

Abstract
We implemented negative electron-transfer dissociation (NETD) on a hybrid ion trap/Orbitrap mass spectrometer to conduct ion/ion reactions using peptide anions and radical reagent cations. In addition to sequence-informative ladders of a•- and x-type fragment ions, NETD generated intense neutral loss peaks corresponding to the entire or partial side-chain cleavage from amino acids constituting a given peptide. Thus, a critical step towards the characterization of this recently introduced fragmentation technique is a systematic study of synthetic peptides to identify common neutral losses and preferential fragmentation pathways. Examining 46 synthetic peptides with high mass accuracy and high resolution analysis permitted facile determination of the chemical composition of each neutral loss. We identified 19 unique neutral losses from 14 amino acids and three modified amino acids, and assessed the specificity and sensitivity of each neutral loss using a database of 1542 confidently identified peptides generated from NETD shotgun experiments employing high-pH separations and negative electrospray ionization. As residue-specific neutral losses indicate the presence of certain amino acids, we determined that many neutral losses have potential diagnostic utility. We envision this catalogue of neutral losses being incorporated into database search algorithms to improve peptide identification specificity and to further advance characterization of the acidic proteome.
Keywords: Negative electron transfer dissociation, Peptide neutral losses, Peptide dissociation, Ion-ion reactions, Electron transfer dissociation, Negative electrospray ionization, FT MS/MS
Introduction
Tandem mass spectrometry (MS/MS) has rapidly evolved over the past decade so that, at present, tens of thousands of peptides are routinely interrogated in a typical shotgun experiment [1–4]. By providing a window through which to monitor global protein abundance and post-translational modification (PTM) dynamics, this methodology has transformed modern biology. Yet because negative ion dissociation methods are limited and underdeveloped, shotgun MS/MS sequencing is almost wholly performed on peptide or protein cations. Compounding this overrepresentation, virtually all proteomic workflows have been tailored to produce cationic species via ionization under acidic conditions. The consequence is that peptides having basic amino acids are preferentially observed. Proteins, of course, often comprise both acidic and basic domains. These acidic domains, along with many acidic PTMs (e.g., phosphorylation, sulfation, glycosylation, etc.) are suppressed and often cloaked by the current methodology.
Negative electron-transfer dissociation (NETD), the ion-ion analog of electron-detachment dissociation (EDD), is one of a few dissociation methods effective for peptide anions [5–14]. The most common positive ion fragmentation scheme—collision-activated dissociation (CAD)—can be performed on peptide anions; however, the spectra are dominated by irregular products ion that are difficult to interpret. NETD is of particular interest due to strong preliminary performance in dissociating peptide anions in a predictable manner, good compatibility with PTMs, and finally, ease of implementation on any ETD-enabled system [9, 15–17]. Ultraviolet photo dissociation (UVPD) is another nascent negative ion fragmentation technique that has recently shown potential for large-scale analysis of the acidic subset of the proteome [18, 19]. Madsen et al. have demonstrated the compatibility of 193 nm UVPD for the dissociation of singly and multiply charged peptide anions from a moderately complex mixture (4 mitogen-activated pathway kinases), offering a potential alternative dissociation mechanism for acidic peptide analysis [20].
NETD results from the gas-phase reaction of multiply charged peptide or protein anions with small atomic or molecular cations. In the first NETD experiment, McLuckey and coworkers effected fragmentation using rare gas cations to extract an electron from multiply deprotonated nucleic acid polymers [21]. Later, radical cations of xenon were shown to initiate a similar process in peptide anions [8]. Since that time, the repertoire of NETD reagents has expanded to include molecular species such as the radical cation of fluoranthene, among others [9, 17]. NETD produces a•- and x-type fragment ions along with abundant neutral loss products in varying proportion, depending on the ionization potential of the reagent [17]. Key to harnessing the utility of NETD is to understand the preferred fragmentation pathways of the method.
Here we systematically identify and catalogue the neutral loss pathways resulting from NETD by analysis of 46 synthetic peptides (Supplementary Table 1). Through these data, we have identified 19 chemically distinct losses from 14 amino acids and 3 PTMs. In pursuing a foundational characterization, we prioritized precision in terms of pathway analysis of side-chain losses from peptides, rather than a comprehensive study of neutral losses arising from varied peptide species at various charge states. We compare our identified NETD fragmentation channels to those previously reported for both positive (e.g., ETD and ECD) and negative (e.g., EDD and negative CAD) fragmentation methods. Next, we examined a set of 1542 NETD MS/MS spectra, which were confidently mapped to peptide sequences, for the presence of these neutral losses. These results will facilitate NETD spectral interpretation, both manual and automated, with the final goal of increasing sequence coverage of the acidic portions of the proteome.
Methods
Samples and Reagents
Forty-six synthetic peptides (Sigma Aldrich, St. Louis, MO, USA and Thermo Scientific, Ulm, Germany), some of which were either phosphorylated (Ser, Thr, or Tyr) or carbamido-methylated (Cys), were directly infused via electrospray (Supplementary Table 1). The doubly charged peptide anions were subsequently analyzed by MS/MS. Solvents and buffers (Sigma Aldrich) were formulated using sodium hydroxide, isopropyl alcohol, acetonitrile, ammonium formate, and ammonium hydroxide (vide infra) for infusion or liquid chromatography (i.e., nanoLC-MS/MS). Yeast cells were cultured, lysed, and extracted for protein as previously described. Protein extracts were digested with trypsin (Promega, Madison, WI, USA) [2].
Mass Spectrometry
A modified hybrid LTQ Orbitrap XL ETD mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) was used to fragment peptide anions via NETD ion/ion reactions and to analyze the resulting fragment ion population. The instrument was modified in the following ways: (1) electron ionization (EI, conducted in the EI/CI source) was used to generate radical fluoranthene reagent cations; (2) the ion optics between the EI/CI source and the LTQ were optimized to transmit reagent cations, and (3) the LTQ firmware was modified to allow for ion/ion reactions between polyanionic peptides and reagent cations [17]. Peptides were ionized via negative ESI at 10−5 M concentrations using an infusion spray solution of 50:50 (vol/vol) 5 mM NaOH:2-propanol as described in Loo et al. [22]. Other instrument parameters, such as ESI conditions, injection pathway voltages, ion/ion reaction time, etc., were optimized on a per-peptide basis. For each synthetic peptide, the doubly deprotonated precursor was isolated and subjected to fragmentation via NETD. The resulting product ion population was analyzed in the Orbitrap mass analyzer at a resolving power of 30,000 at 400 Th. Spectra were averaged and internally recalibrated using the theoretical mass of the doubly deprotonated precursor (software developed in-house).
Large-Scale Analysis
NanoLC-MS/MS data were acquired using a Waters nano-ACQUITY (Waters Corporation, Milford, MA, USA) coupled to the modified hybrid LTQ Orbitrap XL mass spectrometer. Columns were fabricated in-house from 75 μm i.d. × 360 μm o.d. fused silica capillary tubing slurry-packed with a high-pH stable C18 stationary phase (XBridge 3.5 μm, 135 Å; Waters Corporation) to lengths of 5 cm and 12 cm for pre-columns and analytical columns, respectively. Analytical columns utilized an integrated electrospray emitter fabricated by first pulling a tip on an unpacked capillary using a laser puller (P-2000; Sutter Instrument Company, Novato, CA, USA) followed by brief chemical etching of the tip with 40% hydrofluoric acid [23]. Pre-columns were fabricated by inserting the capillary into a nano-volume tee (150 μm bore, VICI Valco Instruments Co. Inc., Houston, TX) housing a 0.2 μm frit and were packed while connected to the tee. Mobile phase A consisted of 5 mM ammonium formate titrated to pH 10 with ammonium hydroxide. Mobile phase B contained 80% acetonitrile with 5 mM ammonium formate from the pH 10 stock. A linear LC gradient from 5% B to 40% to B was performed over 2 h, flowing at 250 nL min−1. A potential of 1.4–1.8 kV was applied for negative mode electrospray ionization. MS1 spectra were acquired at a resolving power of 60,000, followed by data-dependent acquisition of the 10 most intense precursors using NETD, which were subsequently mass analyzed in the ion trap. Precursors having either unassigned or −1 charge states were rejected. A 30 s dynamic exclusion window was employed.
The resulting data files were searched with OMSSA ver. 2.1.8 [24]. Data were queried against the yeast Saccharomyces genome database (SGD), which was concatenated with a reversed version of the same database to allow for target-decoy searching [25, 26]. Full trypsin enzymatic specificity was required, allowing up to three missed cleavages. Carbamidomethylation of Cys (+57 Da) was specified as a fixed modification, while oxidation of Met (+16 Da) was specified as a variable modification. An average mass tolerance of ±2.5 Da was used for precursor ions, and a monoisotopic mass tolerance of ±0.25 Da was used for product ions. The resulting peptide–spectrum matches (PSMs) were filtered to a 1% false-discovery rate (FDR) using both expectation value and precursor mass accuracy with the Coon OMSSA Proteomic Analysis Software Suite (COMPASS) [27].
Results and Discussion
To map the pathways that govern NETD fragmentation and to catalog the preferred neutral loss fragmentation pathways, we subjected a library of 46 synthetic peptides to NETD and analyzed the resulting product ions with high-resolution and mass accuracy Fourier transform Mass Spectrometry (FTMS, Figure 1). Only precursors with charge states of −2 were selected for fragmentation analysis; this is largely because the number of potential pathways through which product ions may be formed increases as charge state increases. Higher charge state precursors may undergo multiple ion/ion reactions, precluding differentiation between product ions that result from sequential ion/ion reactions and from single ion/ion events. However, with −2 precursors, all product ions result from single ion/ion reactions. These FTMS data presented us with a wealth of information to be mined via manual annotation, from which we determined the characteristics and behavior of the NETD process. Figure 2a offers an example NETD MS/MS spectrum acquired from the peptide LIEDAEyTAK. Note extensive backbone cleavage at many Cα – C bonds (a•- and x-type fragments) in addition to numerous other products, mostly neutral losses (Figure 2b). The accurate mass measurements (<2 ppm, by use of internal calibrants) allowed us to generate a limited number of plausible explanations for each of these products (upper row, Figure 2c). For example, analysis of the m/z peak at 1173.459 can confidently be identified as the product of a neutral loss of C4H8, as the next best candidate C3H7N is confidently rejected due its mass error. In cases when the identity of the neutral loss could not be determined from mass error alone, correlation of the chemical composition with the peptide primary sequence permitted facile determination of its identity (lower row, Figure 2c). In this case, examination of the known primary sequence excluded the candidate loss of CH2N3O because that fragment could only be formed via multiple sequential losses and rearrangements. Using this procedure we annotated the NETD MS/MS spectra of the peptide library.
Figure 1.

Instrument diagram of the LTQ Orbitrap XL mass spectrometer depicting the sites of anion and cation injection, ion/ion interaction within the linear ion trap, and a portion of our experimental workflow. Fluoranthene was used as the cation reagent
Figure 2.

(A) NETD spectrum of a synthetic peptide containing phosphotyrosine. (B) Side-chain losses from the charge-reduced molecular ions are represented by their respective molecular formulas. (C) High mass accuracy allows for the facile identification of the chemical composition of the neutral losses (see inset table).The first of the neutral losses was determined purely by relative error. When the relative errors for proposed neutral losses were similar, as in the second example, the neutral loss was compared against the primary peptide sequence to determine if it was possible to observe both proposed neutral losses
Organized by amino acid residue, Table 1 presents a summary of the observed NETD side-chain neutral losses. Ultimately, the losses can be categorized based on the locale of the amino acid side-chain cleavage—entire, partial, and sequential side-chain losses—as denoted with a red line on the neutral amino acid structures. Below we detail each of these pathways and note, from an extensive literature search, whether prior work using various fragmentation techniques produced similar results. Figure 3, a graphical representation of these data, reveals that, overall, both positive (ECD and ETD) and negative (EDD and CAD) mechanisms yield very similar neutral losses to NETD.
Table 1.
Summary Table listing all of the Side-Chain Neutral Losses Observed from the 46 Peptides Analyzed, as well as the Respective Averaged Relative Errors for the Losses. A Detailed Explanation of Average Relative Error Calculations can be Found in the Supplementary Materials. Red Lines on Amino Acid Residues Signify the Site of Cleavage on the Side Chain Giving Rise to an Observed Neutral Loss. LC-MS/MS Results, which Revealed 3180 PSMs, were then Correlated with these Neutral Losses to Yield Sensitivity and Specificity Data. Sensitivity and Specificity Percentages Correspond to the Averaged Results of the Triplicate Whole-Cell Yeast Tryptic Digest. Amino Acid Ionizable Groups have been shown in their Neutral Forms. This gives no Implication of their Predominant Ionic Forms at pH 7.0. In Some Cases, Sensitivity and Specificity Calculations were not Possible or were Impractical to Calculate because: (a) The Neutral Loss is Identical to the Neutral Loss from Another Amino Acid, (b) The Neutral Loss was not able to be Resolved because it was within ±0.25 Da of another Neutral Loss, (c) The Neutral Loss may Potentially come from Sequential Neutral Losses Instead of a Single Loss, or (d) The Neutral Loss Matches a Common Loss from a Peptide Terminal
| Amino Acid Residue | Neutral Structure | Neutral Loss Composition | Neutral Loss Atomic Mass (u) | Average Error (ppm) | Sensitivity (%) | Specificity (%) | |
|---|---|---|---|---|---|---|---|
| PO4 | Ser | 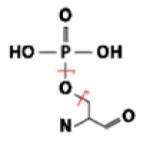 |
H3PO4 | 97.97690 | 0.54 | 7.46 | 95.50 |
| PO3 | 78.95851 | 1.04 | 13.31 | 91.07 | |||
| Thr |  |
H3PO4 | 97.97690 | 0.06 | 14.08 | 88.66 | |
| Nonpolar | Leu | 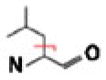 |
C4H8 | 56.06260 | 0.98 | N/Aa | N/Aa |
| lle |  |
C4H8 | 56.06260 | 1.00 | N/Aa | N/Aa | |
| Met | 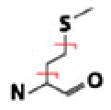 |
C3H6S | 74.01902 | −0.01 | 58.70 | 70.53 | |
| CH2S | 45.98772 | 0.24 | 72.30 | 39.66 | |||
| Phe | 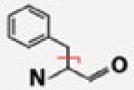 |
C7H7 | 91.05478 | 0.58 | 45.42 | 82.71 | |
| Trp |  |
C9H7N | 129.05785 | 0.16 | 35.73 | 88.37 | |
| Uncharged Polar | Ser |  |
CH2O | 30.01056 | 0.63 | 83.38 | 92.49 |
| Thr |  |
C2H4O | 44.02621 | 1.08 | N/Ab | N/Ab | |
| Gln |  |
C3H5NO | 71.03711 | 0.51 | N/Ab,c | N/Ab,c | |
| CHNO | 43.00581 | 0.38 | 62.25 | 43.82 | |||
| Tyr |  |
C7H7O | 107.04969 | 1.91 | 40.46 | 90.50 | |
| Cys (CAM) |  |
C2H4NOS | 90.00136 | 0.75 | 0.00 | 99.95 | |
| C2H4NO | 58.02929 | 0.10 | 31.11 | 86.34 | |||
| Charged Polar | Lys | 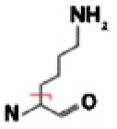 |
C4H9N | 71.07350 | 1.40 | N/Ab | N/Ab |
| Arg | 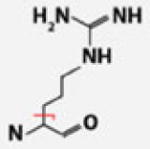 |
C4H9N3 | 99.07965 | 0.46 | 60.94 | 75.92 | |
| His |  |
C4H5N2 | 81.04527 | 0.98 | 64.40 | 91.65 | |
| Asp |  |
CO2 | 43.98983 | 0.95 | N/Ad | N/Ad | |
| Glu |  |
C3H4O2 | 72.02113 | 0.76 | 71.38 | 76.17 | |
| CO2 | 43.98983 | 0.86 | N/Ad | N/Ad |
Figure 3.

A compendium depicting observed major neutral losses from common amino acids with various fragmentation techniques. NETD losses have also been incorporated, revealing preferred fragmentation pathways quite similar to those of other electron-based fragmentation mechanisms. Amino acid ionizable groups have been shown in their neutral forms. This gives no implication of their predominant ionic forms at pH 7.0 [5–7, 9–11, 28–49]
Entire Side-Chain Loss and γ-Radical Formation
As with ETD, ECD, EDD, and negative CAD [5, 28–42], entire amino acid side-chain losses commonly result from NETD reactions (Figure 3). When γ-radicals are introduced via oxidation (i.e., electron abstraction) into amino acid side chains, the Cα–Cβ side-chain bond often cleaves to form the even-electron neutral loss species and the corresponding odd-electron product ion. Our results reveal that NETD induces this cleavage pattern when the precursor peptide contains Ile/Leu, Lys, Arg, Glu, Gln, Thr, and Met residues. For example, the NETD MS/MS spectrum of LIEDAEy-TAK shows that the entire Ile/Leu and Glu side chains are lost (Figure 2). Scheme 1a, adapted from Li et al., illustrates a possible mechanism for the entire side-chain loss using glutamine as an example [39]. Note that while this scheme depicts a particular deprotonation site, other resonance structures could generate the same charge-remote fragmentation (CRF) observed here. Also note that resolving the Val side-chain loss from the CO2 loss is challenging because it falls within the CO2 isotopic envelope. In the case of Leu and Ile, neutral loss occurrence correlated with the presence of either amino acid within the peptide, as the observed side-chain cleavage from both residues yielded neutral losses with identical chemical compositions.
Scheme 1.

(A) Proposed mechanism for the entire side-chain loss resulting from CRF on an amino acid residue with γ-hydrogens by the formation of a γ-radical. (B) Proposed mechanism for the partial side-chain loss from an amino acid residue by CDF. Both were adapted from similar mechanisms proposed by Li et al. for entire and partial side-chain loss resulting from ECD [39]
We often observed entire side-chain losses from amino acids with aromatic moieties. Although the typical representation of aromatic side chains lack γ-hydrogens, many of the amino acids containing these chains exhibit tautomeric forms which do indeed have γ-hydrogens [39]. Though their tautomeric forms vary, the amino acid side chains may undergo CRF, potentially involving γ-radical formation, inducing cleavage of their respective side chains. Such CRF-induced aromatic losses with NETD were observed repeatedly from peptides containing Trp residues (129.05785 Da). Albeit based on a single observation, we postulate that a similar CRF scheme could account for the loss of the entire side chain from Tyr (107.04969 Da), as the neutral loss mass matched the expected mass reported in previous studies utilizing negative CAD [29, 42], and was a hydrogen lighter than in previous studies utilizing fragmentation schemes under positive conditions [5, 30, 34, 35, 37, 39, 41].
We had hypothesized that His residues, exhibiting tautomeric forms containing γ-hydrogens, should present a CRF neutral loss of approximately 80 Da, corresponding to the even-electron loss of the entire side chain. But rather than 80 Da, we observed a loss of approximately 81 Da, corresponding to the odd-electron side-chain loss from His, now containing an extra hydrogen atom relative to the CRF neutral loss. We noted this same odd-electron side-chain loss from Phe residues (91.05478 Da), which, lacking a hydroxyl group on the aromatic moiety, do not have a tautomeric form yielding γ-hydrogens. Other groups utilizing fragmentation techniques such as EDD, ETD, ECD, and negative CAD have obtained similar results (Figure 3) [29–31, 33–35, 37, 39–41].
Partial Side-Chain Loss
Partial side-chain neutral losses, postulated to be a result of charge-directed fragmentation (CDF) rather than CRF, were observed in fewer instances than entire side-chain neutral losses. We observed such losses from Glu and Met residues. Scheme 1b illustrates a possible mechanism for charge-directed, partial side-chain cleavage, again using Glu as an example. With CDF, the relative acidity of certain regions of the peptide (i.e., readily oxidized regions) determines the site of deprotonation and, subsequently, that of cleavage. The postulated correlation between partial side-chain losses and CDF could thus be due to the specific localization of the charged region on the side chain. Notably, as the Glu schemes demonstrate (and is demonstrated by the data), an amino acid can present both entire and partial neutral losses (Scheme 1). Like entire side-chain losses, partial losses can be specific; that is, they can be correlated with amino acid presence within a peptide primary sequence to provide diagnostic utility. Additionally, the same amino acids with partial side-chain losses observed here have been observed following ETD, ECD, EDD, and negative CAD (Figure 3) [5, 7, 29, 32, 34, 36, 37, 39–44].
Interestingly, side-chain modifications can alter the preferred fragmentation pathways in an NETD ion/ion reaction. Take, for example, Ser phosphorylation and Cys carbamidomethylation. NETD frequently results in the loss of CH2O following the CRF of the entire side chain of Ser residues; however, upon phosphorylation of the Ser residue, the same side-chain bond was no longer cleaved. Instead, rather than cleaving off the entire side chain from the amino acid, PO3 is cleaved from the PTM. Similarly, in lieu of the expected fragmentation pathways of the Cys side chain, carbamidomethylation of Cys residues results in the odd-electron neutral losses C2H4NO (58.02929 Da) and C2H4NOS (90.00136 Da).
Sequential Side-Chain Losses
Least common were multiple, sequential neutral losses cleaved from amino acid side chains. We most often observed these sequential losses arising from the departure of two diagnostically less informative neutral species, such as CO2 and H2O. However, such a loss can be coupled with that of an informative, specific amino acid side chain. This is the case with the phosphopeptide LIEDAEyTAK depicted in Figure 2. The loss of C5H8O2 corresponds with the sequential loss of CO2 (i.e., the less informative species) and C4H8 from either Ile or Leu. The sequential loss depends on the locale of the radical site following the cleavage of a neutral loss. In the event that the radical site moves to the peptide following the departure of an entire or partial side chain, the radical may migrate elsewhere and induce further fragmentation, enabling continued side-chain loss through a radical cascade process. Note again, that all of these peptides were doubly charged so none of these sequential losses were the result of multiple ion/ion reactions. However, in cases where the peptide has more than two charges, the occurrences of losses like these should increase with the increased possibility of sequential ion/ion reactions.
Should the sequential losses contain at least one informative, specific neutral loss, they can contribute to post-acquisition diagnostic efforts. We observed such informative sequential losses throughout the collected spectra: CH3NO2 (H2O and CHNO), C4H4O4 (CO2 and C3H4O2), C7H10N (NH3 and C7H7), and C4H5NO3 (CO2 and C3H5NO). Recognize, however, that sequential losses may complicate spectra. For example, the C3H5NO loss from Gln, postulated to be a single-cleavage loss, might actually arise from the sequential loss of CHNO and C2H4; however, no evidence suggesting such a sequential loss has been observed. Similarly, though the C3H6S cleavage we observed can seemingly result from the sequential loss of CH2S coupled with C2H4, we determined it actually resulted from a single side-chain cleavage. The CH2S loss would be derived from carbamidomethylated Cys residues, but these residues were never present in peptides containing amino acids that could produce a loss of C2H4 (such as Ile). Additionally, neutral losses may also occur from a•- and x-type product ions under similar conditions. Thus, when considering the utility of sequential losses, one must keep in mind the concomitant presence of multiple amino acids in a peptide.
Evaluation of Neutral Loss Sensitivity and Specificity
To assess the prospect of including our identified neutral losses into database search algorithms, we queried a large dataset of NETD MS/MS spectra, generated from a triplicate analysis of a tryptic digest of yeast proteins and correlated with high-confidence (1% FDR) to peptide sequences, for the occurrence and specificity of the neutral loss ions. Searching LC-MS/MS data with OMSSA yielded 3180 peptide–spectrum matches (PSMs) and 1542 unique peptide identifications. The PSMs were filtered to a 1% FDR using both expectation value and precursor mass. Using these identifications, we queried every spectrum for the aforementioned neutral losses. This approach yielded sensitivity and specificity statistics for many of the neutral losses we identified (Table 1). In some cases, however, sensitivity and specificity statistics could not be calculated. Briefly, four such cases arose: one, when the neutral loss was identical to the neutral loss from another amino acid; two, when the neutral loss mass was within ±0.25 Da of another neutral loss, thereby making it prohibitively difficult to resolve; three, when the neutral loss potentially came from sequential instead of a single loss; and four, when the neutral loss matched a common loss from a peptide terminal.
For each neutral loss that merited calculation, we generated a receiver operating characteristic (ROC) curve and maximized the sum of specificity and sensitivity using the optimal ion intensity threshold. An example ROC curve is shown in Figure 4 for the loss of CH2O from Ser. As expected, the high specificity of neutral losses from doubly charged precursors is due to the low probability of random noise repeatedly originating at the exact neutral loss site. These data indicate that observed neutral losses are highly predictive of amino acid composition. Conversely, an amino acid sequence was not especially prognostic of the specific neutral losses observed. Therefore, our preliminary investigation suggests that incorporating NETD neutral loss data into search algorithms may enhance PSM confidence and specificity, in line with a previous ECD study [44]; we are thus continuing to probe the diagnostic utility of NETD neutral loss information, furthering the development of this fragmentation technique for high-throughput analyses in shotgun proteomics experiments.
Figure 4.
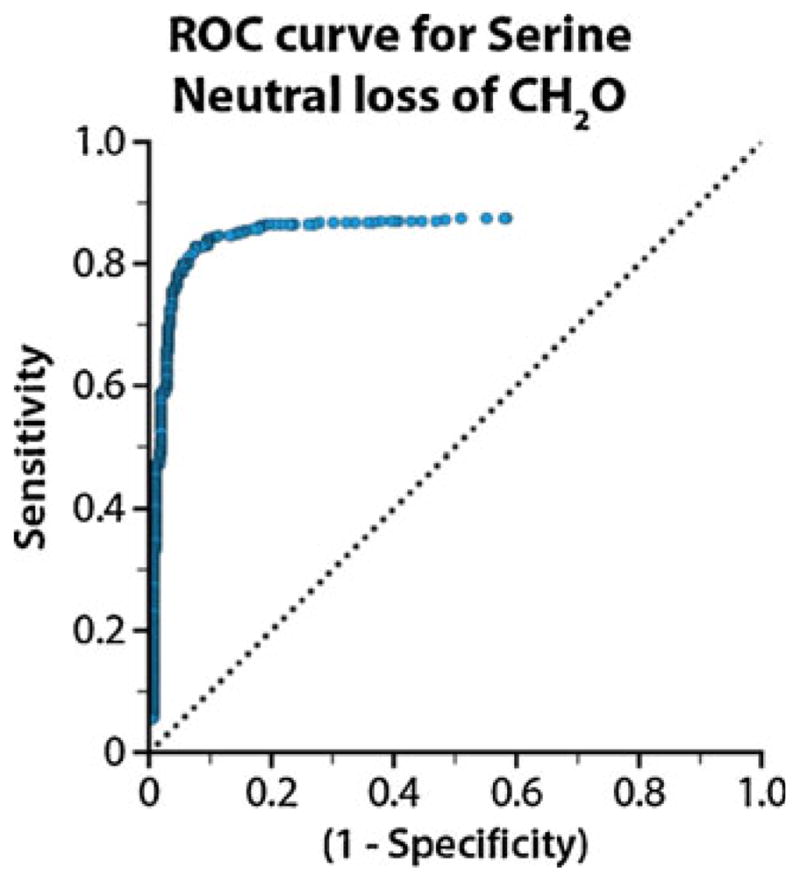
ROC curve, generated by software developed in-house, for the loss of CH2O from Ser residues. The ion-intensity threshold (blue dots) was optimized and selected to maximize the sum of sensitivity and specificity for each neutral loss observed
Conclusions
The acidic subset of the proteome contains a wealth of information that is currently underanalyzed, largely due to the difficulty of implementing current fragmentation techniques and methodologies for efficient and robust analysis of acidic proteins and peptides. NETD, one of only a few fragmentation mechanisms amenable to the analysis of acidic peptide precursors, provides utility in its ability to analyze this sizeable subset of the proteome and probe unexplored protein phenomena. However, before delving into the complexities of the acidic proteome, the elucidation of NETD’s preferential fragmentation pathways is essential. Thorough understanding of the fragmentation technique permits the analysis of a larger diversity of proteins and peptides to interrogate biological and biochemical processes and functions.
Leveraging the high-mass accuracy and resolving power of the Orbitrap, we identified 19 unique neutral losses from a series of infusion experiments using a collection of 46 synthetic peptides. We investigated the potential for each identified neutral loss to be included into database search algorithms by assessing their specificities and sensitivities with a large library of NETD PSMs collected during an LC-MS/MS analysis of yeast lysate. From these results, we conclude that the sensitivity and specificity of some neutral loss ions from a NETD ion/ion reaction can help diagnose peptide primary sequence.
Radical rearrangement ultimately yields three types of neutral losses from the charge-reduced precursor, each occurring with varying frequency. First, losses of the entire side chain, generated by hydrogen abstraction and radical relocation, are quite common and may lead to further rearrangement. Second, and less common, partial side-chain losses arise when the radical localizes to the α-carbon or is charge-directed via acidic amino acid sites of deprotonation. Third, and even less common with doubly charged precursors, sequential neutral losses can occur, typically involving a combination of a diagnostically uninformative and informative neutral loss. If incorporated into database search algorithms, all three types of neutral losses may provide diagnostic utility to improve the specificity and confidence of PSM searches. That is, the observed neutral losses are specific and indicate the presence of the corresponding amino acid within the primary peptide sequence.
This investigation of NETD neutral loss pathways has yielded substantial diagnostic information to assist in acidic peptide precursor identification. Additionally, it has elucidated the vast majority of the remaining, abundant, and previously-unidentifiable product ions resulting from a NETD ion/ion reaction. Incorporating this new information into database searching algorithms can improve the robustness and sensitivity of NETD as a fragmentation method.
Supplementary Material
Acknowledgments
The authors thank A. J. Bureta for software and illustrative assistance, M. Violet Lee for input on figures, and Qiangwei Xia for assistance in neutral loss identification. Thermo Scientific, the Beckman Foundation, the American Society of Mass Spectrometry, Eli Lilly, the NIH (R01GM080148 and P01GM081629), and the NSF (0747990) provided funding for this work.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s13361-011-0331-5) contains supplementary material, which is available to authorized users.
References
- 1.de Godoy LMF, Olsen JV, Cox J, Nielsen ML, Hubner NC, Frohlich F, Walther TC, Mann M. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 2008;455:1251–1254. doi: 10.1038/nature07341. [DOI] [PubMed] [Google Scholar]
- 2.Swaney DL, McAlister GC, Coon JJ. Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat Methods. 2008;5:959–964. doi: 10.1038/nmeth.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swaney DL, Wenger CD, Coon JJ. Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J Proteome Res. 2010;9:1323–1329. doi: 10.1021/pr900863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Washburn MP, Wolters D, Yates JR. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 5.Anusiewicz I, Jasionowski M, Skurski P, Simons J. Backbone and side-chain cleavages in electron eetachment dissociation (EDD) J Phys Chem A. 2005;109:11332–11337. doi: 10.1021/jp055018g. [DOI] [PubMed] [Google Scholar]
- 6.Budnik BA, Haselmann KF, Zubarev RA. Electron detachment dissociation of peptide di-anions: an electron–hole recombination phenomenon. Chem Phys Lett. 2001;342:299–302. [Google Scholar]
- 7.Kjeldsen F, Haselmann KF, Budnik BA, Jensen F, Zubarev RA. Dissociative capture of hot (3–13 eV) electrons by polypeptide polycations: an efficient process accompanied by secondary fragmentation. Chem Phys Lett. 2002;356:201–206. [Google Scholar]
- 8.Coon J, Shabanowitz J, Hunt D, Syka J. Electron transfer dissociation of peptide anions. J Am Soc Mass Spectrom. 2005;16:880–882. doi: 10.1016/j.jasms.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 9.Huzarska M, Ugalde I, Kaplan DA, Hartmer R, Easterling ML, Polfer NC. Negative electron transfer dissociation of deprotonated phosphopeptide anions: Choice of radical cation reagent and competition between electron and proton transfer. Anal Chem. 2010;82:2873–2878. doi: 10.1021/ac9028592. [DOI] [PubMed] [Google Scholar]
- 10.Kjeldsen F, Horning OB, Jensen SS, Giessing AMB, Jensen ON. Towards liquid chromatography time-scale peptide sequencing and characterization of post-translational modifications in the negative-ion mode using electron detachment dissociation tandem mass spectrometry. J Am Soc Mass Spectrom. 2008;19:1156–1162. doi: 10.1016/j.jasms.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 11.Kjeldsen F, Silivra OA, Ivonin IA, Haselmann KF, Gorshkov M, Zubarev RA. C-↦-C backbone fragmentation dominates in electron detachment dissociation of gas-phase polypeptide polyanions. Chem Eur J. 2005;11:1803–1812. doi: 10.1002/chem.200400806. [DOI] [PubMed] [Google Scholar]
- 12.Larraillet V, Vorobyev A, Brunet C, Lemoine J, Tsybin YO, Antoine R, Dugourd P. Comparative dissociation of peptide polyanions by electron impact and photo-induced electron detachment. J Am Soc Mass Spectrom. 2010;21:670–680. doi: 10.1016/j.jasms.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Crizer DM, Xia Y, McLuckey SA. Transition metal complex cations as reagents for gas-phase transformation of multiply deprotonated polypeptides. J Am Soc Mass Spectrom. 2009;20:1718–1722. doi: 10.1016/j.jasms.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 14.Haselmann KF, Budnik BA, Kjeldsen F, Nielsen ML, Olsen JV, Zubarev RA. Electronic excitation gives informative fragmentation of polypeptide cations and anions. Eur J Mass Spectrom. 2002;8:117–121. [Google Scholar]
- 15.Leach FE, Wolff JJ, Xiao ZP, Ly M, Laremore TN, Arungundram S, Al-Mafraji K, Venot A, Boons GJ, Linhardt RJ, Amster IJ. Negative electron transfer dissociation Fourier transform mass spectrometry of glycosaminoglycan carbohydrates. Eur J Mass Spectrom. 2011;17:167–176. doi: 10.1255/ejms.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolff JJ, Leach FE, Laremore TN, Kaplan DA, Easterling ML, Linhardt RJ, Amster IJ. Negative electron transfer dissociation of glycosaminoglycans. Anal Chem. 2010;82:3460–3466. doi: 10.1021/ac100554a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McAlister GC, Russell JD, Rumachik NG, Hebert AS, Swaney DL, Geer LY, Syka JEP, Coon JJ. Negative electron transfer dissociation identifies thousands of acidic peptides from complex mixtures. Proceedings of the 58th ASMS Conference; Salt Lake City, UT. 2010. [Google Scholar]
- 18.Kjeldsen F, Silivra OA, Zubarev RA. Zwitterionic states in gas-phase polypeptide ions revealed by 157-nm ultra-violet photodissociation. Chem Eur J. 2006;12:7920–7928. doi: 10.1002/chem.200600248. [DOI] [PubMed] [Google Scholar]
- 19.Wilson JJ, Brodbelt JS. Ultraviolet photodissociation at 355 nm of fluorescently labeled oligosaccharides. Anal Chem. 2008;80:5186–5196. doi: 10.1021/ac800315k. [DOI] [PubMed] [Google Scholar]
- 20.Madsen JA, Kaoud TS, Dalby KN, Brodbelt JS. 193-nm photodissociation of singly and multiply charged peptide anions for acidic proteome characterization. Proteomics. 2011;11:1329–1334. doi: 10.1002/pmic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herron WJ, Goeringer DE, McLuckey SA. Gas-phase electron-transfer reactions from multiply-charged anions to rare-gas cations. J Am Chem Soc. 1995;117:11555–11562. [Google Scholar]
- 22.Loo JA, Loo RRO, Light KJ, Edmonds CG, Smith RD. Multiply charged negative-ions by electrospray ionization of polypeptides and proteins. Anal Chem. 1992;64:81–88. doi: 10.1021/ac00025a015. [DOI] [PubMed] [Google Scholar]
- 23.Valaskovic GA, Kelleher NL, Little DP, Aaserud DJ, McLafferty FW. Attomole-sensitivity electrospray source for large-molecule mass spectrometry. Anal Chem. 1995;67:3802–3805. doi: 10.1021/ac00116a030. [DOI] [PubMed] [Google Scholar]
- 24.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH. Open mass spectrometry search algorithm. J Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 25.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 26.Cherry JM, Ball C, Weng S, Juvik G, Schmidt R, Adler C, Dunn B, Dwight S, Riles L, Mortimer RK, Botstein D. Genetic and physical maps of Saccharomyces cerevisiae. Nature. 1997;387:67–73. [PMC free article] [PubMed] [Google Scholar]
- 27.Wenger CD, Phanstiel DH, Lee MV, Bailey DJ, Coon JJ. COMPASS: A suite of pre- and post-search proteomics software tools for OMSSA. Proteomics. 2011;11(6):1064–1074. doi: 10.1002/pmic.201000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belyayev MA, Cournoyer JJ, Lin C, O’Connor PB. The effect of radical trap moieties on electron capture dissociation spectra of Substance P. J Am Soc Mass Spectrom. 2006;17:1428–1436. doi: 10.1016/j.jasms.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Bowie JH, Brinkworth CS, Dua S. Collision-induced fragmentations of the (M – H)− parent anions of underivatized peptides: an aid to structure determination and some unusual negative ion cleavages. Mass Spectrom Rev. 2002;21:87–107. doi: 10.1002/mas.10022. [DOI] [PubMed] [Google Scholar]
- 30.Cooper HJ, Hakansson K, Marshall AG, Hudgins RR, Haselmann KF, Kjeldsen F, Budnik BA, Polfer NC, Zubarev RA. Letter: The diagnostic value of amino acid side-chain losses in electron capture dissociation of polypeptides. Comment on: “Can the (M•-X) region in electron capture dissociation provide reliable information on amino acid composition of polypeptides?”. Eur J Mass Spectrom. 2003;9:221–222. doi: 10.1255/ejms.555. [DOI] [PubMed] [Google Scholar]
- 31.Cooper H, Hudgins R, Håkansson K, Marshall A. Characterization of amino acid side chain losses in electron capture dissociation. J Am Soc Mass Spectrom. 2002;13:241–249. doi: 10.1016/S1044-0305(01)00357-9. [DOI] [PubMed] [Google Scholar]
- 32.Cooper HJ, Hudgins RR, Håkansson K, Marshall AG. Secondary fragmentation of linear peptides in electron capture dissociation. Int J Mass Spectrom. 2003;228:723–728. [Google Scholar]
- 33.Falth M, Savitski MM, Nielsen ML, Kjeldsen F, Andren PE, Zubarev RA. Analytical utility of small neutral losses from reduced species in electron capture dissociation studied using SwedECD database. Anal Chem. 2008;80:8089–8094. doi: 10.1021/ac800944u. [DOI] [PubMed] [Google Scholar]
- 34.Fung YME, Chan TWD. Experimental and theoretical investigations of the loss of amino acid side chains in electron capture dissociation of model peptides. J Am Soc Mass Spectrom. 2005;16:1523–1535. doi: 10.1016/j.jasms.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 35.Haselmann KF, Budnik BA, Kjeldsen F, Polfer NC, Zubarev RA. Can the (M• – X) region in electron capture dissociation provide reliable information on amino acid composition of polypeptides? Eur J Mass Spectrom. 2002;8:461–469. doi: 10.1255/ejms.555. [DOI] [PubMed] [Google Scholar]
- 36.Kjeldsen F, Zubarev R. Secondary losses via γ-lactam formation in hot electron capture dissociation: a missing link to complete de novo sequencing of proteins? J Am Chem Soc. 2003;125:6628–6629. doi: 10.1021/ja035042c. [DOI] [PubMed] [Google Scholar]
- 37.Leymarie N, Costello CE, O’Connor PB. Electron capture dissociation initiates a free radical reaction cascade. J Am Chem Soc. 2003;125:8949–8958. doi: 10.1021/ja028831n. [DOI] [PubMed] [Google Scholar]
- 38.Li XJ, Cournoyer JJ, Lin C, O’Connor PB. The effect of fixed charge modifications on electron capture dissociation. J Am Soc Mass Spectrom. 2008;19:1514–1526. doi: 10.1016/j.jasms.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li XJ, Lin C, Han L, Costello CE, O’Connor PB. Charge remote fragmentation in electron capture and electron transfer dissociation. J Am Soc Mass Spectrom. 2010;21:646–656. doi: 10.1016/j.jasms.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun RX, Dong MQ, Song CQ, Chi H, Yang B, Xiu LY, Tao L, Jing ZY, Liu C, Wang LH, Fu Y, He SM. Improved peptide identification for proteomic analysis based on comprehensive characterization of electron transfer dissociation spectra. J Proteome Res. 2010;9:6354–6367. doi: 10.1021/pr100648r. [DOI] [PubMed] [Google Scholar]
- 41.Xia QW, Lee MV, Rose CM, Marsh AJ, Hubler SL, Wenger CD, Coon JJ. Characterization and diagnostic value of amino acid side chain neutral losses following electron-transfer dissociation. J Am Soc Mass Spectrom. 2011;22:255–264. doi: 10.1007/s13361-010-0029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brinkworth CS, Dua S, McAnoy AM, Bowie JH. Negative ion fragmentations of deprotonated peptides: backbone cleavages directed through both Asp and Glu. Rapid Commun Mass Spectrom. 2001;15:1965–1973. doi: 10.1002/rcm.457. [DOI] [PubMed] [Google Scholar]
- 43.Swaney DL, McAlister GC, Wirtala M, Schwartz JC, Syka JEP, Coon JJ. Supplemental activation method for high-efficiency electron-transfer dissociation of doubly protonated peptide precursors. Anal Chem. 2007;79:477–485. doi: 10.1021/ac061457f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Savitski MM, Nielsen ML, Zubarev RA. Side-chain losses in electron capture dissociation to improve peptide identification. Anal Chem. 2007;79:2296–2302. doi: 10.1021/ac0619332. [DOI] [PubMed] [Google Scholar]
- 45.Adamson JT, Hakansson K. Infrared multiphoton dissociation and electron capture dissociation of high-mannose type glycopeptides. J Proteome Res. 2006;5:493–501. doi: 10.1021/pr0504081. [DOI] [PubMed] [Google Scholar]
- 46.Bilusich D, Bowie JH. Fragmentations of (M – H)− anions of underivatised peptides. Part 2: Characteristic cleavages of Ser and Cys and of disulfides and other post-translational modifications, together with some unusual internal processes. Mass Spectrom Rev. 2009;28:20–34. doi: 10.1002/mas.20206. [DOI] [PubMed] [Google Scholar]
- 47.Chalkley R, Brinkworth C, Burlingame A. Side-chain fragmentation of alkylated cysteine residues in electron capture dissociation mass spectrometry. J Am Soc Mass Spectrom. 2006;17:1271–1274. doi: 10.1016/j.jasms.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 48.Kjeldsen F, Haselmann KF, Sorensen ES, Zubarev RA. Distinguishing of Ile/Leu amino acid residues in the PP3 protein by (hot) electron capture dissociation in Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem. 2003;75:1267–1274. doi: 10.1021/ac020422m. [DOI] [PubMed] [Google Scholar]
- 49.Mormann M, Macek B, de Peredo AG, Hofsteenge J, Peter-Katalinic J. Structural studies on protein O-fucosylation by electron capture dissociation. Int J Mass Spectrom. 2004;234:11–21. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.