

Abstract
Endocannabinoid (eCB) signaling is tightly regulated by eCB biosynthetic and degradative enzymes. The eCB 2-arachidonoylglycerol (2-AG) is hydrolyzed primarily by monoacylglycerol lipase (MAGL). Here, we investigated whether eCB signaling, synaptic function, and learning behavior were altered in MAGL knock-out mice. We report that MAGL−/− mice exhibited prolonged depolarization-induced suppression of inhibition (DSI) in hippocampal CA1 pyramidal neurons, providing genetic evidence that the inactivation of 2-AG by MAGL determines the time course of the eCB-mediated retrograde synaptic depression. CB1 receptor antagonists enhanced basal IPSCs in CA1 pyramidal neurons in MAGL−/− mice, while the magnitude of DSI or CB1 receptor agonist-induced depression of IPSCs was decreased in MAGL−/− mice. These results suggest that 2-AG elevations in MAGL−/− mice cause tonic activation and partial desensitization of CB1 receptors. Genetic deletion of MAGL selectively enhanced theta burst stimulation (TBS)-induced long-term potentiation (LTP) in the CA1 region of hippocampal slices but had no significant effect on LTP induced by high-frequency stimulation or long-term depression induced by low-frequency stimulation. The enhancement of TBS-LTP in MAGL−/− mice appears to be mediated by 2-AG-induced suppression of GABAA receptor-mediated inhibition. MAGL−/− mice exhibited enhanced learning as shown by improved performance in novel object recognition and Morris water maze. These results indicate that genetic deletion of MAGL causes profound changes in eCB signaling, long-term synaptic plasticity, and learning behavior.
Introduction
Endocannabinoids (eCBs) regulate mood, emotion, appetite, pain, and cognition via the stimulation of cannabinoid (CB1) receptors (Hill et al., 2009; Bellocchio et al., 2010). Anandamide (Devane et al., 1992) and 2-arachidonoylglycerol (2-AG) (Mechoulam et al., 1995; Sugiura et al., 1995) are eCB ligands that bind the CB1 receptor. Unlike traditional neurotransmitters that are stored in synaptic vesicles, the eCBs are produced and released “on demand” (Marsicano et al., 2003), and their endogenous levels are principally regulated by eCB biosynthetic and degradative enzymes. Anandamide and 2-AG are hydrolyzed by fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), respectively (Cravatt et al., 1996; Blankman et al., 2007). Pharmacological and genetic studies indicate that FAAH and MAGL are key regulators of eCB-dependent signaling and behavior (Cravatt et al., 2001; Kathuria et al., 2003; Long et al., 2009; Chanda et al., 2010; Schlosburg et al., 2010).
A major function of eCBs is to regulate synaptic transmission (Alger, 2005). Depolarization-induced suppression of excitation (DSE) and inhibition (DSI) are forms of retrograde synaptic depression mediated by 2-AG-induced activation of CB1 receptors (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001) because DSE and DSI are abolished in mice lacking the 2-AG biosynthetic enzyme diacylglycerol lipase-α (Gao et al., 2010; Tanimura et al., 2010) and are enhanced by MAGL inhibitors, but not FAAH inhibitors (Makara et al., 2005; Straiker and Mackie, 2005; Safo et al., 2006; Hashimotodani et al., 2007; Pan et al., 2009). MAGL-deficient (MAGL−/−) mice exhibited dramatic elevations in brain 2-AG levels, CB1 receptor desensitization, and a loss of cannabimimetic behavioral effects such as analgesia and hypomotility (Chanda et al., 2010; Schlosburg et al., 2010). We investigated whether DSI and other eCB/CB1 receptor-mediated responses in hippocampal CA1 pyramidal neurons were altered in MAGL−/− mice.
Exposure to cannabis or synthetic cannabinoids produces deficits in memory, attention, and cognition in humans (Solowij et al., 2002; Messinis et al., 2006) and animals (Lichtman et al., 1995; Hampson and Deadwyler, 1999; Boucher et al., 2009; Puighermanal et al., 2009). The hippocampus is a primary brain region responsible for cannabinoid-induced cognitive deficits (Lichtman et al., 1995; Boucher et al., 2009). Long-term potentiation (LTP) is thought to represent a putative cellular model of learning and memory (Bliss and Collingridge, 1993). Synthetic CB1 agonists suppress LTP induction in the CA1 region in hippocampal slices in vitro (Nowicky et al., 1987; Misner and Sullivan, 1999; Hoffman et al., 2007) and in vivo (Hill et al., 2004). What might be the impact of sustained 2-AG elevations on long-term synaptic plasticity and learning behaviors? We examined LTP and long-term depression (LTD) induction in the hippocampus in MAGL+/+ and MAGL−/− mice. We also investigated the impact of genetic deletion of MAGL on novel object recognition and Morris water maze, two hippocampus-dependent learning behavioral tasks (Morris et al., 1982; Reed and Squire, 1997). Surprisingly, we found that both hippocampal LTP and performance in the learning tasks were enhanced in MAGL−/− mice.
Materials and Methods
Animals.
MAGL+/+, MAGL+/−, and MAGL−/− mice on a mixed 129SvEv/C57BL/6J background were generated by the Texas Institute of Genomic Medicine (Schlosburg et al., 2010). Genotyping of MAGL+/+, MAGL+/−, and MAGL−/− mice was performed by PCR using DNA sample obtained from the tail or ear. The MAGL+/+ and MAGL−/− mice used in this study were littermates from second- to fourth-generation intercrosses of 129SvJ-C57BL/6 MAGL+/− mice.
Slice preparation.
All animal use was in accordance with protocols approved by the Institutional Animal Care and Use Committee of Medical College of Wisconsin. Mice (45–60 d of age) of either sex were anesthetized by isoflurane inhalation and decapitated. Transverse hippocampal slices (300 μm thick) were prepared as described in our previous study (Pan et al., 2009). Slices were prepared at 4–6°C in a solution containing the following (in mm): 220 sucrose, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgSO4, 26 NaHCO3, 10 glucose, and 1 sodium ascorbate. The slices were transferred to and stored in artificial CSF (ACSF) containing the following (in mm): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose at room temperature. All solutions were saturated with 95% O2 and 5% CO2.
Electrophysiology.
Whole-cell and field recordings were made using patch-clamp amplifiers (Multiclamp 700B) under infrared–differential interference contrast microscopy. Data acquisition and analysis were performed using digitizers (DigiData 1440A) and analysis software pClamp 10 (Molecular Devices). Signals were filtered at 2 kHz and sampled at 10 kHz. Evoked and spontaneous IPSCs were recorded from CA1 hippocampal pyramidal neurons. Glutamate receptor antagonists 6-cyano-7-nitroquinoxaline-2,3-dione disodium (CNQX) (20 μm) and d-2-amino-5-phosphonovaleric acid (d-AP-5) (20 μm) were present in the ACSF. The pipettes were filled with an internal solution containing the following (in mm): 80 Cs-methanesulfonate, 60 CsCl, 2 QX-314, 10 HEPES, 0.2 EGTA, 2 MgCl2, 4 Mg-ATP, 0.3 Na2GTP, and 10 Na2-phosphocreatine, pH 7.2 with CsOH. To induce DSI, the CA1 pyramidal neurons were depolarized from −60 to 0 mV for 5 s, and IPSCs were evoked at 4 s intervals. IPSCs were evoked at 20 s intervals to examine the effects of (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate (WIN55,212-2), N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251), and 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide (SR141716) on IPSCs. Input–output (I/O) curves of IPSCs were generated using incremental stimulus intensities of 10–60 μA. The stimulating and recording pipettes were placed at the same depth in the slice and the distance between them was kept constant (∼300 μm). Paired-pulse depression of evoked IPSCs was examined at 20, 40, 80, 120, 200, 400, 800, and 1200 ms interstimulus intervals with stimulation strength correlated to ∼40% of the maximal evoked IPSC. Series resistance (15–20 MΩ) was monitored throughout the recordings, and data were discarded if the resistance changed by >20%.
Field recordings were made using glass pipettes filled with the 1 m NaCl (1–2 MΩ) and placed in the stratum radiatum of the CA1 region of the hippocampal slices, field EPSPs (fEPSPs) were evoked by stimulation of the Schaffer collateral/commissural pathway at 0.05 Hz with a bipolar tungsten electrode (WPI). I/O curves were generated by plotting fEPSP slope against presynaptic fiber volley amplitude following incremental stimulus intensities. Paired-pulse facilitation (PPF) was examined at 20, 40, 80, 120, 200, and 400 ms interpulse intervals with stimulation strength correlated to ∼40% of the maximal evoked fEPSP. For LTP and LTD experiments, stable baseline fEPSPs were recorded for at least 15 min at an intensity that induced ∼40% of the maximal evoked response. Theta burst stimulation (TBS) consisted of a series of 5 or 15 bursts, with four pulses per burst at 100 Hz with a 200 ms interburst interval. High-frequency stimulation (HFS) consisted of three trains of 100 Hz for 1 s with 20 s intertrain intervals. Low-frequency stimulation (LFS) (1 Hz for 15 min) was used to induce LTD. All recordings were performed at 32 ± 1°C by using an automatic temperature controller.
Immunohistochemistry.
MAGL−/− and MAGL+/+ mice were deeply anesthetized by intraperitoneal injection of sodium pentobarbital (50 mg/kg; Sigma-Aldrich) and transcardially fixed with 4% paraformaldehyde in 0.1 m phosphate buffer supplemented with 4% sucrose. The brains were sectioned at 30–40 μm thickness with a cryostat after postfix and dehydration. After H2O2 treatment and rinsing three times in PBS, free-floating sections were blocked for 1 h at room temperature with blocking solution (1% bovine serum albumin, 5% normal goat serum, and 1% Triton X-100 in 0.1 m PBS, pH 7.4). Coronal hippocampal sections were incubated with 1:800 GAP-43 (Sigma-Aldrich) at 4°C for 48 h. After rinsing in PBS, the sections were incubated in the secondary antibodies: 1:100 goat anti-mouse IgG-HRP (Bio-Rad) for 4 h at room temperature. After rinsing twice with PBS and once with 0.1 m ammonium phosphate buffer (APB), pH 7.0, immunoreactivity was detected with 0.05% DAB–0.004% H2O2 in APB for 2 min, and the reaction was stopped with APB for 5 min and then rinsed in PBS three times, dehydrated, and coverslipped. NeuroTrace green fluorescent Nissl stain (N21480; Invitrogen) was performed according to the manufacturer's protocol. Sections were analyzed by using a Zeiss Axio Imager Z1 microscope.
Open-field test.
MAGL+/+ and MAGL−/− mice (2–3 months of age) were placed individually in one corner of the open field (45 cm length × 35 cm wide × 30 cm deep box) and allowed to freely explore the arena during a 5 min test session (Wang et al., 2010). Locomotor activities were recorded using an automated video-tracking system (Mobile Datum). Total distance traveled during a 5 min session was calculated.
Novel object recognition.
The procedure was based on published studies with minor modifications (Wang et al., 2004). It comprised three sequential phases including habituation, training, and preference test. MAGL+/+ and MAGL−/− mice (2–3 months of age) were individually habituated to the open-field box for 15 min. Training trials were performed 24 h after habituation. Two wooden block objects were placed at the two corners (8 cm from each adjacent wall) of the box. An animal was placed into the center of the box at an equal distance to the two objects and was allowed 5 min of exploration of the two objects. Mice that showed a clear preference (>60%) for one object during the training phase were excluded from the experiment. Preference test trials were performed 5 min, 1 d, and 3 d after the training trials from the same cohorts of mice. Before each preference test trial, one of the objects was replaced with a novel object that differed in both shape and color. The mouse was returned in the same position as during the training trials and was allowed to explore the familiar object and novel object for 5 min. To avoid spontaneous preference, the objects were randomly assigned as either familiar or novel for each mouse. The positions of the objects and the objects used as novel or familiar were counterbalanced between the mice. Moreover, the open-field box and the objects were thoroughly cleaned with water and ethanol after each session to avoid potential odorant cues. The time spent exploring each object was scored and the preference was expressed as the percentage of time spent on each object (familiar or novel) divided by the total exploring time on the two objects. The exploration behavior was defined as directing the head to the object with a distance <2 cm. Turning around or sitting on the object was not considered as exploratory behavior.
Morris water maze.
The Morris water maze (MWM) test was based on published procedure with minor modifications (Vorhees and Williams, 2006). MAGL+/+ and MAGL−/− mice (2–3 months of age) were placed in a circular tank (135 cm diameter, 60 cm height) filled to a depth of 40 cm with water at 25°C. The water was made opaque by adding nonfat milk powder. A hidden platform (10 cm diameter) was submerged 1 cm below the surface of the water in the center of the target quadrant. The maze was placed in a room with various visual cues. MAGL+/+ and MAGL−/− mice were trained for 6 consecutive days to find the hidden platform. On each day, each animal was subjected to four trials with a 15 min interval between trials. Thirty minutes before the first trial of each day, MAGL+/+ and MAGL−/− mice were randomly divided into two groups for each genotype and were given an intraperitoneal injection of vehicle (10% DMSO in 0.9% NaCl) or AM251 (2 mg/kg). Mice were allowed to search for the platform for 60 s. If the mice did not find the platform within 60 s, they were gently guided to the platform. Mice were allowed to remain on the platform for 15 s. Probe trials were conducted 24 h after the last training without any drug or vehicle treatment. During the probe test, the platform was removed from the tank and the animals were allowed to swim in the pool for 60 s. The navigation of the mice was monitored by a video-tracking system (Mobile Datum). The time spent in each quadrant, swimming speed, and latency to platform were recorded and analyzed.
Chemicals.
All drugs were prepared as concentrated stock solutions and stored at −20 or −80°C before use. CNQX-Na2 (Sigma-Aldrich) and d-AP-5 (Tocris) were dissolved in water. Picrotoxin (Sigma-Aldrich) was dissolved in ACSF through sonication. WIN55,212-2 (Tocris), AM251 (Tocris), and SR141716 (or rimonabant) (Sanofi-Aventis) were dissolved in DMSO. When these drugs were applied to slices, control slices were treated in the same concentration of the respective solvent for similar exposure time. Drug-treated slices were interleaved with control slices from the same animal.
Data analysis and statistics.
Data are presented as the mean ± SEM. The decay time constant (τ) of DSI was measured using a single exponential function of y = y0 + k × exp(−x/τ), in which y is the magnitude of DSI, y0 is the beginning magnitude of DSI (i.e., the first one after depolarization), k is the constant multiplier, and x is the time. The magnitude of DSI was calculated as follows: DSI (%) = 100 × [1 − (mean of two IPSCs after depolarization/mean of five IPSCs before depolarization)]. Values of two to three DSI trials were averaged for each neuron. The magnitude of LTP or LTD (percentage) was calculated as follows: 100 × [mean fEPSP slope during the final 10 min of recording/mean baseline fEPSP slope]. The depression (percentage) of fEPSPs/IPSCs by CB1 agonists or antagonists was calculated as follows: 100 × [mean amplitude of fEPSPs/IPSCs at last 5 min of drug application/mean amplitude of baseline fEPSPs/IPSCs]. Spontaneous IPSCs were analyzed with Mini Analysis Program (Synaptosoft). Results were analyzed with Student's t test, two-way or three-way repeated-measures ANOVA, and two-way or three-way ANOVA followed by Tukey's post hoc test (when necessary). Results were considered to be significant at p < 0.05.
Results
Alterations of eCB/CB1 receptor-mediated physiological responses in MAGL−/− mice
MAGL+/+, MAGL+/−, and MAGL−/− mice were confirmed by PCR genotyping from tail or ear DNA (Fig. 1A). Nissl staining did not detect any overt alterations in brain size or the gross morphology of brains of MAGL+/+ and MAGL−/− mice (Fig. 1B,C). The eCB/CB1 receptor signaling is involved in axonal growth and pathfinding (Berghuis et al., 2007; Harkany et al., 2008). GAP-43 is a neural-specific growth-associated protein and marker of axonal growth and presynaptic terminals (Skene and Willard, 1981). However, there was no detectable difference in GAP-43 staining or cytoarchitecture of the hippocampus between MAGL+/+ and MAGL−/− mice (Fig. 1D). Nevertheless, these findings do not exclude possible changes in brain structure in MAGL−/− mice under other experimental conditions.
Figure 1.
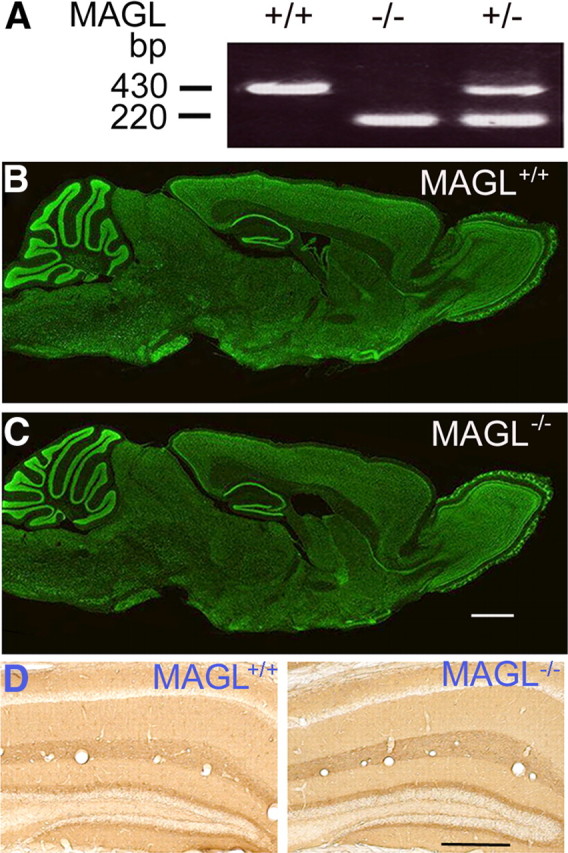
Normal gross brain morphology of MAGL−/− mice. A, Genotyping of MAGL+/+, MAGL+/−, and MAGL−/− mice was performed by PCR using ear DNA. The 430 bp band corresponds to the wild-type allele and the 220 bp band corresponds to the gene-trapped allele. B, C, Nissl staining of parasagittal sections of the brains of MAGL+/+ (B) and MAGL−/− mice (C) show similar gross brain morphology. Scale bars, 1 mm. N = 3 mice each phenotype. D, Immunohistochemical analysis of the hippocampus of MAGL+/+ and MAGL−/− mice using antibody against GAP-43, a marker of axonal growth and presynaptic terminals. No detectable difference of GAP-43 staining or hippocampal cytoarchitecture was found between MAGL+/+ and MAGL−/− mice. Scale bars, 0.5 mm. N = 3 mice each phenotype.
We investigated whether DSI, which is known to be mediated by 2-AG-induced activation of CB1 receptors (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001; Gao et al., 2010; Tanimura et al., 2010), was altered in hippocampal CA1 pyramidal neurons in MAGL−/− mice. IPSCs were evoked every 4 s by stimulating inhibitory synaptic inputs in the presence of glutamate receptor antagonists (CNQX; 20 μm) and d-AP-5 (20 μm). DSI was induced by a 5 s depolarization step from −60 to 0 mV. The decay time constant of DSI was significantly prolonged in MAGL−/− mice (MAGL+/+, 9.5 ± 1.8 s, n = 9; MAGL−/−, 25.6 ± 5.5 s, n = 10; p < 0.05; Fig. 2A,B); however, the magnitude of DSI was significantly decreased in MAGL−/− mice (MAGL+/+, 32.8 ± 3.9%, n = 9; MAGL−/−, 20.5 ± 3.2%, n = 10; p < 0.05; Fig. 2A,B).
Figure 2.

Genetic deletion of MAGL altered eCB/CB1 receptor-mediated responses in CA1 pyramidal neurons. A, Sample traces of IPSCs (top) and averaged DSI (bottom) in CA1 pyramidal neurons in MAGL+/+ and MAGL−/− mice (n = 9–10 cells; N = 3–5 mice). The solid lines are single exponential fitting curves of the decay of DSI. B, Summary of the decay time constant (τ) (left) and magnitude (right) of DSI in MAGL+/+ and MAGL−/− mice (n = 9–10; N = 3–5; *p < 0.05). C, Bath application of the CB1 receptor antagonist AM251 (2 μm) increased the amplitude of evoked IPSCs in hippocampal CA1 pyramidal neurons in MAGL−/− mice but did not significantly affect IPSCs in MAGL+/+ mice (n = 7–8; N = 3–4 mice; p < 0.05). D, Bath application of the CB1 receptor agonist WIN55212-2 (2 μm) induced significantly less depression of IPSCs in MAGL−/− mice than that in MAGL+/+ mice (n = 7–8; N = 3–4; p < 0.05). Error bars indicate SEM.
MAGL−/− mice exhibited 10- to 30-fold increase in 2-AG levels in the brain (Chanda et al., 2010; Schlosburg et al., 2010). The dramatic increase in 2-AG levels could result in tonic activation of CB1 receptors and persistent suppression of synaptic transmission. If this is the case, CB1 receptor antagonists should enhance basal synaptic transmission in MAGL−/− mice. In support of this prediction, we found that bath application of CB1 receptor antagonist AM251 (2 μm) significantly enhanced evoked IPSCs in CA1 pyramidal neurons in hippocampal slices in MAGL−/− mice but did not significantly affect evoked IPSCs in MAGL+/+ mice (MAGL+/+, 106.4 ± 6.1% of baseline, n = 8; MAGL−/−, 141.6 ± 8.1%, n = 7; p < 0.01; Fig. 2C). Bath application of another CB1 receptor antagonist SR141716 (2 μm) produced similar enhancement of IPSCs in MAGL−/− mice without significantly affecting the amplitude of evoked IPSCs in MAGL+/+ mice (MAGL+/+, 109.4 ± 6.6%, n = 7; MAGL−/−, 138.1 ± 8.6%, n = 8; p < 0.05). Similar to the present results, prior studies have shown that bath application of AM251 or SR141716 had no significant effect on baseline IPSCs in rat hippocampal slices (Wilson and Nicoll, 2001; Chevaleyre and Castillo, 2003). Thus, MAGL limits tonic activation of the CB1 receptor by 2-AG in MAGL+/+ mice, while the accumulated 2-AG causes tonic activation of CB1 receptors in MAGL−/− mice.
CB1 receptor binding density is decreased in MAGL−/− mice or mice chronically treated with a MAGL inhibitor (Chanda et al., 2010; Schlosburg et al., 2010). To test possible CB1 receptor desensitization, we examined whether the CB1 receptor agonist WIN55,212-2-induced depression of IPSCs was attenuated in MAGL−/− mice. Bath application of WIN55,212-2 (2 μm) produced significantly less depression of IPSCs in hippocampal CA1 pyramidal neurons in MAGL−/− mice than that in MAGL+/+ mice (MAGL+/+, 41.3 ± 4.0%, n = 7; MAGL−/−, 24.3 ± 5.7%, n = 8; p < 0.05; Fig. 2D). Together, these data indicate that genetic deletion of MAGL causes tonic activation and partial desensitization of the CB1 receptor in the hippocampus.
Selective enhancement of TBS-induced LTP in CA1 region of the hippocampus in MAGL−/− mice
We determined whether basal excitatory synaptic transmission and long-term synaptic plasticity in the hippocampus were altered in MAGL−/− mice. fEPSPs were evoked by Schaffer collateral stimulation at 0.05 Hz, and the recording electrode was placed in the stratum radiatum of the CA1 region of the hippocampal slices prepared from MAGL+/+ and MAGL−/− mice. We first determined the I/O relationship between presynaptic axon fiber volley and fEPSP slopes by stimulating Schaffer collateral with incremental intensities. MAGL−/− mice did not exhibit significant changes in the slope of I/O curves compared with that of MAGL+/+ mice (n = 7–8 slices from 4 mice each; p > 0.05; data not shown). These results indicate that basal synaptic strength was not significantly changed in MAGL−/− mice.
Next, we compared the paired-pulse ratio (PPR), which is used as a measure of the probability of transmitter release. Synapses with high probability of transmitter release often display low PPR, whereas synapses with low probability of transmitter release display high PPR (Zucker and Regehr, 2002). The PPR was measured at a series of interpulse intervals (20, 40, 80, 120, 200, and 400 ms). Stimulation of Schaffer collateral pathway produced PPF at shorter interpulse intervals (from 20 to 200 ms). The corresponding PPR at each interpulse interval was not significantly different between MAGL+/+ (20 ms, 1.34 ± 0.07; 40 ms, 1.45 ± 0.07; 80 ms, 1.38 ± 0.05; 120 ms, 1.31 ± 0.05; 200 ms, 1.19 ± 0.04; 400 ms, 1.02 ± 0.04; n = 7 slices from 4 mice) and MAGL−/− mice (20 ms, 1.27 ± 0.07; 40 ms, 1.37 ± 0.05; 80 ms, 1.32 ± 0.05; 120 ms, 1.25 ± 0.05; 200 ms, 1.17 ± 0.04; 400 ms, 1.04 ± 0.03; n = 8 slices from 5 mice; p > 0.05), suggesting that genetic deletion of MAGL had no significant effect on the probability of glutamate release at the Schaffer collateral-to-CA1 pyramidal cell synapses.
We also examined the effects of genetic deletion of MAGL on LTP in the CA1 region of the hippocampal slices. LTP in the hippocampus is typically induced by either TBS (Larson et al., 1986) or HFS (Bliss and Lomo, 1973; Bliss and Collingridge, 1993). TBS is designed to mimic the in vivo firing patterns of hippocampal neurons during exploratory behavior (Larson et al., 1986). Recordings of fEPSPs were first made in the absence of GABAA receptor blockers. We used two TBS protocols to induce LTP, one consisting of 5 bursts, the other consisting of 15 bursts. In hippocampal slices prepared from MAGL+/+ mice, the five- burst TBS protocol induced a significant increase in the slope of fEPSPs, which gradually returned to baseline levels in 60 min (106.1 ± 5.3% of baseline; n = 8; Fig. 3A). However, the same protocol induced a long-lasting enhancement of fEPSPs, or LTP, in MAGL−/− mice (129.5 ± 6.8% of baseline; n = 9; p < 0.05; Fig. 3A). The 15 burst TBS induced LTP in both MAGL+/+ and MAGL−/− mice; the magnitude of LTP was significantly greater in MAGL−/− mice compared with that of MAGL+/+ mice (MAGL+/+, 130.9 ± 6.9% of baseline, n = 7; MAGL−/−, 163.1 ± 7.6% of baseline, n = 9; p < 0.01; Fig. 3B). In contrast, HFS (100 Hz, 1 s × 3) induced similar LTP in the CA1 region of hippocampal slices prepared from MAGL+/+ and MAGL−/− mice (MAGL+/+, 132.5 ± 6.5% of baseline, n = 8; MAGL−/−, 141.3 ± 6.5% of baseline, n = 9; p > 0.05; Fig. 3C). LTD induced with LFS (1 Hz for 900 s) was not significantly different between MAGL+/+ and MAGL−/− mice (MAGL+/+, 13.7 ± 4.4%, n = 8; MAGL−/−, 17.1 ± 4.3%, n = 7; p > 0.05; Fig. 3D). Thus, genetic deletion of MAGL selectively facilitated TBS-induced LTP in the hippocampus but had no significant effects on HFS-LTP and LFS-LTD.
Figure 3.

Genetic deletion of MAGL selectively facilitates TBS-induced LTP in hippocampal slices but had no significant effects on either HFS-induced LTP or LFS-induced LTD. A, Five-burst TBS protocol induced LTP in MAGL−/− mice (n = 9; N = 5 mice) but not in MAGL+/+ mice (n = 8; N = 4; p < 0.05). B, Fifteen-burst TBS protocol induced LTP with greater magnitude in MAGL−/− mice (n = 9; N = 4) than that in MAGL+/+ mice (n = 7; N = 3; p < 0.01). C, HFS protocol induced similar LTP in MAGL−/− mice (n = 9; N = 6) and MAGL+/+ mice (n = 8; N = 5; p > 0.05). D, There was no significant difference of LFS-induced LTD between MAGL+/+ mice (n = 8; N = 4) and MAGL−/− mice (n = 7; N = 4; p > 0.05).
Why does genetic deletion of MAGL produce differential regulation of these two types of LTP? LTP is often tightly controlled by GABAA receptor-mediated inhibition and GABAA receptor antagonists or pathophysiological stimuli that reduce GABAergic inhibition decrease the threshold for LTP induction (Wigström and Gustafsson, 1983; Meredith et al., 2003; Liu et al., 2005). TBS-induced LTP is more sensitive to changes in GABAergic inhibition than HFS-induced LTP (Chapman et al., 1998). 2-AG-induced tonic suppression of GABAergic inhibition in MAGL−/− mice (Fig. 2C) might account for the difference in TBS-induced LTP between MAGL+/+ and MAGL−/− mice. To test this possibility, we repeated the TBS-LTP experiments in the presence of CB1 receptor antagonist SR141716 or AM251. Slices were treated with SR141716 (2 μm) or AM251 (2 μm) for at least 40 min before the 15 burst TBS was applied. There was no significant difference in TBS-induced LTP between MAGL+/+ and MAGL−/− mice in the presence of SR141716 (MAGL+/+, 127.8 ± 7.3% of baseline, n = 7; MAGL−/−, 129.9 ± 6.9% of baseline, n = 8; p > 0.05; Fig. 4A) or AM251 (MAGL+/+, 131.5 ± 6.7% of baseline, n = 8; MAGL−/−, 134.3 ± 5.9% of baseline, n = 8; p > 0.05; Fig. 4D); this was because SR141716 or AM251 significantly decreased the magnitude of TBS-LTP in MAGL−/− mice (p < 0.01) without affecting TBS-LTP in MAGL+/+ mice (p > 0.05; Fig. 4D).
Figure 4.

The mechanisms for facilitation of TBS-LTP induction in MAGL−/− mice. A, In the presence of the CB1 receptor antagonist SR141716 (2 μm), TBS-induced LTP in the CA1 region of hippocampal slices was not significantly different between MAGL+/+ and MAGL−/− mice (n = 7–8; N = 4–5 mice; p > 0.05). B, Bath application of SR141716 (2 μm) did not significantly affect basal fEPSPs in MAGL+/+ and MAGL−/− mice (n = 6–7; N = 3–4; p > 0.05). C, TBS-LTP induced in the presence of GABAA receptor blocker picrotoxin (50 μm) was not significantly different between MAGL+/+ and MAGL−/− mice (n = 7–8; N = 5 each; p > 0.05). D, Summary of 15 burst TBS-induced LTP under various experimental conditions (*p < 0.05; **p < 0.01). SR, SR141716; PTX, picrotoxin. Error bars indicate SEM.
Next, we examined whether 2-AG elevations in MAGL−/− mice produced tonic suppression of baseline fEPSPs in the CA1 region of the hippocampus. We found that bath application of SR141716 (2 μm) did not significantly affect baseline fEPSPs in either MAGL+/+ or MAGL−/− mice (MAGL+/+, 106.9 ± 5.1% of baseline, n = 6; MAGL−/−, 111.6 ± 4.0% of baseline, n = 7; p > 0.05; Fig. 4B). Together with the findings that the PPR and I/O curve of fEPSPs were not significantly altered in MAGL−/− mice, our data indicate that 2-AG elevations in MAGL−/− mice selectively suppress GABAergic inhibitory synaptic transmission in hippocampal CA1 region but do not significantly affect excitatory synaptic transmission.
To further test the possibility that 2-AG-induced suppression of GABAergic inhibition is responsible for the difference in TBS-induced LTP between MAGL+/+ and MAGL−/− mice, we performed TBS-induced LTP in the presence of GABAA receptor antagonist picrotoxin (50 μm). Under this condition, there was no difference in TBS-induced LTP between MAGL+/+ and MAGL−/− mice (MAGL+/+, 172.8 ± 7.5% of baseline, n = 8; MAGL−/−, 179.5 ± 8.6% of baseline, n = 7; p > 0.05; Fig. 4C,D). Thus, the presence of picrotoxin abrogated the difference in TBS-induced LTP between MAGL+/+ and MAGL−/− mice. Together, these results suggest that GABAergic inhibition to CA1 pyramidal neurons is selectively suppressed in MAGL−/− mice, which may explain why TBS-LTP is facilitated in these mice.
The reduction of GABAergic inhibition onto CA1 pyramidal neurons in MAGL−/− mice
To directly detect possible alterations of GABAA receptor-mediated inhibition, we recorded evoked IPSCs and spontaneous IPSCs (sIPSCs) in CA1 pyramidal neurons of hippocampal slices prepared from MAGL+/+ and MAGL−/− mice. The I/O relationship of evoked IPSCs was determined by plotting the amplitude of the IPSCs against the stimulus intensities. MAGL−/− mice exhibited significant decreases in the mean amplitude of evoked IPSCs at several stimulus intensities compared with that of MAGL+/+ mice (p < 0.05; Fig. 5A), indicating that the strength of inhibitory synaptic transmission is decreased in MAGL−/− mice. The PPR of evoked IPSCs was measured at a number of interpulse intervals (20, 40, 80 120, 200, 400, 800, and 1200 ms). The evoked IPSCs displayed paired-pulse depression at most of these intervals (20–800 ms) in MAGL+/+ mice. The PPR of evoked IPSCs at 20–100 ms intervals was significantly increased in MAGL−/− mice compared with that of MAGL+/+ mice (Fig. 5B). The increase in the PPR suggests a decrease in GABA release in MAGL−/− mice (Zucker and Regehr, 2002).
Figure 5.

The reduction of GABAergic inhibition in hippocampal CA1 pyramidal neurons in MAGL−/− mice. A, Genetic deletion of MAGL decreased the slope of input–output curves of IPSCs evoked from the striatum radiatum (n = 8 each; N = 4–5 mice; *p < 0.05). Top, Examples of IPSCs evoked by a range of stimulation intensities in hippocampal slices from MAGL+/+ and MAGL−/− mice. B, The increase in paired-pulse ratio (IPSC2/IPSC1) at 20, 40, 80, and 120 ms interpulse intervals in MAGL−/− mice (n = 8; N = 4–5; *p < 0.05). Top, Superimposed paired-pulse IPSCs at 20–400 ms interpulse intervals. C, Representative sIPSCs in CA1 pyramidal neurons in MAGL+/+ and MAGL−/− mice. D–F, The amplitude of sIPSCs was not altered in MAGL−/− mice as shown by cumulative probability plots of sIPSC amplitude distribution (D) and histograms of the mean amplitude of sIPSCs (F). The CB1 antagonist SR141716 had no significant effects on sIPSC amplitude (E, F). G, The mean frequency of sIPSCs was decreased in MAGL−/− mice compared with that in MAGL+/+ (n = 9–10; N = 4–5; **p < 0.01), and the decrease was reversed by SR1417716 (n = 8–9; N = 3–4; **p < 0.01). Error bars indicate SEM.
Next, we determined whether sIPSCs in CA1 pyramidal neurons were altered in MAGL−/− mice (Fig. 5C). The amplitude of sIPSCs was not significantly changed in MAGL−/− mice compared with that of MAGL+/+ mice, as shown by the cumulative amplitude plots (Van der Kloot, 1991) (p > 0.05; Fig. 5D) and amplitude histograms (p > 0.05; Fig. 5F). In contrast, the frequency of sIPSCs was significantly decreased in MAGL−/− mice compared with that of MAGL+/+ mice (MAGL+/+, 7.9 ± 0.6 Hz, n = 9; MAGL−/−, 4.9 ± 0.7 Hz, n = 10; p < 0.01; Fig. 5G). These results suggest that GABA release, but not GABAA receptor sensitivity, is decreased in MAGL−/− mice (Zucker and Regehr, 2002).
To test whether the decrease in GABA release was caused by 2-AG-induced CB1 receptor activation, we recorded sIPSCs in the presence of SR141716 (2 μm). SR141716 significantly increased the frequency of sIPSCs in CA1 pyramidal neurons in MAGL−/− mice (SR141716, 8.6 ± 0.7 Hz, n = 8; p < 0.01 vs corresponding control; Fig. 5G) but had no effect on the frequency of sIPSCs in MAGL+/+ mice (SR141716, 8.3 ± 0.8 Hz, n = 9; p > 0.05 vs corresponding control; Fig. 5G). There were no significant differences in the frequency as well as the amplitude of sIPSCs between MAGL+/+ and MAGL−/− mice (p > 0.05; Fig. 5E–G). Thus, genetic deletion of MAGL per se does not alter basal GABAergic synaptic transmission in CA1 pyramidal neurons. However, the associated increase in 2-AG levels in MAGL−/− mice causes tonic activation of CB1 receptors, selective suppression of GABAergic inhibition, and facilitation of TBS-LTP induction.
The enhancement of object recognition memory and spatial learning in MAGL−/− mice
Having shown that genetic deletion of MAGL selectively facilitates TBS-LTP in the hippocampus, we determined whether hippocampus-dependent learning and memory were altered in MAGL−/− mice. Novel object recognition task measures spontaneous preference to a novel object over a familiar object (Ennaceur and Delacour, 1988) and is a form of recognition memory that depends on the hippocampus (Myhrer, 1988; Reed and Squire, 1997). During training, MAGL+/+ and MAGL−/− mice were presented with two objects for 5 min. Both genotypes of mice spent approximately equal time exploring each object; the percentage of time spent for approaching or sniffing the objects was not significantly different (p > 0.05; Fig. 6A). Thus, MAGL+/+ and MAGL−/− mice had comparable visual perception and curiosity to explore the objects. During the sequential retention memory tests, one of the original objects was replaced with a novel object at 5 min, 1 d, and 3 d after the training. If a mouse remembered the original object, it spent more time with the novel object (Ennaceur and Delacour, 1988). Two-way repeated-measures ANOVA shows that the percentage of time spent in the exploration of the novel object was significantly decreased with time (F(2,40) = 48.7; p < 0.001), but no significant main effect of genotype was found (F(1,40) = 1.9; p > 0.05). However, there was significant genotype by time interaction (F(2,40) = 5.6; p < 0.01). Tukey's post hoc analysis indicates that MAGL+/+ and MAGL−/− mice exhibited similar preference to the replaced novel object at 5 min test session (p > 0.05), suggesting that short-term memory was not altered in MAGL−/− mice (Fig. 6B). However, at 1 d session, MAGL−/− mice exhibited significantly greater preference toward the novel object than MAGL+/+ mice (p < 0.01). This result indicates that MAGL−/− mice exhibit improved recognition memory compared with that MAGL+/+ mice. However, no preference for a novel object was observed at 3 d test session for MAGL+/+ and MAGL−/− mice as both types of mice returned to the basal levels (p > 0.05; Fig. 6B).
Figure 6.

MAGL−/− mice exhibited enhanced memory in novel object recognition test. A, In the training session, MAGL+/+ and MAGL−/− mice spent roughly equal time to explore the two objects (A, B). B, In the test sessions, MAGL+/+ and MAGL−/− mice showed similar preference to a novel object 5 min after the training (p > 0.05). One day after the training, MAGL−/− mice showed enhanced recognition memory because of the increased preference to a novel object compared with that of MAGL+/+ mice (**p < 0.01). Three days after the training, neither MAGL+/+ nor MAGL−/− mice showed significant preference to a novel object since they explored the original and novel objects with ∼50% of the time (N = 10–12 mice). Error bars indicate SEM.
We tested whether spatial learning and memory was altered in MAGL+/+ mice using MWM (Vorhees and Williams, 2006). MAGL+/+ and MAGL−/− mice were trained to find the hidden platform four trials per day for 6 d. Thirty minutes before the first trial of each day, mice were given intraperitoneal injection of vehicle or AM251 (2 mg/kg). Three-way repeated-measures ANOVA shows that the latency to escape to the hidden platform (the escape latency) was decreased following the 6 d training sessions (F(5,150) = 72.7; p < 0.001). There were a significant main effect of drug treatment (vehicle vs AM251) on the escape latency (F(1,30) = 8.8; p < 0.01) and a significant genotype by drug treatment interaction (F(1,30) = 5.3; p < 0.05). The escape latency was significantly different between vehicle-treated MAGL+/+ and MAGL−/− mice at the third and fourth days (p < 0.05), suggesting that MAGL−/− mice learned more quickly to find the hidden platform than MAGL+/+ mice (Fig. 7A). AM251 treatment had no significant effect on the escape latency in wild-type mice (p > 0.05), consistent with previous studies showing that CB1 antagonist SR141716 or CB1 receptor knock-out did not affect the escape latency in Morris water maze (Varvel and Lichtman, 2002). However, AM251 treatment significantly increased the escape latency in MAGL−/− mice (p < 0.01) and abrogated the difference in the escape latency between MAGL+/+ and MAGL−/− mice (p > 0.05; Fig. 7A). These results suggest that the decrease in the escape latency in MAGL−/− mice at the third and fourth days is caused by 2-AG-induced tonic activation of the CB1 receptor.
Figure 7.

MAGL−/− mice exhibited enhanced spatial learning in the Morris water maze test. A, MAGL+/+ and MAGL−/− mice were pretreated with vehicle or AM251 and trained for 6 d to find the hidden platform. The escape latency was significantly decreased at the third and fourth days in vehicle-treated MAGL−/− mice (N = 9) compared with that in vehicle-treated MAGL+/+ mice (N = 9; *p < 0.05). There was no significant difference in the escape latency between AM251-treated MAGL+/+ (N = 8) and MAGL−/− mice (N = 8; p > 0.05). B, Vehicle- or AM251-treated MAGL+/+ and MAGL−/− mice exhibited similar swimming speed in the pool during the 6 d training sessions (p > 0.05). C, During probe test, the platform was removed from the pool; all groups of MAGL+/+ and MAGL−/− mice exhibited preference for the targeted quadrant over the other three quadrants (p < 0.001); however, vehicle-treated MAGL−/− mice spent more time searching in the targeted quadrant than vehicle-treated MAGL+/+ mice (**p < 0.01). AM251 treatment significantly decreased the time spent in the targeted quadrant in MAGL−/− mice (p < 0.01). D, During the probe test, the latency to targeted platform location was not significantly different among vehicle- or AM251-treated MAGL+/+ and MAGL−/− mice (p > 0.05). E, Representative swimming tracings of vehicle-treated MAGL+/+ and MAGL−/− mice during the probe test. Error bars indicate SEM.
There was no significant difference of swimming speed among vehicle- and AM251-treated MAGL+/+ and MAGL−/− mice (p > 0.05; Fig. 7B). Previous studies have shown that the locomotor activity in an open field was similar in MAGL+/+ and MAGL−/− mice (Chanda et al., 2010; Schlosburg et al., 2010). Confirming these findings, we found that the total ambulatory distance in a 5 min session in an open field was not significantly different between MAGL+/+ (21.5 ± 2.3 m; n = 12) and MAGL−/− mice (20.5 ± 3.0 m; n = 11; p > 0.05). Thus, the decrease in the escape latency of MAGL−/− mice cannot be attributed to altered movement or swimming ability in these mice.
To further assess spatial learning and memory in MWM, we performed probe trials on the seventh day, ∼24 h after the lasting training session. The platform was removed from the tank and the mice were given 60 s to explore the pool. Three-way ANOVA showed that all groups of MAGL+/+ and MAGL−/− mice exhibited a clear preference for the targeted quadrant in which the platform was previously located (F(3,120) = 45.5; p < 0.001). There was significant genotype by drug treatment by targeted quadrant interaction for the time in the targeted quadrant (F(3,120) = 5.2; p < 0.01). Tukey's post hoc analysis indicates that vehicle-treated MAGL−/− mice spent more time searching in the targeted quadrant than vehicle-treated MAGL+/+ mice (p < 0.01; Fig. 7C,E). AM251 treatment significantly decreased the time spent in the targeted quadrant in MAGL−/− mice (p < 0.01) but had no significant effect in MAGL+/+ mice. There was no significant difference in the time spent in the targeted quadrant between AM251-treated MAGL+/+ and MAGL−/− mice (p > 0.05). During the probe trial, both vehicle- and AM251-treated MAGL+/+ and MAGL−/− mice took similar initial time to find the previous location of the removed platform (“Latency to platform”; p > 0.05; Fig. 7D), suggesting that both genotypes of mice were able to recall the location of the platform.
Discussion
In the present study, we investigated how sustained elevations of 2-AG in MAGL-deficient mice affected eCB signaling, synaptic function, and learning behaviors. We show that genetic deletion of MAGL altered DSI and other CB1 receptor-mediated depression of IPSCs in hippocampal CA1 pyramidal neurons. In addition, we demonstrate that TBS-induced LTP in the hippocampus was facilitated in MAGL−/− mice. Finally, we find that MAGL−/− mice exhibited improved performance in novel object recognition and Morris water maze.
Alterations of eCB/CB1 receptor-mediated responses in the hippocampus
DSE and DSI are forms of retrograde synaptic depression that require the CB1 receptor activation (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001) and the 2-AG biosynthetic enzyme diacylglycerol lipase-α (Gao et al., 2010; Tanimura et al., 2010). MAGL inhibitors, but not FAAH inhibitors, augmented DSE/DSI (Makara et al., 2005; Straiker and Mackie, 2005; Szabo et al., 2006; Hashimotodani et al., 2007; Pan et al., 2009), suggesting that the inactivation of 2-AG by MAGL shapes the duration of DSE/DSI. However, some of the MAGL inhibitors have multiple targets and unspecific effects (Lio et al., 1996; De Petrocellis et al., 1997; Kozak et al., 2000). We demonstrate that the decay of DSI in CA1 pyramidal neurons was prolonged in MAGL−/− mice. DSE in cultured autaptic hippocampal neurons was also prolonged in MAGL−/− mice (Straiker et al., 2011). Together, these studies provide genetic evidence that degradation of 2-AG by MAGL determines the time course of DSE/DSI.
The CB1 receptor antagonists AM251 and SR141716 significantly increased basal IPSCs in CA1 pyramidal neurons in MAGL−/− mice, but not in MAGL+/+ mice. Thus, 2-AG elevations in MAGL−/− mice cause persistent suppression of IPSCs. Conversely, the CB1 receptor agonist WIN55212-2 induced greater depression of IPSCs in MAGL+/+ mice than that in MAGL−/− mice. The magnitude of DSI in CA1 pyramidal neurons was decreased in MAGL−/− mice. Biochemical studies have shown that brain 2-AG levels are dramatically increased, whereas CB1 receptor binding density is significantly decreased in MAGL−/− mice (Chanda et al., 2010; Schlosburg et al., 2010). Together, these studies indicate that genetic deletion of MAGL causes tonic activation and partial desensitization of the CB1 receptor.
Selective facilitation of TBS-induced LTP in MAGL−/− mice
We show that TBS-induced LTP in hippocampal CA1 region was facilitated in MAGL−/− mice, while either HFS-induced LTP or LFS-induced LTD remained unaltered. Although HFS and TBS are common induction protocols for LTP, TBS is perhaps more physiologically relevant than HFS. TBS resembles in vivo firing patterns of hippocampal neurons in animals during exploratory behavior (Kandel and Spencer, 1961; Otto et al., 1991), while it is unlikely that hippocampal CA1 pyramidal neurons in vivo fire at 100 Hz for a full second because of spike frequency adaptation (Alger and Nicoll, 1980; Pedarzani and Storm, 1993). LTP at many excitatory synapses is tightly controlled by GABAA receptor-mediated inhibition (Wigström and Gustafsson, 1983; Huang et al., 1999; Liu et al., 2005). However, HFS-induced LTP is less sensitive to the levels of GABAergic inhibition than TBS-induced LTP (Chapman et al., 1998), perhaps due to HFS-induced intracellular accumulation of Cl− or desensitization of GABAA receptors (Isomura et al., 2003; Wang et al., 2006; Gong et al., 2009). One possibility is that 2-AG-induced selective suppression of GABAergic inhibition might account for the facilitation of TBS-LTP in MAGL−/− mice. In support of this possibility, we find that 2-AG elevations in MAGL−/− mice produced persistent suppression of basal IPSCs in CA1 pyramidal neurons and this effect was blocked by CB1 receptor antagonists. In the hippocampus, CB1 receptors are predominantly expressed on axonal terminals of cholecystokinin-expressing interneurons (Katona et al., 1999; Tsou et al., 1999). The CB1 receptor labeling density in inhibitory axonal terminals was ∼20 times higher than in excitatory terminals in the stratum radiatum of the CA1 region (Kawamura et al., 2006), where our recordings were made. We also find that CB1 receptor antagonists or GABAA receptor antagonist picrotoxin abrogated the difference in TBS-LTP induction between MAGL+/+ and MAGL−/− mice. Together, these results suggest that 2-AG-induced selective suppression of GABAergic inhibition provides a potential mechanism for the facilitation of TBS-LTP in MAGL−/− mice.
Consistent with our findings, previous studies have shown that the induction of DSI or CB1 receptor-mediated LTD at inhibitory synapses facilitated LTP induction (Carlson et al., 2002; Chevaleyre and Castillo, 2003, 2004; Zhu and Lovinger, 2007; Pan et al., 2011). In contrast, Δ9-tetrahydrocannabinol (Δ9-THC) (the active ingredient of marijuana) or other synthetic CB1 agonists blocked LTP in hippocampal slices in vitro (Nowicky et al., 1987; Collins et al., 1994; Terranova et al., 1995; Misner and Sullivan, 1999; Hoffman et al., 2007) and in vivo (Hill et al., 2004). Bath application of anandamide (Terranova et al., 1995) or 2-AG (Stella et al., 1997) blocked LTP in hippocampal slices in a CB1 receptor-dependent manner. Thus, exogenous and endogenous CB1 ligands produce distinct modulation of LTP in the hippocampus. We will discuss possible mechanisms for this differential regulation of LTP induction in the next section.
Improved performance in learning tasks in MAGL−/− mice
Enhanced LTP in a transgenic animal can be associated with improved or impaired learning behaviors (Migaud et al., 1998; Tang et al., 1999). MAGL−/− mice exhibited enhanced LTP as well as enhanced performance in Morris water maze and novel object recognition. MAGL−/− mice learned to find the hidden platform in a water maze more quickly than MAGL+/+ mice. Similar enhanced performance in water maze has also been reported in mice lacking anandamide-degrading enzyme FAAH (Varvel et al., 2007). In contrast with these findings, Δ9-THC and synthetic CB1 agonists impair spatial and cognitive performance tasks, including Morris water maze and novel object recognition, in animals (Lichtman et al., 1995; Hampson and Deadwyler, 1999; Varvel and Lichtman, 2002; Boucher et al., 2009; Puighermanal et al., 2009). Moreover, marijuana use leads to learning and memory deficits in humans (Solowij et al., 2002; Messinis et al., 2006). Why do exogenous and endogenous CB1 ligands exert opposite effects on hippocampal LTP and learning behavior?
Exogenous CB1 agonists depressed both excitatory and inhibitory transmission in the CA1 region (Wilson and Nicoll, 2001; Ohno-Shosaku et al., 2002). Temporal coordination of excitatory and inhibitory synaptic potentials is essential for theta (4–12 Hz), gamma (30–80 Hz), and ripple (100–200 Hz) oscillations, which are important for the formation of hippocampus-dependent memories (Buzsáki et al., 2003). Δ9-THC and a synthetic CB1 agonist disrupt these three types of synchronous, rhythmic action potential firing in the hippocampus (Hájos et al., 2000; Robbe et al., 2006), which may explain why synthetic cannabinoids impair hippocampal LTP and learning and memory. In contrast, endogenous 2-AG does not disrupt hippocampal rhythmic action potential firing (Robbe et al., 2006). 2-AG-mediated selective depression of inhibitory transmission decreases the threshold for LTP induction (Carlson et al., 2002; Chevaleyre and Castillo, 2003, 2004; Zhu and Lovinger, 2007; Pan et al., 2011). These observations may explain why synthetic cannabinoids and endogenous 2-AG exert opposite effects on hippocampal LTP and learning behavior.
Transgenic mice with various mechanisms of enhanced GABAergic inhibition exhibit impairments in both hippocampal LTP and Morris water maze performance (Costa et al., 2002; Cui et al., 2008; Gong et al., 2009); the learning deficits in these transgenic mice or wild-type mice treated with Δ9-THC were reversed by GABAA receptor antagonists (Varvel et al., 2005; Cui et al., 2008). Thus, the decreased GABAergic inhibition in the hippocampus might also underlie the improved performance in learning tasks in MAGL−/− mice.
During the probe trial of Morris water maze, MAGL−/− mice showed stronger place preference for the targeted quadrant than MAGL+/+ mice. Possible explanations include that MAGL−/− mice have better recollection of the previous location of the platform, or alternatively, these mice exhibited retarded “reversal learning” or “extinction.” CB1 knock-out mice exhibited significant deficits in a reversal task in which the location of the hidden platform was moved to the opposite side of the tank (Varvel and Lichtman, 2002) and showed impaired extinction of fear conditioning memories (Marsicano et al., 2002). It remains to be determined whether enhanced learning or impaired extinction of hidden platform memories contributes to the stronger place preference for the targeted quadrant in MAGL−/− mice.
Previous studies have shown that MAGL−/− mice show behavioral tolerance to CB1 agonists and the loss of cannabimimetic behavioral effects, which can be explained by CB1 receptor desensitization (Chanda et al., 2010; Schlosburg et al., 2010). In contrast, some of the electrophysiological and behavioral phenotypes of MAGL−/− mice identified here can be attributable to 2-AG-induced tonic activation of CB1 receptors. Thus, both activation and desensitization of CB1 receptors may underlie the alterations of physiological and behavioral responses in MAGL−/− mice.
While synthetic cannabinoids impair hippocampal LTP and behavioral learning (Lichtman et al., 1995; Hoffman et al., 2007), the present study revealed that sustained 2-AG elevations in MAGL−/− mice enhanced hippocampal LTP and cognitive and spatial learning. Morris water maze performance was also enhanced in FAAH knock-out mice (Varvel et al., 2007). Together, these findings imply that eCB degradation inhibitors and direct-acting CB1 agonists could exert distinct actions on learning and memory, an observation that may have important translational implications.
Footnotes
This work was supported by National Institutes of Health Grants DA017259 (B.F.C.) and DA024741 (Q.-s.L.) and by Extendicare Foundation (Q.-s.L.). We thank Cecilia J. Hillard and Eric Danielson for critical comments on an earlier version of this manuscript.
References
- Alger BE. Endocannabinoid identification in the brain: studies of breakdown lead to breakthrough, and there may be NO hope. Sci STKE. 2005;2005:pe51. doi: 10.1126/stke.3092005pe51. [DOI] [PubMed] [Google Scholar]
- Alger BE, Nicoll RA. Epileptiform burst afterhyperolarization: calcium-dependent potassium potential in hippocampal CA1 pyramidal cells. Science. 1980;210:1122–1124. doi: 10.1126/science.7444438. [DOI] [PubMed] [Google Scholar]
- Bellocchio L, Lafenêtre P, Cannich A, Cota D, Puente N, Grandes P, Chaouloff F, Piazza PV, Marsicano G. Bimodal control of stimulated food intake by the endocannabinoid system. Nat Neurosci. 2010;13:281–283. doi: 10.1038/nn.2494. [DOI] [PubMed] [Google Scholar]
- Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urbán GM, Monory K, Marsicano G, Matteoli M, Canty A, Irving AJ, Katona I, Yanagawa Y, Rakic P, Lutz B, Mackie K, Harkany T. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science. 2007;316:1212–1216. doi: 10.1126/science.1137406. [DOI] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher AA, Vivier L, Metna-Laurent M, Brayda-Bruno L, Mons N, Arnold JC, Micheau J. Chronic treatment with Δ9-tetrahydrocannabinol impairs spatial memory and reduces zif268 expression in the mouse forebrain. Behav Pharmacol. 2009;20:45–55. doi: 10.1097/FBP.0b013e3283242f6a. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Traub RD, Pedley T. The cellular synaptic generation of EEG. In: Ebersole JS, Pedley TA, editors. Current practice of clinical encephalography. Philadelphia: Lippincott-Williams and Wilkins; 2003. pp. 1–11. [Google Scholar]
- Carlson G, Wang Y, Alger BE. Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat Neurosci. 2002;5:723–724. doi: 10.1038/nn879. [DOI] [PubMed] [Google Scholar]
- Chanda PK, Gao Y, Mark L, Btesh J, Strassle BW, Lu P, Piesla MJ, Zhang MY, Bingham B, Uveges A, Kowal D, Garbe D, Kouranova EV, Ring RH, Bates B, Pangalos MN, Kennedy JD, Whiteside GT, Samad TA. Monoacylglycerol lipase activity is a critical modulator of the tone and integrity of the endocannabinoid system. Mol Pharmacol. 2010;78:996–1003. doi: 10.1124/mol.110.068304. [DOI] [PubMed] [Google Scholar]
- Chapman CA, Perez Y, Lacaille JC. Effects of GABAA inhibition on the expression of long-term potentiation in CA1 pyramidal cells are dependent on tetanization parameters. Hippocampus. 1998;8:289–298. doi: 10.1002/(SICI)1098-1063(1998)8:3<289::AID-HIPO10>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron. 2004;43:871–881. doi: 10.1016/j.neuron.2004.08.036. [DOI] [PubMed] [Google Scholar]
- Collins DR, Pertwee RG, Davies SN. The action of synthetic cannabinoids on the induction of long-term potentiation in the rat hippocampal slice. Eur J Pharmacol. 1994;259:R7–R8. doi: 10.1016/0014-2999(94)90666-1. [DOI] [PubMed] [Google Scholar]
- Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Kucherlapati R, Jacks T, Silva AJ. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415:526–530. doi: 10.1038/nature711. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Costa RM, Murphy GG, Elgersma Y, Zhu Y, Gutmann DH, Parada LF, Mody I, Silva AJ. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell. 2008;135:549–560. doi: 10.1016/j.cell.2008.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Melck D, Ueda N, Maurelli S, Kurahashi Y, Yamamoto S, Marino G, Di Marzo V. Novel inhibitors of brain, neuronal, and basophilic anandamide amidohydrolase. Biochem Biophys Res Commun. 1997;231:82–88. doi: 10.1006/bbrc.1997.6000. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, Mark L, Piesla MJ, Deng K, Kouranova EV, Ring RH, Whiteside GT, Bates B, Walsh FS, Williams G, Pangalos MN, et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci. 2010;30:2017–2024. doi: 10.1523/JNEUROSCI.5693-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong N, Li Y, Cai GQ, Niu RF, Fang Q, Wu K, Chen Z, Lin LN, Xu L, Fei J, Xu TL. GABA transporter-1 activity modulates hippocampal theta oscillation and theta burst stimulation-induced long-term potentiation. J Neurosci. 2009;29:15836–15845. doi: 10.1523/JNEUROSCI.4643-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hájos N, Katona I, Naiem SS, MacKie K, Ledent C, Mody I, Freund TF. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci. 2000;12:3239–3249. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- Hampson RE, Deadwyler SA. Cannabinoids, hippocampal function and memory. Life Sci. 1999;65:715–723. doi: 10.1016/s0024-3205(99)00294-5. [DOI] [PubMed] [Google Scholar]
- Harkany T, Mackie K, Doherty P. Wiring and firing neuronal networks: endocannabinoids take center stage. Curr Opin Neurobiol. 2008;18:338–345. doi: 10.1016/j.conb.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Kano M. Presynaptic monoacylglycerol lipase activity determines basal endocannabinoid tone and terminates retrograde endocannabinoid signaling in the hippocampus. J Neurosci. 2007;27:1211–1219. doi: 10.1523/JNEUROSCI.4159-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Froc DJ, Fox CJ, Gorzalka BB, Christie BR. Prolonged cannabinoid treatment results in spatial working memory deficits and impaired long-term potentiation in the CA1 region of the hippocampus in vivo. Eur J Neurosci. 2004;20:859–863. doi: 10.1111/j.1460-9568.2004.03522.x. [DOI] [PubMed] [Google Scholar]
- Hill MN, Hillard CJ, Bambico FR, Patel S, Gorzalka BB, Gobbi G. The therapeutic potential of the endocannabinoid system for the development of a novel class of antidepressants. Trends Pharmacol Sci. 2009;30:484–493. doi: 10.1016/j.tips.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Oz M, Yang R, Lichtman AH, Lupica CR. Opposing actions of chronic Δ9-tetrahydrocannabinol and cannabinoid antagonists on hippocampal long-term potentiation. Learn Mem. 2007;14:63–74. doi: 10.1101/lm.439007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Isomura Y, Sugimoto M, Fujiwara-Tsukamoto Y, Yamamoto-Muraki S, Yamada J, Fukuda A. Synaptically activated Cl− accumulation responsible for depolarizing GABAergic responses in mature hippocampal neurons. J Neurophysiol. 2003;90:2752–2756. doi: 10.1152/jn.00142.2003. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Spencer WA. Electrophysiology of hippocampal neurons. II. After-potentials and repetitive firing. J Neurophysiol. 1961;24:243–259. doi: 10.1152/jn.1961.24.3.243. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y, Fukaya M, Maejima T, Yoshida T, Miura E, Watanabe M, Ohno-Shosaku T, Kano M. The CB1 cannabinoid receptor is the major cannabinoid receptor at excitatory presynaptic sites in the hippocampus and cerebellum. J Neurosci. 2006;26:2991–3001. doi: 10.1523/JNEUROSCI.4872-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak KR, Rowlinson SW, Marnett LJ. Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J Biol Chem. 2000;275:33744–33749. doi: 10.1074/jbc.M007088200. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Larson J, Wong D, Lynch G. Patterned stimulation at the theta frequency is optimal for the induction of hippocampal long-term potentiation. Brain Res. 1986;368:347–350. doi: 10.1016/0006-8993(86)90579-2. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Dimen KR, Martin BR. Systemic or intrahippocampal cannabinoid administration impairs spatial memory in rats. Psychopharmacology (Berl) 1995;119:282–290. doi: 10.1007/BF02246292. [DOI] [PubMed] [Google Scholar]
- Lio YC, Reynolds LJ, Balsinde J, Dennis EA. Irreversible inhibition of Ca2+-independent phospholipase A2 by methyl arachidonyl fluorophosphonate. Biochim Biophys Acta. 1996;1302:55–60. doi: 10.1016/0005-2760(96)00002-1. [DOI] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makara JK, Mor M, Fegley D, Szabó SI, Kathuria S, Astarita G, Duranti A, Tontini A, Tarzia G, Rivara S, Freund TF, Piomelli D. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat Neurosci. 2005;8:1139–1141. doi: 10.1038/nn1521. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutiérrez SO, van der Stelt M, López-Rodriguez ML, Casanova E, Schütz G, Zieglgänsberger W, Di Marzo V, Behl C, Lutz B. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–88. doi: 10.1126/science.1088208. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Meredith RM, Floyer-Lea AM, Paulsen O. Maturation of long-term potentiation induction rules in rodent hippocampus: role of GABAergic inhibition. J Neurosci. 2003;23:11142–11146. doi: 10.1523/JNEUROSCI.23-35-11142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messinis L, Kyprianidou A, Malefaki S, Papathanasopoulos P. Neuropsychological deficits in long-term frequent cannabis users. Neurology. 2006;66:737–739. doi: 10.1212/01.wnl.0000201279.83203.c6. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Misner DL, Sullivan JM. Mechanism of cannabinoid effects on long-term potentiation and depression in hippocampal CA1 neurons. J Neurosci. 1999;19:6795–6805. doi: 10.1523/JNEUROSCI.19-16-06795.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O'Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Myhrer T. Exploratory behavior and reaction to novelty in rats with hippocampal perforant path systems disrupted. Behav Neurosci. 1988;102:356–362. doi: 10.1037//0735-7044.102.3.356. [DOI] [PubMed] [Google Scholar]
- Nowicky AV, Teyler TJ, Vardaris RM. The modulation of long-term potentiation by Δ9-tetrahydrocannabinol in the rat hippocampus, in vitro. Brain Res Bull. 1987;19:663–672. doi: 10.1016/0361-9230(87)90052-9. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Tsubokawa H, Mizushima I, Yoneda N, Zimmer A, Kano M. Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J Neurosci. 2002;22:3864–3872. doi: 10.1523/JNEUROSCI.22-10-03864.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T, Eichenbaum H, Wiener SI, Wible CG. Learning-related patterns of CA1 spike trains parallel stimulation parameters optimal for inducing hippocampal long-term potentiation. Hippocampus. 1991;1:181–192. doi: 10.1002/hipo.450010206. [DOI] [PubMed] [Google Scholar]
- Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, Liu QS. Blockade of 2-arachidonoylglycerol hydrolysis by selective monoacylglycerol lipase inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) enhances retrograde endocannabinoid signaling. J Pharmacol Exp Ther. 2009;331:591–597. doi: 10.1124/jpet.109.158162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Zhong P, Sun D, Liu QS. Extracellular signal-regulated kinase signaling in the ventral tegmental area mediates cocaine-induced synaptic plasticity and rewarding effects. J Neurosci. 2011;31:11244–11255. doi: 10.1523/JNEUROSCI.1040-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- Puighermanal E, Marsicano G, Busquets-Garcia A, Lutz B, Maldonado R, Ozaita A. Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nat Neurosci. 2009;12:1152–1158. doi: 10.1038/nn.2369. [DOI] [PubMed] [Google Scholar]
- Reed JM, Squire LR. Impaired recognition memory in patients with lesions limited to the hippocampal formation. Behav Neurosci. 1997;111:667–675. doi: 10.1037//0735-7044.111.4.667. [DOI] [PubMed] [Google Scholar]
- Robbe D, Montgomery SM, Thome A, Rueda-Orozco PE, McNaughton BL, Buzsaki G. Cannabinoids reveal importance of spike timing coordination in hippocampal function. Nat Neurosci. 2006;9:1526–1533. doi: 10.1038/nn1801. [DOI] [PubMed] [Google Scholar]
- Safo PK, Cravatt BF, Regehr WG. Retrograde endocannabinoid signaling in the cerebellar cortex. Cerebellum. 2006;5:134–145. doi: 10.1080/14734220600791477. [DOI] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu QS, Lichtman AH, Cravatt BF. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene JH, Willard M. Axonally transported proteins associated with axon growth in rabbit central and peripheral nervous systems. J Cell Biol. 1981;89:96–103. doi: 10.1083/jcb.89.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marijuana Treatment Project Research Group. Solowij N, Stephens RS, Roffman RA, Babor T, Kadden R, Miller M, Christiansen K, McRee B, Vendetti J. Cognitive functioning of long-term heavy cannabis users seeking treatment. JAMA. 2002;287:1123–1131. doi: 10.1001/jama.287.9.1123. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Straiker A, Mackie K. Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol. 2005;569:501–517. doi: 10.1113/jphysiol.2005.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Wager-Miller J, Hu SS, Blankman JL, Cravatt BF, Mackie K. COX-2 and FAAH can regulate the time course of depolarization induced suppression of excitation. Br J Pharmacol. 2011 doi: 10.1111/j.1476-5381.2011.01486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Szabo B, Urbanski MJ, Bisogno T, Di Marzo V, Mendiguren A, Baer WU, Freiman I. Depolarization-induced retrograde synaptic inhibition in the mouse cerebellar cortex is mediated by 2-arachidonoylglycerol. J Physiol. 2006;577:263–280. doi: 10.1113/jphysiol.2006.119362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, Sakimura K, Kano M. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron. 2010;65:320–327. doi: 10.1016/j.neuron.2010.01.021. [DOI] [PubMed] [Google Scholar]
- Terranova JP, Michaud JC, Le Fur G, Soubrié P. Inhibition of long-term potentiation in rat hippocampal slices by anandamide and WIN55212-2: reversal by SR141716 A, a selective antagonist of CB1 cannabinoid receptors. Naunyn Schmiedebergs Arch Pharmacol. 1995;352:576–579. doi: 10.1007/BF00169393. [DOI] [PubMed] [Google Scholar]
- Tsou K, Mackie K, Sañudo-Peña MC, Walker JM. Cannabinoid CB1 receptors are localized primarily on cholecystokinin-containing GABAergic interneurons in the rat hippocampal formation. Neuroscience. 1999;93:969–975. doi: 10.1016/s0306-4522(99)00086-x. [DOI] [PubMed] [Google Scholar]
- Van der Kloot W. The regulation of quantal size. Prog Neurobiol. 1991;36:93–130. doi: 10.1016/0301-0082(91)90019-w. [DOI] [PubMed] [Google Scholar]
- Varvel SA, Lichtman AH. Evaluation of CB1 receptor knockout mice in the Morris water maze. J Pharmacol Exp Ther. 2002;301:915–924. doi: 10.1124/jpet.301.3.915. [DOI] [PubMed] [Google Scholar]
- Varvel SA, Anum E, Niyuhire F, Wise LE, Lichtman AH. Δ9-THC-induced cognitive deficits in mice are reversed by the GABAA antagonist bicuculline. Psychopharmacology (Berl) 2005;178:317–327. doi: 10.1007/s00213-004-1988-2. [DOI] [PubMed] [Google Scholar]
- Varvel SA, Wise LE, Niyuhire F, Cravatt BF, Lichtman AH. Inhibition of fatty-acid amide hydrolase accelerates acquisition and extinction rates in a spatial memory task. Neuropsychopharmacology. 2007;32:1032–1041. doi: 10.1038/sj.npp.1301224. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ferguson GD, Pineda VV, Cundiff PE, Storm DR. Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat Neurosci. 2004;7:635–642. doi: 10.1038/nn1248. [DOI] [PubMed] [Google Scholar]
- Wang W, Gong N, Xu TL. Downregulation of KCC2 following LTP contributes to EPSP-spike potentiation in rat hippocampus. Biochem Biophys Res Commun. 2006;343:1209–1215. doi: 10.1016/j.bbrc.2006.03.038. [DOI] [PubMed] [Google Scholar]
- Wang W, Sun D, Pan B, Roberts CJ, Sun X, Hillard CJ, Liu QS. Deficiency in endocannabinoid signaling in the nucleus accumbens induced by chronic unpredictable stress. Neuropsychopharmacology. 2010;35:2249–2261. doi: 10.1038/npp.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigström H, Gustafsson B. Facilitated induction of hippocampal long-lasting potentiation during blockade of inhibition. Nature. 1983;301:603–604. doi: 10.1038/301603a0. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM. Persistent synaptic activity produces long-lasting enhancement of endocannabinoid modulation and alters long-term synaptic plasticity. J Neurophysiol. 2007;97:4386–4389. doi: 10.1152/jn.01228.2006. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]