

Abstract
A number of immunological functions are ascribed to cell-surface expressed forms of the endoplasmic reticulum (ER)2 chaperone calreticulin. Here we examined impacts of ER stress-inducing drugs upon cell-surface calreticulin induction, and resulting immunological consequences. We show that cell-surface expression of calreticulin and secretion of calreticulin, BiP, gp96 and PDI are induced by thapsigargin treatment, which depletes ER calcium, but not by tunicamycin treatment, which inhibits protein glycosylation. Surface expression of calreticulin in viable, thapsigargin-treated fibroblasts correlates with their enhanced phagocytic uptake by bone marrow-derived dendritic cells (BMDC). Incubation of BMDC with thapsigargin-treated fibroblasts enhances sterile IL-6 production, and LPS-induced generation of IL-1β, IL-12, IL-23 and TNF-α. However, extracellular calreticulin is not required for enhanced pro-inflammatory responses. Furthermore, the pattern of pro-inflammatory cytokine induction by thapsigargin-treated cells and cell supernatants resembles that induced by thapsigargin itself, and indicates that other ER chaperones present in supernatants of thapsigargin-treated cells also do not contribute to inducing the innate immune response. Thus, secretion of various ER chaperones, including calreticulin, is induced by ER calcium depletion. Calreticulin, previously suggested as an eat-me signal in dead and dying cellular contexts, can also promote phagocytic uptake of cells subject to ER calcium depletion. Finally there is a strong synergy between calcium-depletion in the ER and sterile IL-6 as well as LPS-dependent IL-1β, IL-12, IL-23 and TNF-α innate responses, findings that have implications for understanding inflammatory diseases that originate in the ER.
INTRODUCTION
The endoplasmic reticulum (ER) is an important site of protein folding, calcium storage, and intracellular signaling (1). A number of ER chaperones are important in the functions of the ER. Calreticulin is a chaperone that maintains quality control of glycoprotein folding by binding mono-glucosylated proteins in the ER. Calreticulin also contributes to calcium storage in the ER (reviewed in (2)). Several recent studies have shown that the cell-surface expression of calreticulin can be induced in different cell types and by different cell treatments (reviewed in (3, 4)). Cell death stimuli suggested to induce cell-surface calreticulin on dying cells include UV light, gamma-irradiation, anthracyclin chemotherapeutics like mitoxantrone, and platinum-based chemotherapeutics like oxaliplatin (5-8). The presence of calreticulin on the surface of dead and dying tumor cells has been suggested to stimulate therapeutic and protective anti-tumor immune responses in mice (6-8). Calreticulin on the surface of apoptotic cells and dying tumor cells is also suggested to function as an eat-me signal in the phagocytosis of these cells (5, 7, 9), and by this mechanism, calreticulin could promote the presentation of antigens derived from dying cells to T cells to induce anti-tumor immunity. Other mechanisms could also account for the immunostimulatory effects of cell-surface calreticulin. Purified ER chaperones such as heat shock protein 90 (HSP90), calreticulin (10) and gp96 (HSPC4) have been implicated in induction of co-stimulatory molecule expression and cytokine production by dendritic cells (reviewed in ((11)). While it has been suggested that TLR ligand contamination could account for the reported immunogenicity of these and other soluble HSP’s, studies demonstrating immunogenicity of cell-associated HSPs, including HSP90 and gp96, suggest that HSPs can be immunomodulatory independently of microbial contaminants (12-14).
A number of studies have also suggested links between ER calcium depletion and detection of ER-resident proteins, including calreticulin, in the extracellular space. Treatment of NIH3T3 cells with the calcium ionophore A23167, that depletes intracellular calcium stores (15), resulted in secretion of gp96, an ER-resident chaperone, and reduced intracellular levels of calreticulin (referred to in the paper as CRP55) (16). Furthermore, treatment of NIH3T3 cells with the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitor thapsigargin (17) resulted in a decrease in calreticulin ER staining intensity and an increase in the co-localization of calreticulin with wheat germ agglutinin in a non-ER cellular compartment suggested to be the Golgi (16). Another study showed that thapsigargin increased levels of surface calreticulin in the SH-SY5Y neuroblastoma cell line (18). In Hela cells, a direct correlation between an agent’s ability to deplete ER calcium and its ability to induce surface calreticulin expression was also shown (18). Furthermore, two other groups showed that ER calcium depletion by thapsigargin results in secretion and surface expression of BiP (19, 20). Finally, we recently showed that amino acid residues that contribute to the polypeptide-specific chaperone activity of calreticulin also influence its surface expression in thapsigargin-treated mouse embryonic fibroblasts (MEFs) (21).
Calcium depletion in the ER impairs protein folding (1, 22) due to impacts on the functional activities of calcium-dependent chaperones (23). In calcium-depleted cells, the unfolded protein response (UPR) pathway is induced, a cellular stress response pathway activated by the presence of misfolded proteins in the ER (24). Different arms of the UPR pathway function to stop protein translation, increase syntheses of molecular chaperones, and induce other cellular changes towards the restoration of the folding capacity of the ER (reviewed in (24)). Thapsigargin is a commonly used drug to induce the UPR pathway, and as noted above, thapsigargin induces calcium depletion (17). Another drug that is commonly used to induce the UPR pathway is tunicamycin, which inhibits protein glycosylation thereby causing protein misfolding in the ER (25). In the present studies, we compare the impacts of these two ER stress-inducing drugs as well as the chemotherapeutic mitoxantrone, on cell-surface expression of calreticulin in different cell types. We show that cell-surface expression of calreticulin, and secretion of other ER proteins including calreticulin, is specific to the ER calcium depletion-induced form of ER stress. Cell-surface expression of calreticulin in thapsigargin treated cells involves a failure in effective ER retention mechanisms for a number of ER-resident proteins. This mode of calreticulin release from the ER is distinct from that described for anthracyclin-induced surface calreticulin expression, where co-translocation with ERp57 is suggested (26, 27). Nonetheless, surface calreticulin induced by thapsigargin treatment does enhance the phagocytic uptake of cells. The presence of various extracellular chaperones, including calreticulin, allowed us to examine functions of the extracellular forms of calreticulin and other ER chaperones in inducing an innate immune response. We found that incubation of BMDC with thapsigargin-treated fibroblasts or conditioned media from the treated cells enhances production of IL-6 under sterile conditions, but not IL-1β, IL-12 or TNF-α, cytokines previously shown to be induced by macrophages following exposure to extracellular gp96 (14) or a calreticulin fragment (10). Furthermore, direct thapsigargin treatment of BMDC enhances IL-6 production under sterile conditions as previously shown in macrophages (28), and also enhances various pro-inflammatory responses to LPS more broadly and significantly than BMDC treatment with tunicamycin. Together the findings point to strong synergies between ER calcium depletion and innate immune responses and suggest that the combined presence of several ER chaperones in the extracellular environment per se is not strongly pro-inflammatory.
MATERIALS and METHODS
Animals
Mice were maintained in Specific Pathogen Free (SPF) facility and cared for according to the University Committee on Use and Care of Animals’ (UCUCA) approved protocols. Mice were euthanized by CO2 asphyxiation.
Drugs and antibodies
Tunicamycin and mitoxantrone were purchased from Calbiochem or Sigma, and used at 10 μg/ml or 1 μM, respectively unless indicated otherwise. Thapsigargin was purchased from both Sigma and Alexis Biochemicals (now Enzo Life Sciences) and used at 5 μM, unless indicated otherwise. Mitoxantrone, thapsigargin and tunicamycin were all dissolved in DMSO and stored in single use aliquots at -20° C at stock concentrations of 1 mM, 5 mM and 10 mg/ml, respectively. Frozen aliquots and freshly dissolved drugs did not show differences in measured activities. Chicken-anti-calreticulin or rabbit-anti-calreticulin were purchased from Affinity Bioreagents, now part of Thermo Scientific (PA1-902A), and Abcam (ab2907), respectively and used for flow cytometry at a concentration of 2.5 μg/ml and a dilution of 1:200, respectively. Annexin-V (AnnV)-FITC was purchased from Biovision and BD biosciences. 7AAD was purchased from Sigma. All other fluorescent secondary antibodies were purchased from Jackson Immunoresearch. For immunoblots, goat-anti-calreticulin (Santa Cruz, sc-7431) was used at a dilution of 1:2500, and secondary antibodies conjugated to horseradish peroxidase were purchased from Jackson Immunoresearch and used at a dilution of 1:30,000. Mouse-anti-BIP (BD biosciences, 610978) and rabbit-anti-calreticulin (Abcam, ab2907) were used in immunoprecipitations at a concentration of 1:1000 or 1:1666 respectively. Rabbit-anti-PDI (1:2500, santa cruz, sc-20132), mouse-anti-BIP (BD biosciences, 610978), rabbit-anti-HMGB1 (Abcam, ab18256) and rabbit-anti-GP96 (1:1000, cell signaling technologies, 2104S) were used in immunoblots.
Cell culture and dendritic cell preparations
All cell lines, and primary thymocytes and splenocytes were maintained in RPMI 1640 media (Gibco) supplemented with 100 μg/ml streptomycin, 100 U/ml of penicillin (some media additionally contained 0.25 ug/ml of the anti-mycotic amphotericin B) and 10% FBS (v/v) (Invitrogen). One exception was that during experiments in which immunoblotting analyses of ER proteins in the supernatants or conditioned media of drug-treated cells were undertaken, the samples were generated in 1% serum instead of 10% serum to minimize background bands from serum proteins. Only cells testing negative for mycoplasma using a PCR-based detection assay (Sigma, VenorGem®) were used in experiments measuring cytokine production. Mycoplasma negative lines were maintained in media containing a prophylactic concentration of the anti-mycoplasma antibiotic, plasmocin (Invivogen, 5 ug/ml). Calreticulin deficient (CRT-/- K42) and WT (K41) MEFs were a kind gift from Dr. Marek Michalak. The CT26 mouse colon cancer cell line was purchased from American Tissue Culture Collection. Primary BMDC were derived by culturing mouse femur and tibia bone marrow in granulocyte macrophage colony-stimulating factor (GMCSF)-containing media for 5-7 days in 24 well plates after treating with red cell lysis buffer for 2 minutes on ice (Sigma). BMDC media was made up of RPMI supplemented with 100 μg/ml streptomycin, 100 U/ml of penicillin, 10% FBS (v/v), 1 mM HEPES, 1 mM sodium pyruvate, 0.1 mM MEM Non-Essential Amino Acids (Gibco) and 50 μM β-mercaptoethanol. BMDC media was changed every other day, and non-adherent and loosely adherent cells were harvested by pipeting. Calreticulin deficient MEFs expressing wild type or mutant calreticulin or an expression vector with no inserted gene were generated as described (29). Human multiple myeloma lines NCI H929 (H929) and RPMI8226 were the kind gift of Dr. Malathi Kandarpa and Andrzej Jakubowiak. Murine thymi from C57BL/6 or BALB/c mice were homogenized with a syringe plunger and cell strainer. Murine spleens were prepared as the thymi, with an additional 2-minute treatment with red cell lysis buffer and subsequent wash before being plated. All cells were maintained at 37° C and 5% CO2.
Flow cytometry
For calreticulin surface analysis by flow cytometry, cell lines were detached with PBS + 8 mM EDTA, washed in PBS and resuspended in PBS containing 2% FBS (flow cytometry buffer). 1.5-2×105 cells were stained in 70 μl flow cytometry buffer containing the respective antibody. For calreticulin + block stains, the same dilution of chicken anti-calreticulin was pre-incubated for 15-30 minutes at room temperature with a saturating concentration of peptide (KEQFLDGDAWTNRWVESKHK) corresponding to the sequence in calreticulin that the antibody was generated against and affinity purified with. Cells were washed two times with flow cytometry buffer, resuspended in 70 μl buffer containing donkey anti-chicken conjugated to PE (1:250) and incubated for 30-60 minutes rocking at 4° C. Cells were then washed two times. Finally, cells were resuspended in a 1x dilution of AnnV-FITC and 7AAD in AnnV binding buffer (10 mM HEPES (pH 7.4), 140 mM NaCl, and 2.5 mM CaCl2), and data for each sample was collected on a FACSCanto flow cytometer (BD). Analysis was performed using Flowjo 8.8.6 (Treestar Inc.).
Immunoprecipitations
The day before drug treatment, 1-2×106 cells were seeded in a 10 cm dish and allowed to grow overnight. The next day the media was removed and cells were treated with different drugs for 5 hours as described above. Supernatants from each treatment were centrifuged to remove cell debris, transferred to fresh tubes, and incubated overnight with indicated antibodies at 4° C, with gentle agitation. The following day, samples were centrifuged to remove precipitated proteins, transferred to new tubes and incubated for at least 5 hours with Protein G beads (GE Healthcare). The beads were washed three times with 1 ml of 1% NP-40 in PBS and then boiled in the presence of SDS and DTT. The samples were analyzed by SDS-PAGE and immunoblotting with indicated antibodies. In parallel, cells from each plate were harvested and lysed in triton X-100 lysis buffer (10 mM Na2HPO4, 10 mM Tris, 130 mM NaCl, 1% Triton X-100, complete EDTA-free protease inhibitors, pH 7.5) on ice for at least 1 hour, followed by a 30-minute centrifugation at 4° C to remove cell debris. Protein concentrations of cleared lysates were determined with Pierce bicinchoninic acid (BCA) assay and these lysates were included in the immunoblotting analysis of the immunoprecipitated proteins.
Cytokine profiling of BMDC co-incubated with drug, drug-treated MEFs, or MEF-derived conditioned media
For data in Figure 5, MEFs were drug-treated as indicated for 5-6 hours, following which, cells were harvested, washed 2-3 times (first in 25 ml media, then either twice in 10 ml or once in 25-40 ml media) and incubated alone or with BMDC as described below. For data shown in Figure 6, MEFs were treated with indicated drugs for 5-6 hours, supernatants collected (CM1), cells washed 3 times in 7 ml of PBS, following which 4 ml of BMDC media were added to the cells. After a 5-6 hour collection period, conditioned media, called conditioned media 2 (CM2), were harvested. CM1 and CM2 were centrifuged to clear any floating cells or cellular debris. For Figure 6A, a portion of CM2 was added to BMDC (MEF CM2+DC) and another portion directly tested for the presence of specific cytokines (MEF CM2). For Figure 6B, CM1 or CM2 were added directly to BMDC as in 6A. For Figure S3 CM1 and CM2 were first dialyzed using 3 kDa or 10 kDa centricon filters as indicated (using at least 3 successive concentrations and dilutions), before adding retentate or flow-through to BMDC. Separate wells of dendritic cells were directly treated with the indicated concentrations of the described drugs (Direct treatment), or left untreated. In parallel analyses, a second set of MEF plates were treated as described above for generation of the CM2. MEFs were then harvested and stained with AnnV and 7AAD (to measure cell viability at the end of the MEF CM2 collection). Protocols for BMDC incubations with treated cells, CM or drug were adapted from Torchinsky et al. 2009 (30). Day 5 or 6 BMDCs (5×105) were mixed with 1×106 washed target cells, co-sedimented and incubated for 19-23.5 hours (Fig. 5), or treated separately with drugs for 21-23 hours (Fig. 6B) or with MEF CM for 15-23 hours (Fig. 6A and B) in 24 well plates in the presence or absence of described amounts of LPS before centrifuging the plates for 5 min at 1200 rpm and harvesting the resulting supernatants. Supernatants were finally submitted to the University of Michigan Cancer Center Immunology Core (UMCCIC) to detect the indicated cytokines by ELISA. Duoset ELISA development systems were purchased from R&D systems (Minneapolis, MN).
FIGURE 5. Co-culture of thapsigargin-treated target cells with BMDC induces and enhances pro-inflammatory cytokine production in a calreticulin-independent manner.
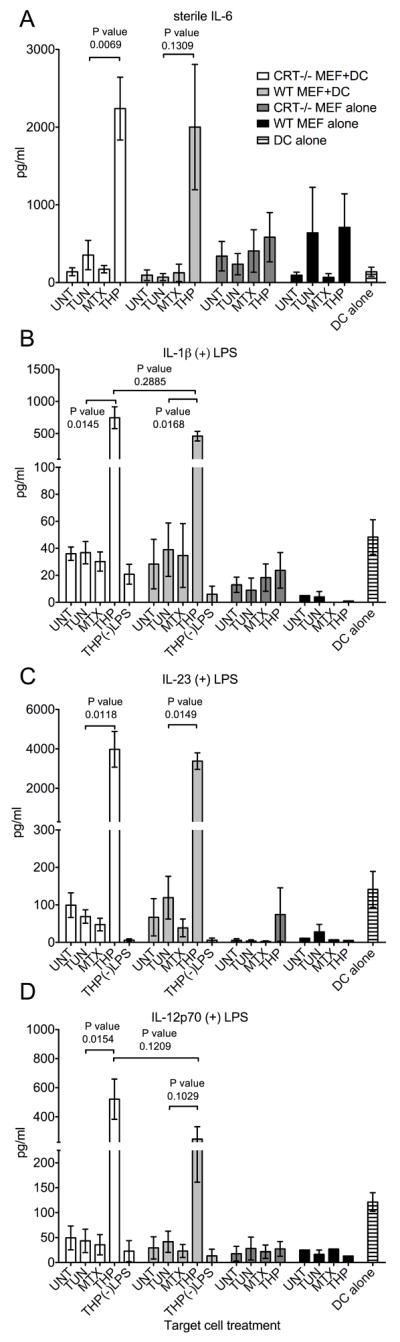
(A-D) Cytokine production by DCs, MEFs, and DC+MEF co-cultures. WT or CRT-/- MEF target cells were treated with TUN, MTX or THP for 5.5 or 6.5 hours or left untreated. Adherent target cells were then harvested, washed and incubated in media alone (MEF alone) or co-incubated with BMDC (MEF+DC) in the absence (A) or presence (B-D) of 0.5-2 ng/ml LPS for 18.5-23.5 hours and the concentrations of the indicated cytokine were measured in duplicate by ELISA. Wells of DC incubated in the presence (B-D) or absence (A) of LPS were included in each experiment as an additional control (DC alone). The THP(-)LPS bars indicate conditions where thapsigargin-treated WT MEF or CRT-/- MEFs were incubated with DC, but in the absence of LPS, to illustrate the LPS dependence of enhanced production of indicated cytokines. The data from co-incubations of DC and MEFs show the mean and standard error of 3 (WT) or 5 (CRT-/-) experiments. The data from MEFs alone or DC alone show the mean and standard error of 1-3 experiments in the presence of LPS, or 2-4 experiments in the absence of LPS. The p-values from two-tailed, paired t-tests are indicated.
FIGURE 6. Differential induction of pro-inflammatory cytokines by drugs and by conditioned media (CM) from drug-treated MEFs.

BMDC were treated with MEF CM or drugs either under sterile conditions (top panels for IL-6 measurements) or in the presence of LPS (lower panels for IL-1β, IL-23 and IL-12p70 measurements; 1 ng/ml LPS (A) or 1-10 ng/ml LPS (B) were used) for 21-23 (A) or 15-22 (B) hours. Indicated cytokine measurements followed CM+DC, drug+DC or DC-media (DC alone) co-incubations in the presence or absence of LPS. (A) Concentrations of the indicated cytokines in MEF CM2+DC co-incubations, or DC alone were measured in duplicate by ELISA. The THP(-)LPS bars indicate conditions where CM2 from thapsigargin-treated WT or CRT-/- MEFs were incubated with DC. CRT-/- MEF CM2 and WT MEF CM2 bars indicate direct cytokine measurements of the indicated CM2 samples without further treatment with LPS. The results with WT MEFs show the average of three independent experiments, and the results with the CRT-/- MEFs are from a single experiment. (B) THP 200 nM CM represents CM prepared as other CM, but by incubating MEFs with 200 nM THP instead of 5 μM THP. Direct DC treatment represents cytokine production by BMDC that were directly treated with thapsigargin (black bars), or tunicamycin (vertically striped bars) at their specified concentrations or left untreated (horizontally striped bars). The (-)LPS bars represent measurements from DC treated with THP CM1 or 5 μM thapsigargin as indicated, but in the absence of LPS. Data are averages of four (direct treatments of BMDC with drugs) or eight (CM2) experiments, or from a single experiment (CM1).
Phagocytosis assays
Target cells were labeled with CellTracker™ Green CMFDA (5-chloromethylfluorescein diacetate) (Invitrogen) in 10 cm plates that were seeded with 2-2.5×106 target MEFs per plate 12-20 hours before labeling. Each plate was labeled as directed with 0.1-0.35 μM CMFDA diluted in optimem serum free media for 20-40 minutes at 37° C. Media was removed and replaced with previously described tissue culture media (RPMI with 10% serum) and incubated at 37° C for 30-60 minutes. After this, the media was removed and replaced with fresh media. Next the plates were then exposed to 3 minutes of UV light on a UV light box or left untreated and all plates were incubated overnight in tissue culture incubator. The following morning, the plates were then treated with 5 μM thapsigargin or 10 μg/ml tunicamycin for 5-6 hours or left untreated as indicated. Cells were finally harvested (UV) or washed twice in 10-12 ml PBS (untreated and drug-treated), then harvested with 8 mM EDTA in PBS and diluted in RPMI for counting (a third wash) before pelleting and resuspending at the appropriate concentration. 1×105 Day 5 or 6 BMDC were then mixed with 4×105 targets in 96 well plates and the plates were then incubated at 37° C or 4° C for 60 minutes. Cells were then fixed for 10 minutes with formalin (fisher PROTOCOL) diluted 1:10 in flow cytometry buffer (PBS+2% FBS), washed twice, stained with anti-CD11c-APC (1:220, BD-biosciences) for 20-40 minutes at 4° C. Cells were washed twice and data for each sample was collected on the FACS Canto flow cytometer (BD). Analysis was performed using Flowjo 8.8.6 (Treestar Inc.). CMFDA was read on the FITC channel.
RESULTS
Differential impacts of ER-stress inducing drugs on cell-surface calreticulin
Thapsigargin (THP) inhibits sarco-endoplasmic reticulum Ca2+-ATPase pumps and causes a reduction in levels of ER calcium (17). We recently showed that thapsigargin treatment of MEFs for short times (5-6 hours) induces cell-surface expression of calreticulin (21), as previously shown in neuroblastoma cells (18), and additionally that thapsigargin treatment of the MEFs also induced calreticulin secretion (21). In the present studies, we asked whether treatment of cells with another drug commonly used to induce ER stress, tunicamycin (TUN), would also result in cell-surface and secreted calreticulin. Additionally, the effect of treating cells with the anthracyclin chemotherapeutic mitoxantrone (MTX) was also tested, as previous studies have suggested that mitoxantrone is a strong inducer of cell-surface calreticulin in pre-apoptotic and apoptotic cells (7, 8, 26).
We first examined cells subject to short drug exposures, which left the large majority of cells intact. To distinguish the apoptotic population that had exposed phosphatidylserine on the outer leaflet of the plasma membrane (31), annexin-V (AnnV) was used. Furthermore, 7-Aminoactinomycin D (7AAD) was used, which is impermeable to live and early apoptotic cells, but stains late-apoptotic/secondary necrotic cells that have lost membrane integrity. The analyses were initially conducted with fibroblasts derived from wild-type (WT MEFs) or calreticulin deficient mouse embryos (CRT-/- MEFs) to control for the specificity of antibody binding. Untreated cells, or cells exposed to tunicamycin, thapsigargin or mitoxantrone for 5-7 hours were triple stained with AnnV-FITC, chicken-anti-calreticulin followed by anti-chicken-PE, and 7AAD, and analyzed by flow cytometry. As previously shown (21), in the AnnV(-), 7AAD(-) gate (which comprised 80-90% of cells in all treatment groups (Fig. 1A)), thapsigargin treatment induced a significant enhancement in surface calreticulin relative to untreated cells (Fig. 1B). Thapsigargin-induced increase in calreticulin surface staining of treated cells relative to untreated cells was observed in WT MEFs but not in calreticulin deficient MEFs, demonstrating specific staining for calreticulin (Fig. 1B). The thapsigargin-induced increase in calreticulin stain was also inhibited by pre-treatment of the anti-calreticulin antibody with the calreticulin-derived, peptide epitope (peptide block) that had been used as an immunogen for generating the anti-calreticulin antibody (anti-CRT+block), demonstrating that the staining was specific to the Fab region of the antibody. Consistent with a previously published study by Obeid et al. (7), tunicamycin did not induce surface calreticulin expression in viable cells. In contrast with the same study, however, we saw little pre-apoptotic induction of surface calreticulin in response to short mitoxantrone treatments relative to the calreticulin up-regulation induced by short thapsigargin treatments of cells (Fig. 1B).
FIGURE 1. A specific form of ER stress induces cell-surface calreticulin.
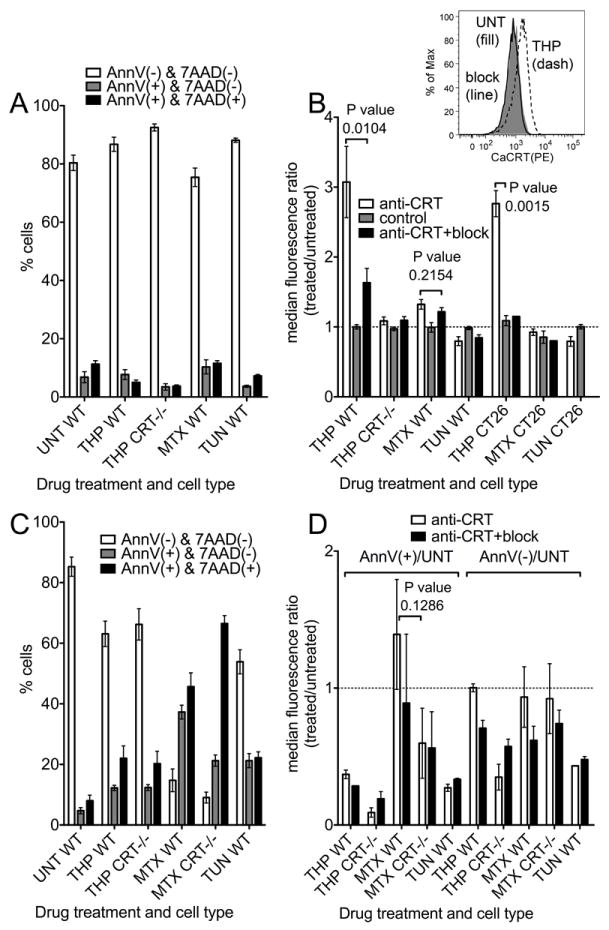
Cell death profiles (A and C) and surface calreticulin expression profiles (B and D) of different treatment conditions were analyzed by flow cytometry. WT or calreticulin deficient (CRT-/-) MEFs were incubated in 5 μM thapsigargin (THP), 1 μM mitoxantrone (MTX), 10 μg/ml tunicamycin (TUN), or media alone (UNT) for 5-7 hours (A-B) or 17 hours (C-D). Cells were then stained with anti-calreticulin (anti-CRT), anti-calreticulin pre-incubated with the calreticulin-derived peptide used as the immunogen and used to affinity purify the antibody (anti-CRT+block), or secondary antibody alone (control). All cells were also stained with AnnV and 7AAD and analyzed by flow cytometry. The median fluorescence values of anti-calreticulin staining of treated cells were measured for the AnnV(-)7AAD(-) (B and D as indicated) or the AnnV(+)7AAD(-) populations (D as indicated) and plotted as a ratio relative to the median fluorescence of AnnV(-)7AAD(-), untreated cells stained under the same condition (B and D). In B, inset shows representative histogram overlays of untreated (UNT, (fill)) or THP-treated (THP (dashed)) CT26 cells stained with anti-calreticulin antibody. THP-treated cells were stained in the presence (block (line)) or absence (THP (dashed)) of the peptide block. (A and C) Percentage of cells in the AnnV(-)/7AAD(-) (viable/pre-apoptotic), AnnV(+)/7AAD(-) (early apoptotic) and AnnV(+)/7AAD(+) (late apoptotic/secondary necrotic) populations are shown. Graphs show averages of 5-10 (A), 5-7 (B), 3-5 (C), or 2-3 (D) experiments. The p-values from two-tailed, paired t-tests are indicated.
Because of the contrast of our data with the findings presented by Obeid et al. (7), we measured changes in surface calreticulin levels in the CT26 mouse colon cancer cell line used in that study. The strong induction of cell-surface calreticulin seen in thapsigargin-treated WT MEFs was also seen in CT26 cells. In contrast, mitoxantrone-treatment did not expose surface calreticulin in CT26 cells, similar to the results obtained with WT MEFs (Fig. 1B). It is possible that expression of cell-surface calreticulin in mitoxantrone–treated cells occurs with different kinetics than those in thapsigargin-treated cells, or in cells that are at a different stage of cell death. To examine impacts of longer time-points of each drug treatment upon surface calreticulin induction, we next examined cells treated with each drug for 17 hours, which induced approximately 2-5 times more apoptotic cells (Fig. 1C). This allowed us to compare surface calreticulin levels on AnnV+ apoptotic cells and AnnV(-) pre-apoptotic cells to those on viable, AnnV(-), untreated cells (Fig. 1D). None of the long drug treatments induced a significant increase in calreticulin staining in apoptotic or pre-apoptotic MEFs although the mitoxantrone-treated, apoptotic cells had a trend suggesting a slight up-regulation of surface calreticulin relative to viable, untreated cells (Fig. 1D).
However, interpretation of some of the data presented in Figure 1D may be complicated by the fact that apoptotic cells are smaller than viable cells (Fig. 2A). To further examine this point, we compared the level of surface calreticulin staining in the presence or absence of the anti-calreticulin peptide block within a given viable or apoptotic cell population (Fig. 2B-C). Figures 2B-C include some of the same data shown in Figure 1B and D, but analyzed differently as described, and also include analyses of additional cell types. These analysis showed similar trends of induction in AnnV(-) cells as measured in Figure 1B. Interestingly, mitoxantrone-treated AnnV(-) cells had slightly higher levels of calreticulin than untreated AnnV(-) WT MEFs. This difference was statistically significant, and specific to the WT MEFs (Fig. 2B). These data analyses also confirmed that apoptotic cells resulting from long thapsigargin treatments do not have elevated levels of cell surface calreticulin, suggesting that the association of calreticulin with the cell surface is transient or unstable (Fig. 2C). Consistent with the data analyses from Figure 1D, the analyses of Figure 2C suggest that apoptotic cells resulting from longer mitoxantrone treatments have elevated levels of calreticulin, although the differences did not achieve statistical significance. Overall, it appears that mitoxantrone-induced surface calreticulin is lower in AnnV(-) cells at early time points relative to that induced by thapsigargin, and quite variable in AnnV(+) cells at later time points. Additionally, whereas thapsigargin-induced calreticulin surface expression was observable in WT MEFs and CT26 cells following short time points of drug treatment, similar treatments of other cell types including primary murine splenocytes, thymocytes, and other cancer cell lines did not strongly induce cell-surface calreticulin (Fig. 2B-C). It is possible that the cancer cell lines display cell surface calreticulin in the absence of drug treatments as recently suggested (32), which accounts for the lower levels of thapsigargin-induced surface calreticulin (Fig. 2B). Further investigations will be needed to understand the cell type specific effects of thapsigargin treatments.
FIGURE 2. Impacts of cell size, cell type and drug treatment on surface calreticulin expression and detection.
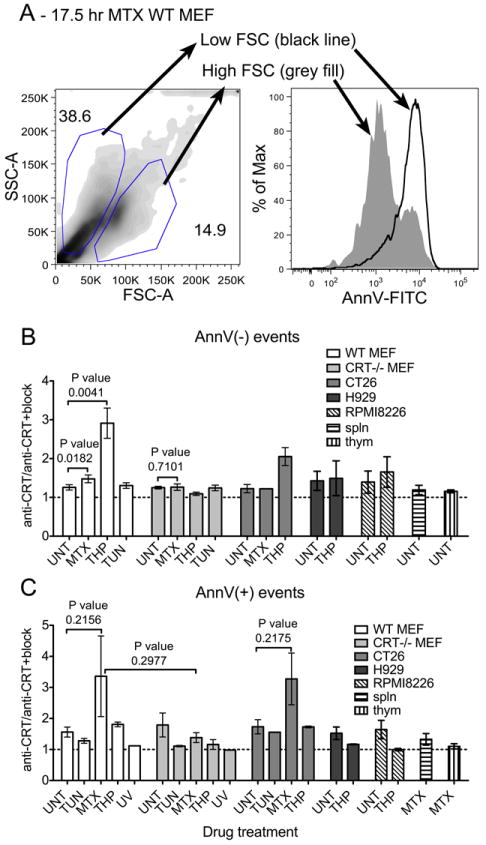
(A) Representative forward scatter (FSC-A) and side-scatter (SSC-A) density plot of MTX-treated apoptotic WT MEFs showing the gating of larger, higher forward scatter cells and smaller, lower forward scatter cells which are AnnV low (larger cells represented by the grey filled peak) and high (smaller cells represented by the black line), respectively, in the adjacent AnnV histogram overlay. (B-C) Graphs show cell surface calreticulin expression of indicated cell types subjected to the indicated treatments for short (B) or long (C) time points unless described otherwise. Levels of cell surface CRT are represented here by the median fluorescence of the anti-CRT stain relative to that of the anti-CRT+block stain. WT MEF, CRT-/- MEF and CT26 bars show data from Figure 1 (with the exception of CT26 long time point/AnnV+ data not shown in Figure 1D which is shown here) re-analyzed in this graph as the CRT/CRT+block ratio for a given cell population. Drug treatment times for the data not shown in Figure 1 were (B and C) THP (H929 & RPMI8226), 4.5 or 6 hrs and (C) TUN (CT26) 17 hrs, THP (CT26) 17 or 21 hrs, MTX (CT26) 14-21 hrs, MTX (Splenocytes and thymocytes), 7-15 hrs. The average cell death profiles (AnnV&7AAD double negative/AnnV single positive) and n values for cell types not described in Figure 1 were: (B and C) H929 (n=2) UNT-82/3.6, THP-56/25; RPMI8226 (n=2) UNT-63/4.3, THP-51/10; (B) splenocytes UNT-58/16.5 (n=5); thymocytes UNT-72/17.9 (n=11) (C) CT26, UNT 94/0.5 (n=4), TUN 93/0.3 (n=1), MTX 68/2.9 (n=3), THP 76/4.3 (n=2); Splenocytes MTX-14.4/17.8 (n=4); thymocytes MTX-7.3/62 (n=7). Unless indicated otherwise, long drug treatments analyzed the combination of non-adherent and adherent cells and short drug treatments analyzed adherent cells only. RPMI8226 & H929 are naturally non-adherent cells. (C) In one experiment, only adherent CT26 cells were analyzed following a 14-hour MTX treatment. The p-values from two-tailed, paired t-tests are indicated.
Several ER resident proteins are released in thapsigargin but not tunicamycin-treated cells and calreticulin surface expression is independent of ERp57 co-translocation
We examined secretion of ER resident proteins in response to 5-hour treatments of WT MEFs with different drugs. Supernatants from the different drug-treated cells were separated by SDS-PAGE and immunoblotting analyses were performed with antibodies directed against gp96 and PDI. In addition, as neither calreticulin nor BiP were detectable by direct immunoblotting analyses, proteins present in supernatants were immunoprecipitated with antibodies against calreticulin and BiP, followed by immunoblotting analyses to detect those proteins. Thapsigargin, but not tunicamycin or mitoxantrone induced secretion of at least four proteins that are normally retained within the ER: calreticulin, gp96, PDI and BiP (Fig. 3A). Cells treated with 200 nM or 5 μM thapsigargin secreted similar levels of gp96 and PDI (Fig. 3B) and induced similar elevations of surface calreticulin expression (data not shown). Although depletion of ER calcium is expected to occur almost immediately following thapsigargin treatment (17), thapsigargin-induced secretion of ER chaperones was not detectable at 1.25 or 2.5 hours following thapsigargin treatment (Fig. 3C). These findings suggest that ER calcium depletion alone is insufficient to induce a detectable loss of ER retention of chaperones. Previous findings from other cell types indicate that BiP (33, 34) and calreticulin (35, 36) mRNA levels are upregulated following 2-5 hours of tunicamycin and thapsigargin treatment and that BiP protein levels are increased within 5 hours of tunicamycin treatment (34). Consistent with these kinetics of UPR induction, an examination of changes in BiP and calreticulin protein levels revealed small increases in calreticulin and BiP expression at 3 hours, and stronger induction 5 hours post-tunicamycin treatment (Fig. 3D). Over the same time-frame, thapsigargin treatment decreased the levels of cellular calreticulin and BiP, with stronger reduction seen at 5 hours compared to 3 hours (Fig. 3D), consistent with the observation of increases in ER chaperone secretion over this time-frame (Fig. 3C). Taken together, these findings suggest that both depletion of ER calcium and increases in ER chaperone expression are required for chaperones to escape ER retention in thapsigargin-treated cells.
FIGURE 3. Various ER chaperones are released from thapsigargin-treated fibroblasts and thapsigargin-induced surface calreticulin expression is independent of calreticulin-ERp57 binding.
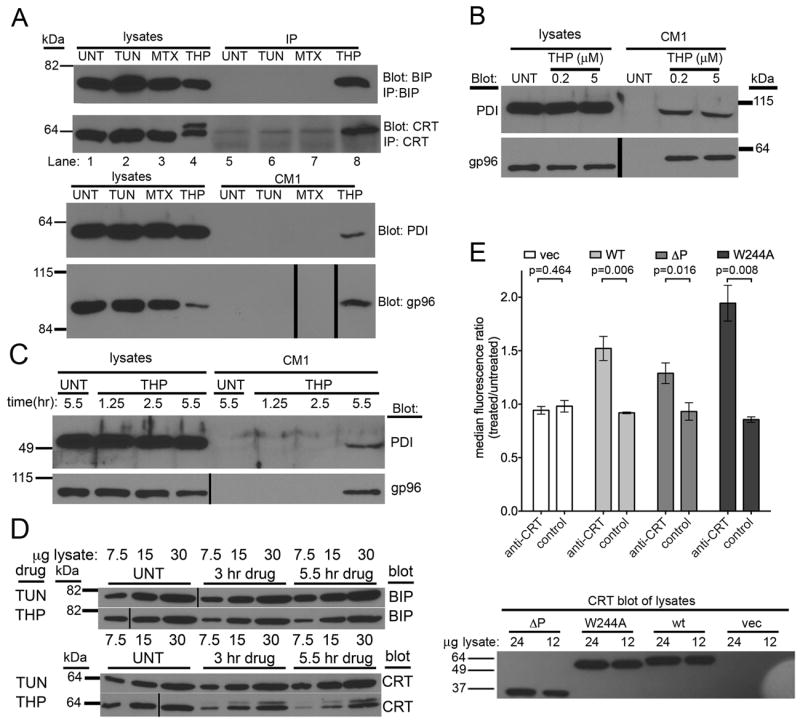
(A) Immunoblotting of cell supernatants and lysates for the presence of various ER chaperones. Supernatants (CM1) from cells treated with TUN, MTX or THP or untreated cells were harvested in parallel with the corresponding cell lysates. Direct immunoblotting analyses of each fraction were performed for PDI and gp96. For CRT and BiP, proteins in cell supernatants were first immunoprecipitated (IP) with anti-CRT or anti-BiP, prior to immunoblotting analyses. (B) Comparison of PDI & gp96 release in response to low or high thapsigargin dose (200 nM or 5 μM). (C) Kinetics of PDI and gp96 secretion. Analyses were performed as in A, but immunoblotting analyses were undertaken at the indicated time points. (D) Kinetics and magnitude of BiP and CRT induction in response to TUN & THP. Indicated microgram amounts of total cell lysates were analyzed as in A at the indicated time-points post-drug treatments. (E) Flow cytometric assessments of ERp57-binding dependence of calreticulin surface expression. Cell-surface calreticulin measured as in Figure 1 before and after thapsigargin (5-7 hrs of 5 μM THP) treatment of CRT-/- MEFs infected with retroviruses encoding wild calreticulin (WT), a calreticulin construct lacking the P-domain (ΔP), a calreticulin point mutant that is unable to interact with ERp57 (W244A), or a virus lacking calreticulin (vec). Representative blots of 3-4 experiments are shown in A. Data are based on (B and C) 1 experiment, (D) 1 experiment shown, representative of 2 total experiments (E) 3-5 experiments. (A) Approximately 8500 cells per lane contributed to the CM1 used to blot for PDI and gp96. The blots of corresponding lysates show approximately 30,000 cells worth of lysate. (A-D) Vertical black lines in blots indicate places where lanes of the blot were re-arranged to match the labeling scheme used in adjacent immunoblots or to digitally remove lanes containing size standard (relevant bands from size standard are shown digitally for each blot). The p-values from two-tailed, paired t-tests are indicated.
The data of Figure 3A indicate that thapsigargin-treatment does not result in a specific release of calreticulin, but rather in a generalized loss of ER retention of various ER proteins. This mechanism of calreticulin release from the ER is distinct from that suggested for anthracyclin-induced calreticulin surface expression, where the specific co-translocation of calreticulin-ERp57 complexes was indicated (26, 27). In the ER, calreticulin and ERp57 interact via the P-domain of calreticulin, and amino acid W244 within the P-domain of calreticulin is important for binding to ERp57 (29, 37, 38). To test the requirement the calreticulin:ERp57 interaction for calreticulin surface expression in thapsigargin-treated cells, wild-type calreticulin, or calreticulin mutants unable to bind to ERp57 (29) (a W244A point mutant and a delta P (ΔP) truncation mutant lacking the P-domain) were expressed in calreticulin deficient MEFs using retroviral infections. Both mutant calreticulin proteins were induced on the cell surface in response to thapsigargin treatment at levels that correlated with their total expression in the lysates, rather than with their abilities to interact with ERp57 (Fig. 3E). Thus, calreticulin does not require association with ERp57 to be expressed on the cell surface in response to ER calcium depletion. Whether ERp57 is present in supernatants of thapsigargin-treated cells is unclear as ERp57 was not detectable in cell supernatants by immunoblotting analyses (data not shown); however ERp57 migrates in close proximity to background bands present in the cell supernatants, and low levels of secreted ERp57 may be masked by the background signals.
In thapsigargin-treated cells, ER chaperones follow the secretory route to gain access to the extracellular compartment
As previously described (39), analysis of cell lysate-derived calreticulin by immunoblotting revealed that thapsigargin treatment of cells induced calreticulin glycosylation (Fig. 3A, calreticulin blot, lane 4) in a subset of calreticulin molecules present in the cell. The glycosylation site of calreticulin appears to be buried under normal conditions, but becomes partly exposed under calcium-depleting conditions (21). It was of note that the glycosylated calreticulin species was absent from cell supernatants, suggesting that the glycosylated form of calreticulin was either ER retained, or preferentially retained on the cell surface (Fig. 3A) (21).
Since calreticulin glycosylation is partial and induced by specific cellular conditions, we assessed the glycosylation status of gp96, a constitutively glycosylated protein, in order to further understand the cellular exit route for ER chaperones in thapsigargin-treated cells. A cytosolic route has recently been described for calreticulin release from apoptotic cells (40). Since cytosolic localization of proteins induces their de-glycosylation, we assessed the glycosylation status of extracellular gp96 in order to further understand whether a secretory or cytosolic route was used in the trafficking of gp96 (and by inference, other ER proteins) to the extracellular space in thapsigargin-treated cells. Previous findings indicate that gp96 secreted in response to cellular calcium perturbation exits the cell through the secretory pathway and becomes differentially glycosylated as it travels through the golgi (16). Consistent with these findings, gp96 from cell supernatants migrated more slowly than gp96 in cell lysates (Fig. 4A). When supernatants from thapsigargin-treated cells were digested with Peptide:N-Glycosidase F (PNGase F), which cleaves carbohydrate residues from glycoproteins, and analyzed by immunoblotting with an anti-gp96 antibody, a decrease in molecular weight was observed, indicating that secreted gp96 is indeed glycosylated (Fig. 4B, gp96 blot, lanes 8 and 9). Finally, the induction of cell-surface calreticulin in response to thapsigargin was reduced when the cells were pre-treated with brefeldin-A (BFA), which blocks ER-golgi trafficking (Fig. 4C). Together, these findings indicate that thapsigargin treatment of cells perturbs normal ER retention mechanisms, and that ER proteins exit the cell via a secretory route in response to thapsigargin treatment.
FIGURE 4. Release of ER chaperones from thapsigargin-treated cells involves the secretory pathway.
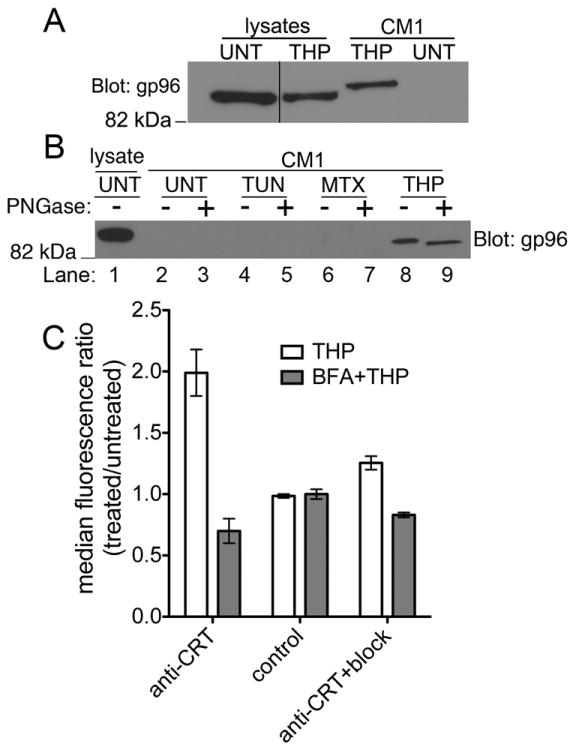
A and B) Immunoblotting analyses with anti-gp96 of supernatants (CM1) and lysates from THP-treated or untreated cells to analyze the presence of glycosylated forms of gp96 in cell supernatants following THP treatment. (A) For this immunoblot, SDS-PAGE-based protein separation was undertaken on an 8% polyacrylamide gel, and separation optimized compared to that shown in 3A, to allow for resolution of different forms of gp96. The supernatant sample (CM1) was also run adjacent to the lysate sample to better resolve alterations in electrophoretic mobility resulting from post-translational modifications. The vertical black line in the gp96 blot denotes lanes that were cut and pasted from the same blot. B) Indicated cell supernatant samples were first digested with Peptide:N-Glycosidase F (+) or left undigested (-), prior to the immunoblotting analyses. C) BFA blockade of the secretory pathway inhibits thapsigargin-induced surface calreticulin expression. Cells were pre-treated with BFA for 45 minutes followed by addition of thapsigargin for 6-7 hours. Cells were harvested and surface calreticulin expression was measured in the AnnV(-) and 7AAD(-) population by flow cytometry as described. Graph shows ratio of surface CRT median fluorescence of BFA+THP-treated/untreated cells. The blot shown in B is representative of 3 experiments and the data in C are the average of 2 experiments.
In a calreticulin-independent manner, thapsigargin-treated cells, cell supernatants and thapsigargin itself induce and enhance innate immune responses more broadly and significantly than corresponding tunicamycin treatments
Using thapsigargin-treated WT or calreticulin deficient MEFs, we investigated whether cell surface and/or extracellular calreticulin were linked to enhanced pro-inflammatory cytokine production. We first examined production of IL-6, a cytokine previously shown to be induced by thapsigargin (28) and tunicamycin (41) treatments of murine macrophages. WT or calreticulin deficient MEFs were treated with tunicamycin, mitoxantrone & thapsigargin for short (5-6.5 hr) or long (20.5 hr) times as described in Fig. 1, harvested, washed extensively with PBS, co-incubated with BMDC or alone, and cytokines in the supernatants of these cultures were quantified by ELISA. BMDC co-incubations with MEFs treated with thapsigargin resulted in higher levels of IL-6 compared to BMDC co-incubations with other target cells (Fig. 5A); however, these effects were independent of calreticulin expression. Thapsigargin-induced increase in IL-6 production was also seen with MEFs subjected to longer time points of drug treatments (Fig. S1B), although the levels of IL-6 measured were lower (Fig. S1B compared to 5A). These findings are consistent with higher levels of MEF apoptosis following long drug treatments (Fig. S1A compared to Fig. 1A), and the expected immunosuppressive effects of apoptotic cells (42). It is noteworthy that mitoxantrone-treated, pre-apoptotic (Fig. 5) or apoptotic (Fig. S1) cells did not induce significant pro-inflammatory cytokine production by BMDC, despite being reported to induce strong anti-tumor immune responses in mice (7).
We also examined production of IL-12, IL-1β, and TNF-α, cytokines that were previously shown to be induced by a membrane-linked extracellular form of gp96 (14) or a bacterially-expressed calreticulin fragment (10). Additionally, we compared induction of IL-23, as both thapsigargin and tunicamycin were recently reported to enhance expression of this cytokine in the presence of TLR agonists (43). Co-incubating thapsigargin-treated MEFs with BMDCs strongly enhanced TLR-dependent (0.5-2 ng/ml LPS) production of IL-1β, IL-12p70 and IL-23 compared to co-incubating other drug-treated or untreated MEFs with BMDC (Fig. 5B-D). Calreticulin was not required for enhanced cytokine production in response to thapsigargin-treated cells, and in fact, calreticulin-deficiency in the thapsigargin-treated MEFs appeared to correlate with higher levels of cytokine production. However, observed differences between wild type and calreticulin-deficient cells were not statistically significant (Fig. 5B-5D). Compared to other drug treatments, thapsigargin-treated cells subjected to long (20.5 hours) drug treatments also stimulated IL-1β and IL-23 (Fig. S1C-D), although at reduced levels compared to co-incubations with MEFs subjected to short drug treatments (Fig. 5). Immunostimulatory effects of tunicamycin-treated apoptotic cells upon IL-23 production were also measurable following longer drug treatments, but at reduced levels compared to THP-treated cells (Fig. S1C).
To better understand the molecular basis for the immunostimulatory effects of thapsigargin-treated cells (Fig. 5), we next examined the impacts of conditioned media (CM) from various drug-treated cells as well as direct drug treatments of BMDC on induction or enhancement of pro-inflammatory cytokine production (Fig. 6). We were particularly interested in asking whether secreted ER chaperones present in supernatants of thapsigargin-treated cells or residual thapsigargin itself might be responsible for the observed pro-inflammatory effects of thapsigargin-treated cells. For these analyses, following each drug treatment of 5-6.5 hours, media containing drugs (CM1) were removed, and cells were washed three times with at least 7 ml PBS to remove soluble drug present in the media. Following these wash steps, fresh media were collected from the washed cells over a 5-6 hour period to generate different MEF CM (CM2). We noted that secretion of PDI and gp96 was detectable for several hours following thapsigargin removal from target cells, although at significantly reduced levels compared to those detected during drug treatment (Fig. S2A, CM1 compared to CM2). Nonetheless, thapsigargin CM2 was able to induce pro-inflammatory cytokine production by BMDC with patterns resembling those induced by thapsigargin-treated cells (Fig. 5 and 6A). Only trace amounts of cytokines, if any, were directly detectable in CM2 from various drug-treated, washed MEFs (Fig. 6A). BMDC mixed with thapsigargin CM2, but not BMDC co-incubated with other CM2 produced IL-6 under sterile conditions (Fig. 6A). Additionally, LPS-dependent production of IL-12p70, IL-23 and IL-1β strongly synergized with stimulation by thapsigargin CM2 to yield more IL-1β, IL-12p70, and IL-23 than other BMDC treatments (Fig. 6A). However, BMDC cytokine production in response to thapsigargin CM2 was clearly independent of calreticulin expression in MEFs (Fig. 6A). The different drug-treated, washed cells used to generate CM2 were also harvested and stained with AnnV and 7AAD and analyzed by flow cytometry. These analyses indicated that, compared to other treatments, the thapsigargin CM2 generation protocol did not enhance levels of apoptotic/secondary necrotic cells (Fig. S2B). Therefore, necrosis-related factors were not a likely explanation for the immunostimulatory properties of thapsigargin-treated cell supernatants. Additionally, there was no correlation between the level of secondary necrotic cells in a given treatment group and its ability to stimulate the BMDC (Fig. S2B and 6A).
It is noteworthy that IL-12p70, IL-23 and IL-1β were not significantly induced by thapsigargin CM2 or thapsigargin-treated cells under sterile conditions (Fig. 5B-D, Fig. 6A, THP(-)LPS bars). On the other hand, IL-12p40 and IL-1β were both previously reported to be induced under sterile conditions by a membrane-linked form of gp96 (14). Furthermore, a recombinant, N-terminally truncated calreticulin fragment purified from E. coli induced TNF-α production by mouse peritoneal macrophages and human PBMC in the absence of a TLR agonist (10). However, TNF-α was also not induced by thapsigargin CM2 or thapsigargin-treated cells (wild type or calreticulin-deficient) under sterile conditions (Fig. S2C-D). Thapsigargin CM2 did enhance TNF-α production mediated by LPS but it was independent of calreticulin expression by the target cells (Fig. S2C-D). Thus, despite containing various extracellular ER chaperones, thapsigargin-treated cell supernatants and cells induced only the production of IL-6 under sterile conditions, and therefore were not as broadly immunostimulatory as predicted by the presence of extracellular gp96 or calreticulin (10, 14).
Previous studies have shown that treatment of cells with tunicamycin or thapsigargin stimulates activation of NF-κB in HeLa cells (44), production of reactive oxygen species (ROS) in MEFs (45), and significantly enhanced production of type I IFN, TNF-α (46), ISG15 and IL-6 (41) by LPS-treated macrophages. Furthermore, thapsigargin and tunicamycin were both shown to enhance IL-23 but not IL-12 production by human monocyte-derived DC in response to a mixture of LPS and a TLR-8 agonist (CL-097) (43). We asked whether direct addition of tunicamycin or thapsigargin to murine BMDC could stimulate production of any of the cytokines measured in Figures 5, and 6A. The levels of cytokines resulting from drug treatment of BMDC and CM treatments of BMDC were compared within the same experiment (Fig. 6B). Thapsigargin and tunicamycin treatment of BMDC both stimulated production of IL-6 in the absence of LPS, although thapsigargin stimulated on average ~9.8-fold more IL-6 than tunicamycin at the drug concentrations and time points used in the analyses (Fig. 6B, 9.8 fold was the average enhancement from five experiments, of which four are shown). Thapsigargin and tunicamycin treatments of BMDC stimulated production of IL-1β and IL-23 (Fig. 6B) in response to 1-10 ng/ml LPS. However, under the conditions tested, thapsigargin was a stronger inducer of the cytokines, particularly of IL-1β. As noted previously with human monocyte-derived DC (43), lower concentrations of thapsigargin resulted in higher levels of IL-23 compared to higher concentrations of thapsigargin. In contrast to the previous studies with human myeloid DCs, thapsigargin (but not tunicamycin) treatment also enhanced IL-12p70 production in response to LPS (Fig. 6B). Importantly, the patterns of cytokine induction by thapsigargin-treated cell supernatants were strikingly similar to those induced by direct thapsigargin treatment of BMDC (Fig. 6B). As noted above, in generating thapsigargin CM2 for Figure 6A, thapsigargin was removed from the MEFs, and MEFs were washed sufficiently to reduce the extracellular, soluble drug concentration to < 15 pM, prior to collection of CM2. It was possible, however, that thapsigargin was sequestered within cell membranes or intracellularly and subsequently partitioned into the media. In support of this possibility, we found that when CM1 (cell supernatants from drug-treated cells) and CM2 (cell supernatants from drug-treated, washed cells) were extensively dialyzed in centricons (3 KDa or 10 KDa) to remove free soluble drug that was present, the majority of cytokine-inducing activity was present in the retentates rather than in the flow through, even though thapsigargin has a molecular weight of 650 Da (Fig. S3). Based on these findings, it is likely that the activity of thapsigargin-treated cell supernatants (CM1 and CM2) (Figs. 6 and S3) and their resemblance to direct thapsigargin treatment of BMDC (Fig. 6B) result from sequestration of thapsigargin within cell membranes or intracellularly during drug treatment of MEFs, and subsequent partitioning of active drug from MEFs into the CM within higher molecular weight membrane vesicles or protein complexes. This would allow thapsigargin to directly stimulate BMDC. Thapsigargin is hydrophobic and requires organic solvents (such as DMSO) for its solubilization, and thus its partitioning into membranes is perhaps not unexpected.
If residual drug in thapsigargin-treated cells was responsible for the BMDC stimulating activity, we expected that reducing the concentration of thapsigargin used to treat MEFs would reduce the immunostimulatory capacity of thapsigargin-treated cells. Thus, we compared abilities of 5 μM or 200 nM thapsigargin CM1 and CM2 to induce production of various pro-inflammatory cytokines. Whereas treatments of BMDC with 5 μM thapsigargin CM1 or CM2 induced IL-6 production, treatments of BMDC with 200 nM thapsigargin CM1 or CM2 did not induce sterile IL-6 production (Fig. 6B). Furthermore, reduced levels of LPS-induced IL-1β and IL-23 (Fig. 6B) were observed when the 200 nM thapsigargin CM1 or CM2 were compared to the 5 μM thapsigargin CM1 or CM2. Notably levels of chaperone secretion were very similar in 200 nM and 5 μM thapsigargin CM1 (Fig. 3B).
Together, these findings indicate that production of pro-inflammatory cytokines induced by thapsigargin-treated cells or CM was not enhanced by calreticulin expression by target cells (Fig. 5, 6 S2C-D and S3). Notably, the profiles of pro-inflammatory cytokine induction by thapsigargin-treated cells and dialyzed CM closely resembled those induced by thapsigargin itself, suggesting that residual thapsigargin within protein/lipid complexes was responsible for enhanced cytokine production (Fig. 6B). Significantly, the effects of direct BMDC treatment with thapsigargin and tunicamycin were different, with direct thapsigargin treatment being more strongly pro-inflammatory.
Calreticulin expression enhances phagocytic clearance of thapsigargin-treated cells, but not of tunicamycin or UV-treated cells
Because cell-surface calreticulin is an eat-me signal in the context of apoptotic cells (5), pre-apoptotic tumor cells (7), and live tumor cells subject to CD47 blockade (32) we asked if thapsigargin-treated cells would also be phagocytosed in a calreticulin-dependent manner. A previously published flow-cytometry-based assay (7) was used to measure phagocytosis. WT or calreticulin deficient MEFs were labeled with a flourophore, then drug or UV-treated, washed and incubated with BMDC. The co-cultures were then fixed and stained with an antibody specific for the DC-specific marker CD-11c. The percentage of CD-11c+ events that were also positive for the target-cell marker were recorded as a measure of target-cell:DC association. The ratio of DC:target cell association in co-cultures incubated at 37° C relative to that of co-cultures incubated at 4° C is reported as the phagocytic index (the 37 °C readings quantify adhesion and phagocytosis, whereas the 4 °C readings quantify adhesion alone). Uptake of UV-treated MEFs was significantly greater than untreated MEFs, but was independent of calreticulin expression by the MEFs (Fig. 7A). The latter finding was surprising in light of a previously suggested role for calreticulin in the phagocytic uptake of apoptotic UV-treated fibroblasts using fibroblasts or a macrophage cell line as the phagocyte (5). Therefore surface calreticulin expression was tested in the UV-treated fibroblasts, which were triple stained for calreticulin as well as markers of apoptosis and plasma membrane integrity, as defined in the analyses of Figure 1. Our analyses indicated that calreticulin is not present or significantly induced on the surface of AnnV+PI- (Fig. 7B) or AnnV-PI- (data not shown) MEFs in response to UV treatment, consistent with the absence of significant, calreticulin-dependent uptake of UV-treated cells. Several differences in protocol may explain these discrepancies. These include the use of a different phagocyte (BMDC (Fig. 7) rather than macrophages or fibroblast (5)), a different phagocytosis assay (flow cytometry (Fig. 7) rather than microcopy (5)), and the use of both adherent and non-adherent UV-treated target cells (Fig. 7), rather than non-adherent UV-treated target cells (5).
FIGURE 7. Calreticulin enhances phagocytic uptake of thapsigargin-treated target cells, but not of UV or tunicamycin-treated target cells.
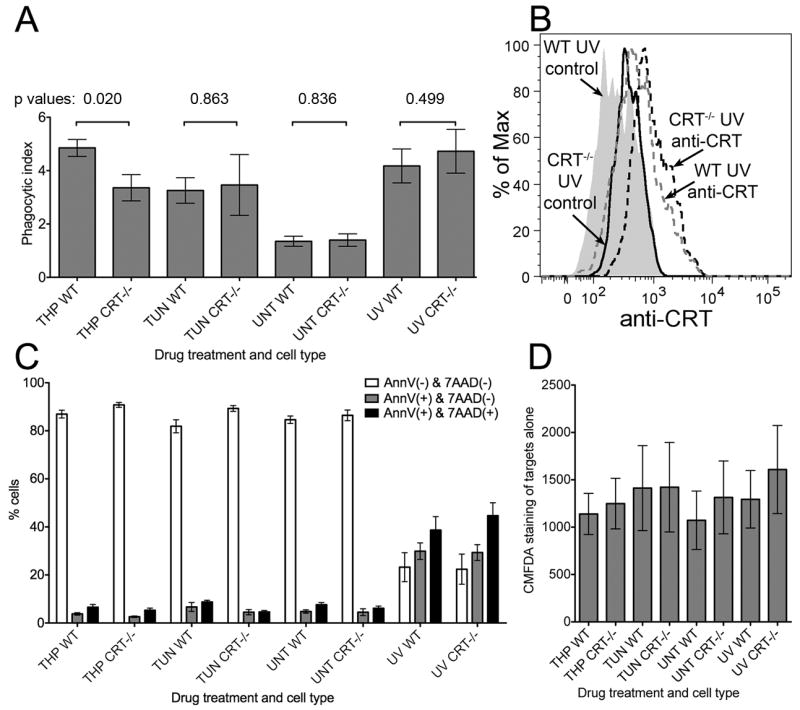
Cell Tracker Green (5-chloromethylfluorescein diacetate; CMFDA) labeling of MEFs, cell death profiles, phagocytic uptake, and surface calreticulin expression following different treatments. WT or CRT-/- MEFs were labeled with Cell Tracker Green, exposed to UV light for 3 minutes and then cultured for an additional 16-24 hrs, or exposed to thapsigargin or tunicamycin for 5-6 hours, or left untreated. (A) Phagocytic uptake: Labeled and treated cells were washed 3 times, mixed with BMDC at an effector to target ratio of 1:5, and incubated for 1 hour at 37° C or 4° C. Co-cultures were fixed, stained with anti-CD11c and analyzed by flow cytometry. For each cell type and treatment condition, the percentages of CMFDA+ events in the CD11c+ gate were measured for the 4° C and 37° C plates, and phagocytic index is plotted as the 37 °C/4 °C ratio of those percentages. The phagocytic index data are averages of 8 total experiments for all conditions with the exception of TUN, which shows the average of four experiments. On average 3-6% of the total added target cells were phagocytosed (B) Surface calreticulin expression was measured in UV-treated WT & CRT-/- MEFs as described in Fig. 1, and data are representative of 3 experiments. C) Cell death profiles: CMFDA-labeled MEFs subject to indicated treatments were stained with AnnV-PE and 7AAD to quantify the percentages of MEFs in the indicated populations. Graphs show the averages of 6 experiments for all conditions except TUN treatments, which show the average of 3 experiments. AnnV/7AAD data were not collected in 2/8 experiments. (D) CMFDA fluorescences of various treated MEFs were measured on the FITC channel. Averages of 8 total experiments are shown for all conditions except TUN treatment, which shows the average of 4 experiments. The p-values from two-tailed, paired t-tests are indicated.
In contrast to UV-treated MEFs, thapsigargin-treated WT MEFs were phagocytosed with significantly higher efficiency than thapsigargin-treated calreticulin deficient MEFs and untreated MEFs (Fig. 7A). The levels of target cell death were similar between WT and calreticulin deficient MEF target cells within a treatment group (Fig. 7C). Additionally, the level of dead target cells in the untreated group was least as high as that in the thapsigargin-treated group (Fig. 7C), a result which indicated that cell death in the thapsigargin-group was insufficient to account for the level of phagocytic uptake observed. Finally, although tunicamycin treatment enhanced phagocytic uptake of target cells relative to untreated cells, there was no calreticulin-dependence to this effect, consistent with the inability to observe cell-surface calreticulin in tunicamycin-treated cells (Fig. 1B and 2B). The fluorescence intensities of the various labeled target cells were similar, and could not explain the observed differences in the phagocytic indices (Fig. 7D). Taken together, these results suggest that thapsigargin-treated cells with measurable cell-surface calreticulin are phagocytosed in a manner that is partially calreticulin-dependent.
DISCUSSION
Cell-surface calreticulin has been characterized as an important factor in inducing anti-tumor immune responses (7, 8, 26, 47). Anti-cancer regimens that induce an anti-tumor immune response are likely to be more effective than non-immunogenic regimens with similar cancer cell cytotoxicity (4). Identifying conditions that induce cell-surface calreticulin, and understanding mechanisms and functional consequences of cell-surface calreticulin expression could help design more effective anti-tumor therapies. Our findings indicate that thapsigargin induces transient, pre-apoptotic, cell-surface calreticulin exposure in some cell types (Fig. 1 and Fig. 2), that thapsigargin treatment of target cells increases pro-inflammatory cytokine production by BMDC in a manner independent of target cell-derived calreticulin (Fig. 5), that extracellular calreticulin either in a cell-surface bound (Fig. 5 and S2D) or soluble form (Fig. 6, S2C) is unable to stimulate production of IL-6, IL-1β, TNF-α, IL-23 or IL-12p70, and that thapsigargin treatment of cells impacts their phagocytic uptake in a calreticulin-dependent manner (Fig. 7).
Two mechanisms are previously described for cell-surface calreticulin exposure (8, 40). The first pathway is dependent on the PERK arm of the UPR, and results in the specific co-translocation of calreticulin-ERp57 complexes to the cell surface (8). The second suggested pathway, relevant to apoptotic cells, involves the binding of cytosolic calreticulin to phosphatidylserine on the inner leaflet of the plasma membrane prior to apoptotic exposure of phosphatidylserine-calreticulin complexes on the cell surface (40). Neither of these pathways seems related to the mechanisms of surface exposure of calreticulin in viable cells subjected to ER calcium depletion, which display a general loss of ER retention of various ER chaperones. The thapsigargin-induced mode of calreticulin surface expression is independent of calreticulin-ERp57 binding (Fig. 3E). Rather, we have recently shown that induction of a generic polypeptide-binding site on calreticulin facilitates its cell-surface expression under calcium-depleting conditions (21).
Our data indicate a secretory route for ER proteins (Fig. 4), as also suggested for the calreticulin-ERp57 co-translocation mechanism (8). It is noteworthy that several of the ER resident proteins detectable in supernatants of thapsigargin-treated cells (Fig. 3) are known calcium binding proteins of the ER (48). As is the case with calreticulin (49), ER retention of BiP, PDI and gp96 may in part involve calcium-dependent processes. However, calcium-depletion alone appears insufficient to induce secretion of the ER chaperones (Fig 3C-D). Rather, the combined effects of KDEL receptor saturation and calcium-depletion appear to be the mechanism that drives secretion of the ER chaperones in thapsigargin-treated cells. Similar mechanisms may be relevant to the extracellular localization of ER chaperones under physiological protein misfolding conditions, known to induce the UPR and alter ER calcium homeostasis. To investigate this possibility, we compared chaperone secretion in wild type and cog/cog mice thyroids, which express a misfolded, mutant thyroglobulin that accumulates in the ER (50). In preliminary findings, we observed enhanced secretion of calreticulin, PDI and gp96 into the apical follicular lumen of cog/cog mice thyroids compared to their wild type counterparts, correlating with significant enhancement in total chaperone levels in the cog/cog thyroid lysates (Jeffery, Kellogg, Arvan and Raghavan, unpublished observations).
Previous studies in HeLa cells have shown that NF-κB activation in response to agents that induce ER stress involves the release of calcium from the ER and the subsequent generation of ROS (reviewed in (51)). This mechanism likely accounts for sterile IL-6 production in response to direct thapsigargin treatments of BMDC (Fig. 6B). Thapsigargin is a stronger activator of the sterile IL-6 response than tunicamycin (Fig. 6B) likely due to the rapid of effect of thapsigargin on cytosolic calcium elevation, whereas elevation of cytosolic calcium in response to tunicamycin treatment may be more modest as well as kinetically delayed. Many recent studies have shown synergies between TLR-derived signals and transcription factors induced by the UPR, including XBP-1 and C/EBP homologous protein (CHOP). Of the cytokines measured in Figures 5 and 6, XBP-1 has been previously shown to synergize with TLR signals in enhancing IL-6 production by murine macrophages (41). In human, monocyte-derived DC, CHOP binding to the IL-23p19 promoter enhanced IL-23 production in response to TLR ligation (43). Similar mechanisms could account for the synergy of ER stress signals and LPS in IL-23 production by BMDC (Fig. 6B). We found that thapsigargin also strongly synergizes with LPS in the generation of IL-1β and IL-12p70. Recent findings have shown that high avidity ligation of ITAM-containing receptors such as DAP12 can synergize with TLR signals to activate pro-inflammatory gene expression (reviewed in (52, 53)). High avidity ligation of DAP12 triggers an acute, transient calcium increase (reviewed in (52)). Here we show that, compared to tunicamycin, thapsigargin treatment strongly enhances various pro-inflammatory responses to LPS (Fig. 6B), consistent with similar findings from previous studies (45, 46). These results point towards direct synergies between cytosolic calcium and TLR responses, and broader synergy between intracellular calcium and TLR signaling compared to those between the UPR and TLR signaling. Interestingly, it has been suggested that ER calcium depletion can occur under certain conditions of ER overload and protein polymerization, even in the absence of classical UPR induction (54). Thus, findings described here may be relevant to a better understanding of inflammatory protein folding disorders that do not induce a classical UPR pathway.
A truncated bacterially-expressed calreticulin construct was shown to induce TNF-α production by murine macrophages and human PBMC (10). N-terminal truncation of calreticulin induces self-association likely via the exposure of hydrophobic surfaces (10, 29), and it is possible that protein truncation induces enhanced binding to bacterial pathogen-associated molecular patterns. We show here that full-length extracellular calreticulin derived from an endogenous source does not enhance the production of various pro-inflammatory cytokines in a thapsigargin-treated BMDC context (Figs. 5, 6, S2C-D and S3). These findings are consistent with a previous report that a secreted form of calreticulin does not induce pro-inflammatory cytokine production by BMDC (55). Calreticulin is known to be present in the extracellular environment under a number of physiological conditions, and a number of extracellular functions are described for calreticulin (reviewed in (3)). Given these extracellular roles of the protein, the lack of innate immune induction by endogenously-derived, extracellular calreticulin is perhaps not surprising.
Calreticulin-high, thapsigargin-treated, target cells were phagocytosed more efficiently than untreated cells, and enhanced phagocytosis was partly dependent on calreticulin expression by the target cells (Fig. 7). Correlating with undetectable levels of surface calreticulin in UV-treated cells, we did not observe an effect of calreticulin on the phagocytosis of UV-treated cells (Fig. 7). In the UV-treated apoptotic cell context, other changes are expected that promote phagocytic uptake (reviewed in (56)), including the exposure of phosphatidylserine, which likely explain why significant uptake was observed in the absence of any calreticulin expression by the target cells (Fig. 7). Correspondingly, UV-treated target cells had a higher level of total association with BMDC than any other target cell group (Fig. S4). This is likely the result of molecules that are upregulated on the surface of apoptotic cells and responsible for bridging apoptotic phagocytic cargo to potential phagocytes (reviewed in (56)). It is important to consider that increases in surface calreticulin by a magnitude measurable by flow cytometry were, in general, difficult to detect in apoptotic cells (Figs. 1-2). Stronger non-specific protein binding to apoptotic cells could render a specific signal more difficult to detect by flow cytometry. Microscopic observations could allow for calreticulin redistribution or upregulation to be more readily visualized on apoptotic cells, as described in other studies (5, 9, 56). It is also important to note that cell-surface calreticulin functions in the context of several other key eat-me and don’t-eat-me signals in viable and apoptotic cells (5, 56, 57). The data of Figure 7 show that tunicamycin-treated cells displayed enhanced phagocytic uptake compared to untreated cells, although this uptake was not calreticulin-dependent. This observation correlates with the absence of detectable surface calreticulin in tunicamycin-treated cells (Fig. 1B). The same level of uptake was seen with thapsigargin-treated, calreticulin-deficient cells (Fig. 7A). It is possible that interference with proper protein folding resulting from tunicamycin and thapsigargin treatments also cause decreased surface expression of don’t-eat-me signals like CD47 (5) and plasminogen activator inhibitor-1 (PAI-1) (57). This potential decreased surface expression of don’t-eat-me signals may explain the increased level of uptake of tunicamycin-treated and thapsigargin-treated, calreticulin deficient cells by BMDC.
In contrast to some previous findings (7), we show here that thapsigargin induces cell-surface calreticulin, and that thapsigargin promotes phagocytic uptake of cells via mechanisms that are, in part, calreticulin-dependent. Thus the inability of thapsigargin-treated cells to induce anti-tumor immunity with sufficient potency (7, 47), is likely not related to an absence of surface calreticulin per se. Other immunogenic signals absent in thapsigargin-treated cells (Fig. S2E-F), including HMGB1 (58) and elevated levels of ATP (59), may be required to confer stronger immunogenicity to tumor cells. Consequently, although thapsigargin has some potentially desirable attributes for a chemotherapeutic, including the abilities to induce stronger phagocytic uptake of treated cells by DC (Fig. 7) and elicit stronger cytokine production by DC (Fig. 5 and 6), these appear to be insufficient signals to confer anti-tumor immune protection. These observations are highly relevant to the ongoing clinical trials evaluating thapsigargin pro-drugs for the treatment of advanced solid tumors (ClinicalTrials.gov Identifier: NCT01056029, (60)). Since phagocytic uptake and cytokine levels can influence T cell priming steps, our results highlight the importance of further research investigating the impacts of different forms of ER stress on the priming of CD8 and CD4 T cell responses.
Supplementary Material
Acknowledgments
We would like to thank the transgenic mouse core for providing mouse bone marrow for dendritic cell preparations, Joanne Sonstein, Natasha Del Cid and Dr. Jeff Curtis for assistance with the development of phagocytosis assays, Dr. Yasmina Laouar for advice on protocols for BMDC preparations and flow cytometry, Joel Whitfield for performing the ELISA assays, Elise Jeffery for assistance with the project, and Drs. Malathi Kandarpa and Andrzej Jakubowiak for the indicated cell lines.
Footnotes
This work was supported by NIH grant AI066131 (to MR), by a pilot grant from the University of Michigan Rheumatic Disease core center (to MR) and by Immunology and Cellular Biotechnology Training Program fellowships (to LRP). This work utilized the Hybridoma Core(s) of the Michigan Diabetes Research and Training Center funded by NIH5P60 DK20572 from the National Institute of Diabetes & Digestive & Kidney Diseases.
List of abbreviations used in this paper: mouse embryonic fibroblast (MEF), bone marrow derived dendritic cell (BMDC), Brefeldin-A (BFA), conditioned media (CM), thapsigargin (THP), tunicamycin (TUN), mitoxantrone (MTX), 5-chloromethylfluorescein diacetate (CMFDA), endoplasmic reticulum (ER), calreticulin (CRT), wild type (WT), heat shock protein (HSP), unfolded protein response (UPR), 7-Aminoactinomycin D (7AAD), Annexin-V (AnnV), reactive oxygen species (ROS), High Mobility Group Box-1 (HMGB1), and Figure (Fig.).
References
- 1.Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal. 2006;8:1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- 2.Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J. 2009;417:651–666. doi: 10.1042/BJ20081847. [DOI] [PubMed] [Google Scholar]
- 3.Gold LI, Eggleton P, Sweetwyne MT, Van Duyn LB, Greives MR, Naylor SM, Michalak M, Murphy-Ullrich JE. Calreticulin: non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010;24:665–683. doi: 10.1096/fj.09-145482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353–363. doi: 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 6.Obeid M, Panaretakis T, Joza N, Tufi R, Tesniere A, van Endert P, Zitvogel L, Kroemer G. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007;14:1848–1850. doi: 10.1038/sj.cdd.4402201. [DOI] [PubMed] [Google Scholar]
- 7.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 8.Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, Durchschlag M, Joza N, Pierron G, van Endert P, Yuan J, Zitvogel L, Madeo F, Williams DB, Kroemer G. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28:578–590. doi: 10.1038/emboj.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuraishi T, Manaka J, Kono M, Ishii H, Yamamoto N, Koizumi K, Shiratsuchi A, Lee BL, Higashida H, Nakanishi Y. Identification of calreticulin as a marker for phagocytosis of apoptotic cells in Drosophila. Exp Cell Res. 2007;313:500–510. doi: 10.1016/j.yexcr.2006.10.027. [DOI] [PubMed] [Google Scholar]
- 10.Hong C, Qiu X, Li Y, Huang Q, Zhong Z, Zhang Y, Liu X, Sun L, Lv P, Gao XM. Functional analysis of recombinant calreticulin fragment 39-272: implications for immunobiological activities of calreticulin in health and disease. J Immunol. 2010;185:4561–4569. doi: 10.4049/jimmunol.1000536. [DOI] [PubMed] [Google Scholar]
- 11.Henderson B, Calderwood SK, Coates AR, Cohen I, van Eden W, Lehner T, Pockley AG. Caught with their PAMPs down? The extracellular signalling actions of molecular chaperones are not due to microbial contaminants. Cell Stress Chaperones. 2010;15:123–141. doi: 10.1007/s12192-009-0137-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007;109:4839–4845. doi: 10.1182/blood-2006-10-054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsan MF, Gao B. Heat shock proteins and immune system. J Leukoc Biol. 2009;85:905–910. doi: 10.1189/jlb.0109005. [DOI] [PubMed] [Google Scholar]
- 14.Liu B, Dai J, Zheng H, Stoilova D, Sun S, Li Z. Cell surface expression of an endoplasmic reticulum resident heat shock protein gp96 triggers MyD88-dependent systemic autoimmune diseases. Proc Natl Acad Sci U S A. 2003;100:15824–15829. doi: 10.1073/pnas.2635458100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drummond IA, Lee AS, Resendez E, Jr, Steinhardt RA. Depletion of intracellular calcium stores by calcium ionophore A23187 induces the genes for glucose-regulated proteins in hamster fibroblasts. J Biol Chem. 1987;262:12801–12805. [PubMed] [Google Scholar]
- 16.Booth C, Koch GL. Perturbation of cellular calcium induces secretion of luminal ER proteins. Cell. 1989;59:729–737. doi: 10.1016/0092-8674(89)90019-6. [DOI] [PubMed] [Google Scholar]
- 17.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tufi R, Panaretakis T, Bianchi K, Criollo A, Fazi B, Di Sano F, Tesniere A, Kepp O, Paterlini-Brechot P, Zitvogel L, Piacentini M, Szabadkai G, Kroemer G. Reduction of endoplasmic reticulum Ca2+ levels favors plasma membrane surface exposure of calreticulin. Cell Death Differ. 2008;15:274–282. doi: 10.1038/sj.cdd.4402275. [DOI] [PubMed] [Google Scholar]
- 19.Delpino A, Castelli M. The 78 kDa glucose-regulated protein (GRP78/BIP) is expressed on the cell membrane, is released into cell culture medium and is also present in human peripheral circulation. Biosci Rep. 2002;22:407–420. doi: 10.1023/a:1020966008615. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem. 2010;285:15065–15075. doi: 10.1074/jbc.M109.087445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeffery E, Peters LR, Raghavan M. The polypeptide binding conformation of calreticulin facilitates its cell-surface expression under conditions of endoplasmic reticulum stress. J Biol Chem. 2011;286:2402–2415. doi: 10.1074/jbc.M110.180877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevens FJ, Argon Y. Protein folding in the ER. Semin Cell Dev Biol. 1999;10:443–454. doi: 10.1006/scdb.1999.0315. [DOI] [PubMed] [Google Scholar]
- 23.Sambrook JF. The involvement of calcium in transport of secretory proteins from the endoplasmic reticulum. Cell. 1990;61:197–199. doi: 10.1016/0092-8674(90)90798-j. [DOI] [PubMed] [Google Scholar]
- 24.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Panaretakis T, Joza N, Modjtahedi N, Tesniere A, Vitale I, Durchschlag M, Fimia GM, Kepp O, Piacentini M, Froehlich KU, van Endert P, Zitvogel L, Madeo F, Kroemer G. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008;15:1499–1509. doi: 10.1038/cdd.2008.67. [DOI] [PubMed] [Google Scholar]
- 27.Obeid M. ERP57 membrane translocation dictates the immunogenicity of tumor cell death by controlling the membrane translocation of calreticulin. J Immunol. 2008;181:2533–2543. doi: 10.4049/jimmunol.181.4.2533. [DOI] [PubMed] [Google Scholar]
- 28.Bost KL, Mason MJ. Thapsigargin and cyclopiazonic acid initiate rapid and dramatic increases of IL-6 mRNA expression and IL-6 secretion in murine peritoneal macrophages. J Immunol. 1995;155:285–296. [PubMed] [Google Scholar]
- 29.Del Cid N, Jeffery E, Rizvi SM, Stamper E, Peters LR, Brown WC, Provoda C, Raghavan M. Modes of calreticulin recruitment to the major histocompatibility complex class I assembly pathway. J Biol Chem. 2010;285:4520–4535. doi: 10.1074/jbc.M109.085407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torchinsky MB, Garaude J, Martin AP, Blander JM. Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature. 2009;458:78–82. doi: 10.1038/nature07781. [DOI] [PubMed] [Google Scholar]
- 31.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 32.Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, Weiskopf K, Willingham SB, Raveh T, Park CY, Majeti R, Weissman IL. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med. 2010;2:63ra94. doi: 10.1126/scitranslmed.3001375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okada T, Haze K, Nadanaka S, Yoshida H, Seidah NG, Hirano Y, Sato R, Negishi M, Mori K. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J Biol Chem. 2003;278:31024–31032. doi: 10.1074/jbc.M300923200. [DOI] [PubMed] [Google Scholar]
- 34.Winnay JN, Boucher J, Mori MA, Ueki K, Kahn CR. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat Med. 2010;16:438–445. doi: 10.1038/nm.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Llewellyn DH, Kendall JM, Sheikh FN, Campbell AK. Induction of calreticulin expression in HeLa cells by depletion of the endoplasmic reticulum Ca2+ store and inhibition of N-linked glycosylation. Biochem J. 1996;318(Pt 2):555–560. doi: 10.1042/bj3180555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waser M, Mesaeli N, Spencer C, Michalak M. Regulation of calreticulin gene expression by calcium. J Cell Biol. 1997;138:547–557. doi: 10.1083/jcb.138.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frickel EM, Riek R, Jelesarov I, Helenius A, Wuthrich K, Ellgaard L. TROSY-NMR reveals interaction between ERp57 and the tip of the calreticulin P-domain. Proc Natl Acad Sci U S A. 2002;99:1954–1959. doi: 10.1073/pnas.042699099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin V, Groenendyk J, Steiner SS, Guo L, Dabrowska M, Parker JM, Muller-Esterl W, Opas M, Michalak M. Identification by mutational analysis of amino acid residues essential in the chaperone function of calreticulin. J Biol Chem. 2006;281:2338–2346. doi: 10.1074/jbc.M508302200. [DOI] [PubMed] [Google Scholar]
- 39.Rizvi SM, Mancino L, Thammavongsa V, Cantley RL, Raghavan M. A polypeptide binding conformation of calreticulin is induced by heat shock, calcium depletion, or by deletion of the C-terminal acidic region. Mol Cell. 2004;15:913–923. doi: 10.1016/j.molcel.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 40.Tarr JM, Young PJ, Morse R, Shaw DJ, Haigh R, Petrov PG, Johnson SJ, Winyard PG, Eggleton P. A mechanism of release of calreticulin from cells during apoptosis. J Mol Biol. 2010;401:799–812. doi: 10.1016/j.jmb.2010.06.064. [DOI] [PubMed] [Google Scholar]
- 41.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 43.Goodall JC, Wu C, Zhang Y, McNeill L, Ellis L, Saudek V, Gaston JS. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci U S A. 2010;107:17698–17703. doi: 10.1073/pnas.1011736107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pahl HL, Baeuerle PA. A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by transcription factor NF-kappa B. EMBO J. 1995;14:2580–2588. doi: 10.1002/j.1460-2075.1995.tb07256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsieh YH, Su IJ, Lei HY, Lai MD, Chang WW, Huang W. Differential endoplasmic reticulum stress signaling pathways mediated by iNOS. Biochem Biophys Res Commun. 2007;359:643–648. doi: 10.1016/j.bbrc.2007.05.154. [DOI] [PubMed] [Google Scholar]
- 46.Smith JA, Turner MJ, DeLay ML, Klenk EI, Sowders DP, Colbert RA. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur J Immunol. 2008;38:1194–1203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, Michaud M, Menger L, Gdoura A, Tajeddine N, Tesniere A, Zitvogel L, Kroemer G. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene. 2011;30:1147–1158. doi: 10.1038/onc.2010.500. [DOI] [PubMed] [Google Scholar]
- 48.Coe H, Michalak M. Calcium binding chaperones of the endoplasmic reticulum. Gen Physiol Biophys. 2009;28:F96–F103. Spec No Focus. [PubMed] [Google Scholar]
- 49.Sonnichsen B, Fullekrug J, Nguyen Van P, Diekmann W, Robinson DG, Mieskes G. Retention and retrieval: both mechanisms cooperate to maintain calreticulin in the endoplasmic reticulum. J Cell Sci. 1994;107(Pt 10):2705–2717. doi: 10.1242/jcs.107.10.2705. [DOI] [PubMed] [Google Scholar]
- 50.Kim PS, Hossain SA, Park YN, Lee I, Yoo SE, Arvan P. A single amino acid change in the acetylcholinesterase-like domain of thyroglobulin causes congenital goiter with hypothyroidism in the cog/cog mouse: a model of human endoplasmic reticulum storage diseases. Proc Natl Acad Sci U S A. 1998;95:9909–9913. doi: 10.1073/pnas.95.17.9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pahl HL, Baeuerle PA. The ER-overload response: activation of NF-kappa B. Trends Biochem Sci. 1997;22:63–67. doi: 10.1016/s0968-0004(96)10073-6. [DOI] [PubMed] [Google Scholar]
- 52.Ivashkiv LB. A signal-switch hypothesis for cross-regulation of cytokine and TLR signalling pathways. Nat Rev Immunol. 2008;8:816–822. doi: 10.1038/nri2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turnbull IR, Colonna M. Activating and inhibitory functions of DAP12. Nat Rev Immunol. 2007;7:155–161. doi: 10.1038/nri2014. [DOI] [PubMed] [Google Scholar]
- 54.Davies MJ, Miranda E, Roussel BD, Kaufman RJ, Marciniak SJ, Lomas DA. Neuroserpin polymers activate NF-kappaB by a calcium signaling pathway that is independent of the unfolded protein response. J Biol Chem. 2009;284:18202–18209. doi: 10.1074/jbc.M109.010744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bak SP, Amiel E, Walters JJ, Berwin B. Calreticulin requires an ancillary adjuvant for the induction of efficient cytotoxic T cell responses. Mol Immunol. 2008;45:1414–1423. doi: 10.1016/j.molimm.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krysko DV, Vandenabeele P. From regulation of dying cell engulfment to development of anti-cancer therapy. Cell Death Differ. 2008;15:29–38. doi: 10.1038/sj.cdd.4402271. [DOI] [PubMed] [Google Scholar]
- 57.Park YJ, Liu G, Lorne EF, Zhao X, Wang J, Tsuruta Y, Zmijewski J, Abraham E. PAI-1 inhibits neutrophil efferocytosis. Proc Natl Acad Sci U S A. 2008;105:11784–11789. doi: 10.1073/pnas.0801394105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 59.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Genin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, Andre F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 60.Janssen S, Rosen DM, Ricklis RM, Dionne CA, Lilja H, Christensen SB, Isaacs JT, Denmeade SR. Pharmacokinetics, biodistribution, and antitumor efficacy of a human glandular kallikrein 2 (hK2)-activated thapsigargin prodrug. Prostate. 2006;66:358–368. doi: 10.1002/pros.20348. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.