburn S, Walsh C, Jain S, Paykel ES. Genetic association between monoamine oxidase A microsatellite and RFLP alleles and bipolar affective disorder: analysis and meta-analysis. Hum Mol Genet. 1996;5:779–782. doi: 10.1093/hmg/5.6.779. [DOI] [PubMed] [Google Scholar]
- Ruchkin VV, Koposov RA, af Klinteberg B, Oreland L, Grigorenko EL. Platelet MAO-B, personality, and psychopathology. J Abnorm Psychol. 2005;114:477–482. doi: 10.1037/0021-843X.114.3.477. [DOI] [PubMed] [Google Scholar]
- Saba A, Chiellini G, Frascarelli S, Marchini M, Ghelardoni S, Raffaelli A, Tonacchera M, Vitti P, Scanlan TS, Zucchi R. Tissue distribution and cardiac metabolism of 3-iodothyronamine. Endocrinology. 2010;151(10):5063–5073. doi: 10.1210/en.2010-0491. [DOI] [PubMed] [Google Scholar]
- Sabelli HC, Javaid JI. Phenylethylamine modulation of affect: therapeutic and diagnostic implications. J Neuropsychiatry Clin Neurosci. 1995;7(1):6–14. doi: 10.1176/jnp.7.1.6. [DOI] [PubMed] [Google Scholar]
- Sabol SZ, Hu S, Hamer D. A functional polymorphism in the monoamine oxidase A gene promoter. Hum Genet. 1998;103:273–279. doi: 10.1007/s004390050816. [DOI] [PubMed] [Google Scholar]
- Salichon N, Gaspar P, Upton AL, Picaud S, Hanoun N, Hamon M, De Maeyer E, Murphy DL, Mossner R, Lesch KP, Hen R, Seif I. Excessive activation of serotonin (5-HT) 1B receptors disrupts the formation of sensory maps in monoamine oxidase a and 5-ht transporter knock-out mice. J Neurosci. 2001;21:884–896. doi: 10.1523/JNEUROSCI.21-03-00884.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samochowiec J, Lesch KP, Rottmann M, Smolka M, Syagailo YV, Okladnova O, Rommelspacher H, Winterer G, Schmidt LG, Sander T. Association of a regulatory polymorphism in the promoter region of the monoamine oxidase A gene with antisocial alcoholism. Psychiatry Res. 1999;86(1):67–72. doi: 10.1016/s0165-1781(99)00020-7. [DOI] [PubMed] [Google Scholar]
- Sanders MJ, Dietrich WD, Green EJ. Behavioral, electrophysiological, and histo-pathological consequences of mild fluid-percussion injury in the rat. Brain Res. 2001;904(1):141–144. doi: 10.1016/s0006-8993(01)02424-6. [DOI] [PubMed] [Google Scholar]
- Sandler M, Glover V, Clow A, Jarman J. Monoamine oxidase-B, monoamine oxidase-B inhibitors, and Parkinson’s disease. A role for superoxide dismutase? Adv Neurol. 1993;60:238–241. [PubMed] [Google Scholar]
- Saura J, Richards JG, Mahy N. Age-related changes on MAO in Bl/C57 mouse tissues: a quantitative radioautographic study. J Neural Transm. 1994;41(Suppl):89–94. doi: 10.1007/978-3-7091-9324-2_11. [DOI] [PubMed] [Google Scholar]
- Schilling B, Lerch K. Cloning, sequencing and heterologous expression of the monoamine oxidase gene from Aspergillus niger. Mol Gen Genet. 1995;247(4):430–438. doi: 10.1007/BF00293144. [DOI] [PubMed] [Google Scholar]
- Schuback DE, Mulligan EL, Sims KB, Tivol EA, Greenberg BD, Chang SF, Yang SL, Mau YC, Shen CY, Ho MS, Yang NH, Butler MG, et al. Screen for MAOA mutations in target human groups. Am J Med Genet. 1999;88:25–28. [PMC free article] [PubMed] [Google Scholar]
- Scott AL, Bortolato M, Chen K, Shih JC. Novel monoamine oxidase A knock out mice with human-like spontaneous mutation. Neuroreport. 2008;19:739–743. doi: 10.1097/WNR.0b013e3282fd6e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setini A, Pierucci F, Senatori O, Nicotra A. Molecular characterization of monoamine oxidase in zebrafish (Danio rerio) Comp Biochem Physiol B Biochem Mol Biol. 2005;140:153–161. doi: 10.1016/j.cbpc.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Shabanov PD, Lebedev AA, Meshcherov ShK, Strel’tsov VF. The effects of neuro-chemical lesioning of dopaminergic terminals in early ontogenesis on behavior in adult rats. Neurosci Behav Physiol. 2005;35(5):535–544. doi: 10.1007/s11055-005-0089-y. [DOI] [PubMed] [Google Scholar]
- Sharp T, Gartside SE, Umbers V. Effects of co-administration of a monoamine oxidase inhibitor and a 5-HT1A receptor antagonist on 5-hydroxytryptamine cell firing and release. Eur J Pharmacol. 1997;320:15–19. doi: 10.1016/s0014-2999(96)00968-5. [DOI] [PubMed] [Google Scholar]
- Shekim WO, Bylund DB, Alexson J, Glaser RD, Jones SB, Hodges K, Perdue S. Platelet MAO and measures of attention and impulsivity in boys with attention deficit disorder and hyperactivity. Psychiatry Res. 1986;18:179–188. doi: 10.1016/0165-1781(86)90029-6. [DOI] [PubMed] [Google Scholar]
- Shih JC, Eiduson S. Multiple forms of monoamine oxidase in the developing brain. Nature. 1969;224:1309–1310. doi: 10.1038/2241309a0. [DOI] [PubMed] [Google Scholar]
- Shih JC, Thompson RF. Monoamine oxidase in neuropsychiatry and behavior. Am J Hum Genet. 1999;65:593–598. doi: 10.1086/302562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson GM, Shih JC, Chen K, Flowers C, Kumazawa T, Spring B. Schizophrenia, monoamine oxidase activity, and cigarette smoking. Neuropsychopharmacology. 1999;20:392–394. doi: 10.1016/S0893-133X(98)00119-5. [DOI] [PubMed] [Google Scholar]
- Sims KB, de la Chapelle A, Norio R, Sankila EM, Hsu YP, Rinehart WB, Corey TJ, Ozelius L, Powell JF, Bruns G, et al. Monoamine oxidase deficiency in males with an X chromosome deletion. Neuron. 1989a;2:1069–1076. doi: 10.1016/0896-6273(89)90231-6. [DOI] [PubMed] [Google Scholar]
- Sims KB, Ozelius L, Corey T, Rinehart WB, Liberfarb R, Haines J, Chen WJ, Norio R, Sankila E, de la Chapelle A, et al. Norrie disease gene is distinct from the monoamine oxidase genes. Am J Hum Genet. 1989b;45:424–434. [PMC free article] [PubMed] [Google Scholar]
- Sotnikova TD, Budygin EA, Jones SR, Dykstra LA, Caron MG, Gainetdinov RR. Dopamine transporter-dependent and -independent actions of trace amine beta-phenyl-ethylamine. J Neurochem. 2004;91(2):362–373. doi: 10.1111/j.1471-4159.2004.02721.x. [DOI] [PubMed] [Google Scholar]
- Steckler T, Rammes G, Sauvage M, van Gaalen MM, et al. Effects of the monoamine oxidase A inhibitor moclobemide on hippocampal plasticity in GR-impaired transgenic mice. J Psychiatr Res. 2001;35(1):29–42. doi: 10.1016/s0022-3956(00)00040-6. [DOI] [PubMed] [Google Scholar]
- Straube T, Schmidt S, Weiss T, Mentzel HJ, et al. Dynamic activation of the anterior cingulate cortex during anticipatory anxiety. Neuroimage. 2009;44(3):975–981. doi: 10.1016/j.neuroimage.2008.10.022. [DOI] [PubMed] [Google Scholar]
- Surguladze S, Russell T, Kucharska-Pietura K, Travis MJ, et al. A reversal of the normal pattern of parahippocampal response to neutral and fearful faces is associated with reality distortion in schizophrenia. Biol Psychiatry. 2006;60(5):423–431. doi: 10.1016/j.biopsych.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Svensson S, Some M, Lundsjo A, Helander A, Cronholm T, Hoog JO. Activities of human alcohol dehydrogenases in the metabolic pathways of ethanol and serotonin. Eur J Biochem. 1999;262:324–329. doi: 10.1046/j.1432-1327.1999.00351.x. [DOI] [PubMed] [Google Scholar]
- Szymanski HV, Naylor EW, Karoum F. Plasma phenylethylamine and phenylalanine in chronic schizophrenic patients. Biol Psychiatry. 1987;22:194–198. doi: 10.1016/0006-3223(87)90230-7. [DOI] [PubMed] [Google Scholar]
- Tariot PN, Cohen RM, Sunderland T, Newhouse PA, Yount D, Mellow AM, Weingartner H, Mueller EA, Murphy DL. L-deprenyl in Alzheimer’s disease. Preliminary evidence for behavioral change with monoamine oxidase B inhibition. Arch Gen Psychiatry. 1987;44:427–433. doi: 10.1001/archpsyc.1987.01800170041007. [DOI] [PubMed] [Google Scholar]
- Thompson AM. Serotonin immunoreactivity in auditory brainstem neurons of the postnatal monoamine oxidase-A knockout mouse. Brain Res. 2008;1228:58–67. doi: 10.1016/j.brainres.2008.06.091. [DOI] [PubMed] [Google Scholar]
- Thompson AM, Thompson GC. Serotonin-immunoreactive neurons in the postnatal MAO-A KO mouse lateral superior olive project to the inferior colliculus. Neurosci Lett. 2009;460:47–51. doi: 10.1016/j.neulet.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Tolbert SR, Fuller MA. Selegiline in treatment of behavioral and cognitive symptoms of Alzheimer disease. Ann Pharmacother. 1996;30:1122–1129. doi: 10.1177/106002809603001012. [DOI] [PubMed] [Google Scholar]
- Udenfriend S, Titus E, Weissbach H, Peterson RE. Biogenesis and metabolismof 5-hydroxyindole compounds. J Biol Chem. 1956;219:335–344. [PubMed] [Google Scholar]
- Upadhyay AK, Wang J, Edmondson DE. Comparison of the structural properties of the active site cavities of human and rat monoamine oxidase A and B in their soluble and membrane-bound forms. Biochemistry. 2008;47:526–536. doi: 10.1021/bi7019707. [DOI] [PubMed] [Google Scholar]
- Upton AL, Salichon N, Lebrand C, Ravary A, Blakely R, Seif I, Gaspar P. Excess of serotonin (5-HT) alters the segregation of ispilateral and contralateral retinal projections in monoamine oxidase A knock-out mice: possible role of 5-HT uptake in retinal ganglion cells during development. J Neurosci. 1999;19:7007–7024. doi: 10.1523/JNEUROSCI.19-16-07007.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gaalen MM, Brueggeman RJ, Bronius PF, Schoffelmeer AN, Vanderschuren LJ. Behavioral disinhibition requires dopamine receptor activation. Psychopharmacology (Berl) 2006;187(1):73–85. doi: 10.1007/s00213-006-0396-1. [DOI] [PubMed] [Google Scholar]
- Vitalis T, Cases O, Callebert J, Launay JM, Price DJ, Seif I, Gaspar P. Effects of monoamine oxidase A inhibition on barrel formation in the mouse somatosensory cortex: determination of a sensitive developmental period. J Comp Neurol. 1998;393:169–184. doi: 10.1002/(sici)1096-9861(19980406)393:2<169::aid-cne3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- von Knorring L, Oreland L, Winblad B. Personality traits related to monoamine oxidase activity in platelets. Psychiatry Res. 1984;12:11–26. doi: 10.1016/0165-1781(84)90134-3. [DOI] [PubMed] [Google Scholar]
- Whibley A, Urquhart J, Dore J, Willatt L, Parkin G, Gaunt L, Black G, Donnai D, Raymond FL. Deletion of MAOA and MAOB in a male patient causes severe developmental delay, intermittent hypotonia and stereotypical hand movements. Eur J Hum Genet. 2010;18:1095–1099. doi: 10.1038/ejhg.2010.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker-Azmitia PM, Zhang X, Clarke C. Effects of gestational exposure to mono-amine oxidase inhibitors in rats: preliminary behavioral and neurochemical studies. Neuropsychopharmacology. 1994;11(2):125–132. doi: 10.1038/npp.1994.42. [DOI] [PubMed] [Google Scholar]
- Williams LM, Gatt JM, Kuan SA, Dobson-Stone C, et al. A polymorphism of the MAOA gene is associated with emotional brain markers and personality traits on an antisocial index. Neuropsychopharmacology. 2009;34(7):1797–1809. doi: 10.1038/npp.2009.1. [DOI] [PubMed] [Google Scholar]
- Williams RB, Marchuk DA, Gadde KM, Barefoot JC, Grichnik K, Helms MJ, Kuhn CM, Lewis JG, Schanberg SM, Stafford-Smith M, Suarez EC, Clary GL, Svenson IK, Siegler IC. Serotonin-related gene polymorphisms and central nervous system serotonin function. Neuropsychopharmacology. 2003;28(3):533–541. doi: 10.1038/sj.npp.1300054. [DOI] [PubMed] [Google Scholar]
- Wu HF, Chen K, Shih JC. Site-directed mutagenesis of monoamine oxidase A and B: role of cysteines. Mol Pharmacol. 1993;43:888–893. [PubMed] [Google Scholar]
- Wylie CJ, Hendricks TJ, Zhang B, Wang L, Lu P, Leahy P, Fox S, Maeno H, Deneris ES. Distinct transcriptomes define rostral and caudal serotonin neurons. J Neurosci. 2010;30(2):670–684. doi: 10.1523/JNEUROSCI.4656-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youdim MB, Collins GG, Sandler M. Multiple forms of rat brain monoamine oxidase. Nature. 1969;223:626–628. doi: 10.1038/223626a0. [DOI] [PubMed] [Google Scholar]
- Yu YW, Tsai SJ, Hong CJ, Chen TJ, Chen MC, Yang CW. Association study of a monoamine oxidase a gene promoter polymorphism with major depressive disorder and antidepressant response. Neuropsychopharmacology. 2005;30(9):1719–1723. doi: 10.1038/sj.npp.1300785. [DOI] [PubMed] [Google Scholar]
- Zucchi R, Chiellini G, Scanlan TS, Grandy DK. Trace amine-associated receptors and their ligands. Br J Pharmacol. 2006;149:967–978. doi: 10.1038/sj.bjp.0706948. [DOI] [PMC free article] [PubMed] [Google Scholar]