

Abstract
Terminal deletion of the long arm of chromosome 4, (4q) is a rare event. It is characterized by spectral phenotypic manifestations, depending upon the site and quantity of chromatin lost. The chromosomal loss which span 4 (q31-q35) segment often manifests as craniofacial anomalies, mental retardation with ocular, cardiac, genitourinary defects and pelvic/limb dysmorphism. These abnormalities are usually unilateral. We report a female child (46, XX), aged 11 months, born to nonconsanguineous parents, bearing chromosomal deletion of 4 (q31.2-35.2) segment, which has manifested as craniofacial hypoplasia of left side of face, ipsilateral ptosis, erythroderma and bilateral thumb anomalies.
Keywords: Erythroderma, genetic defects, developmental defects
Introduction
Chromosomes are small filamentous structures within nuclei of all living cells that carry genetic information.[1] Their numbering begins from the largest to the smallest in size. Each chromosome has a long (q) and a short (p) arm, joined together at centromere.[2] Chromosome 4q deletion is a rare event. The deletion between 4(q11-q31) is called as proximal (interstitial) deletion while the deletion of 4(q31-q35) segment is termed as distal (terminal) deletion. The severity of phenotypic manifestations depends upon the site and quantity of chromatin lost.[2–6] We report a case (karyotype 46, XX) carrying terminal deletion of 4 (q31.2-q35.2) segment which manifested as craniofacial dysmorphism on left side, ipsilateral ptosis, mental retardation, erythroderma and bilateral thumb anomalies. Such rare phenotypic association of 4(q) terminal deletion has never been reported from India, so far.
Case Report
A female child aged 11 months was born at full term by manually assisted breech delivery. Her parents were nonconsanguineous life partners. The child was brought to us for generalized erythema and scaling, craniofacial hypoplasia (left side) [Figure 1]. However, she also had ipsilateral ptosis of eye, mental retardation, bilateral thumb anomalies and poor motor functions. Her parent narrated history of transparent membrane all around the baby at the time of birth; which gradually sheds off. Her family history suggested similar phenotypic characteristics in four cases (proband-index patient, her father and two half sisters) out of total 16 family members belonging to three generations [Figure 2]. There was no history of seizures or deafness.
Figure 1.
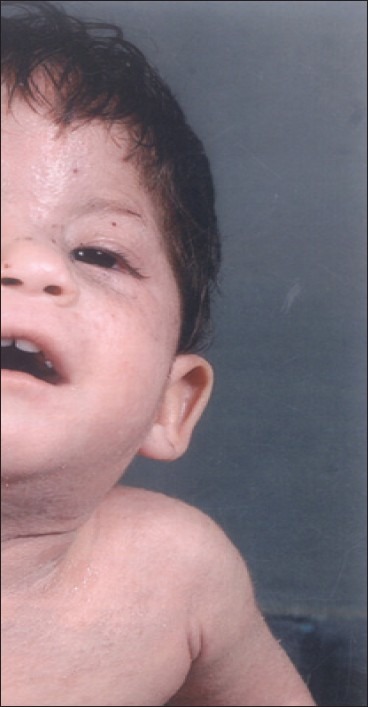
Child showing craniofacial hypoplasia (left side) and ipsilateral ptosis
Figure 2.
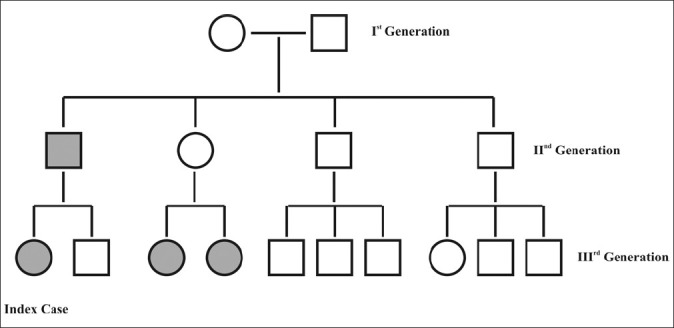
Pedigree chart
Physical examination revealed body weight 4.2 kg, head circumference 40 cm and vertical length 62 cm. She was average built and nourished. The major developmental defect include hypoplasia of left side of face, ipsilateral ptosis and small eyes, anteverted small nose and philtrum, small ear lobules and rudimentary left thumb while right thumb was absent. Her mental functions were suboptimal, and she could speak only monosyllable words. There was no focal sensory loss but motor system development was poor. She had not started crawling even at the age of 11 months. Dermatological examination revealed generalized erythema and fine scaling with interspersed follicular eruptions. Scalp, hair, nails, palms, soles and external genitalia were normal. Vital parameters and other visceral organs were apparently normal.
Her hematological investigations suggested lymphocytic leucocytosis with eosinophilia (TLC-31300/μl, P-18%, L-57%, E-22% and M-03%). Serology against Rubella, CMV and HSV showed raised serum IgG levels (35, 21 and 4.4 IU/ml, respectively), though, serum IgM against them was normal. Karyotyping of peripheral blood lymphocytes in early metaphase showed 46, XX pattern. Fluorescent leveled in situ hybridization technique (FISH) using specific DNA probe for 4(q) evidenced deletion at distal part of chromosome 4 (4q31.2-4q35.2) [Figure 3]. Serum biochemistry and CPK level were normal. Bone marrow examination could not be done. Although skiagram of chest and long bones were normal, but there was poor development of both thumbs. CT scan using contrast and MRI of skull suggested microcephaly with mild enlargement of ventricles. Other tests like, EEG, ECG, EMG, echocardiography, ultrasonography of abdomen were normal. Her IQ was 80. Histopathology of skin biopsy from left upper arm evidenced mild hyper keratosis, focal acanthosis and spongiosis with moderate lymphocytic infiltrate in upper dermis.
Figure 3.
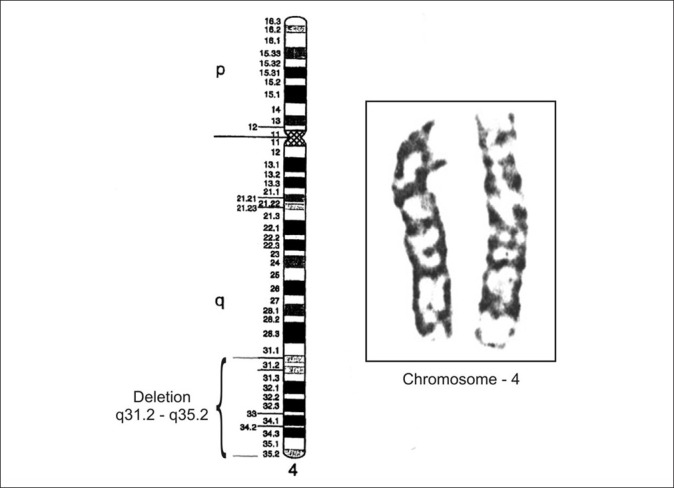
Enlargement of chromosome 4 with ideogram showing terminal deletion of band q31.2 – q35.2
Discussion
Congenital malformations often arise from chromosomal or genetic aberrations; however, gonadal mosaicism may be implicated at rare occasions. Chromosome 4 aberrations of various kinds have been recognized so far; which includes- monosomy or trisomy of either arm (short arm or long arm), translocation, inversion or ring formation, and partial or complete deletion of either arm (4p/4q).[1] The chromosomal deletion at 4 (q11-q31) segments is known as proximal (interstitial) deletion, while the deletion between 4(q31-q35) segment may be either interstitial or terminal deletion. A small interstitial deletion at 4 (q31) has been proposed as the “Minimal critical region” responsible for (4q) syndrome.[2–5] However, subtelomeric deletion of chromosome 4(q) involving the segment 4(q31.2-q35.2) is a rare event. Not only this event but familial occurrence of such aberration is very rare. The sequence of gene loci over this segment 4(q31.2-q35.2) includes pseudogene 8 (argino-succinate synthetase) at 4q31.2, LPS responsive vesicle trafficking (beach and anchor containing gene) at 4q31.3, Aspartyl-glucosamine gene at 4q32-q33, Caspase-3 (Apoptosis-related cystiene peptidase) gene at 4q34, adenine nucleotide translocator at 4q35 and other variants of adenine nucleotide translocator (Psuedogene 6 and 7) at 4q35.2 locus. These gene deletions occur during meiosis (Metaphase-1) of gametogenesis or mitosis of zygote, which is probably due to unknown causes.[2] But some researchers have described the role of tobacco,[7] ionizing radiations,[7] viral infections,[8] alcohol and mental stress[9] in congenital malformations.
Chromosomal deletions at distal end of 4(q) should essentially correlate to the specific genes loss. International Human Genome Sequencing Consortium (2001) mentioned specific properties of most of the genes. The characteristic of the gene located at 4q31.2 is (Pseudo gene 8), 4q31.3 is LPS responsible vesicle trafficking beach and anchor containing, 4q32-q33 are related to aspartyl-glucosamine, 4q34-Caspase-3 (apoptosis-related cystiene peptidase), 4q35-adenine nucleotide translocator and gene at 4q35.2 locus is pseudo gene.[1]
The severity of phenotypic/clinical characteristics depends up on the site of chromosomal deletion and quantity of chromatin lost. Subtelomeric deletion of chromosome 4 (q31-q35) segment is likely to manifest with typical phenotypic abnormalities, like dysmorphism of skull and face (hypertelorism, cleft palate, short nose with broad bridge, small eyes), mental retardation and anomaly of fifth finger with hooked/volar nail.[7–15] Sometimes, there may be cardiac (valvular) defects, piebaldism, genitourinary malformations, adnexal tumors and other anomalies. Chromosome 4q (terminal) deletion have been reported in 92% patients of cancer cervix, in a study by Sherwood, et al.[12] Recent reports have mentioned facio-scapulo-humeral dystrophy (FSHD) in people bearing deletion of D4Z4 (3.2 unit repeat) at 4q35 loci.[13] These patients often manifest as unilateral weakness of face, neck and shoulder muscle, hearing defect, cardiac dysrhythmia and foot drop. So most of children with 4q deletion die before 2 years or they have to face various physical and social challenges, due to multiple deformities and mental retardation.
The index case had craniofacial dysmorphism, mental retardation (IQ 80) and hypoplasia of both thumbs besides underlying 4q deletion (q31-q35). However, icthyosiform erythroderma is an additional finding which worth reporting the case. Other clinical entities, like CHILD syndrome, Conradi-Hünermann-Happle syndrome, Neu-Loxova syndrome, Wolf-Hirschhorn syndrome, fragile X-syndrome and Trisomy 21/18 may be masquerading the clinical picture of 4q deletion syndrome. Hence, karyotyping with high resolution FISH technique or southern blot test is essential for proper diagnosis of congenital malformations. Familial occurrence may need prenatal diagnostic procedures to minimize post-natal complications.
Cutaneous manifestation of chromosomal disorders
Chromosomal disorders may be due to abnormalities of chromosome number or structure. They may involve autosomes or sex chromosomes. Approximately 7.5% of all conceptions have a chromosomal disorder, but most of these are spontaneously aborted. Chromosomal abnormalities generally cause multiple congenital malformations. Children with more than one physical abnormality should undergo chromosomal analysis as part of their investigation. Presence of collodion membrane, cutaneous xerosis, hypertrichosis, café-au-lait spots more than 6 in numbers, ash leaf macules, cutaneous haemangiomas, erythrodermas associated abnormalities of hair, nail and teeth at birth or in early infancy should lead a general practitioner to suspect a chromosomal disorders.
Parent counseling
Genetic counseling depends upon the recurrence risk to parents of having an affected child. Both parents should be investigated thoroughly. If the abnormal character is determined by an autosomal dominant gene and one parent is affected, 50% of the offspring will be affected. Some parents fail to understand that the risk remains constant for every pregnancy, and that an affected first child does not guarantee a normal second child. If the parents of an affected child have no manifestations of genetic abnormality, the recurrence risk is likely to be small, as the child would have the genetic disorder as a fresh mutation. Autosomal recessive disorders are homozygous for the mutant gene. The recurrence risk for the carrier parents is 1 in 4, but offspring risk for those who are affected is small. Most recessive conditions are rare and it is unlikely that the affected person will marry another carrier.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
References
- 1.Breg WR. Abnormalities of chromosome 4 and 5. In: Garner LI, editor. Endocrine and Genetic disease of childhood and adolescence. Philadelphia: W B Saunders; 1975. pp. 101–319. [Google Scholar]
- 2.International Human Genome Sequencing Consortium: Initial sequencing of human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell JA, Packman S, Loughman WD, Fineman RM, Zackai E, Patil SR, et al. Deletions of different segments of the long arm of chromosome 4. Am J Med Genet. 1981;8:73–89. doi: 10.1002/ajmg.1320080110. [DOI] [PubMed] [Google Scholar]
- 4.Sijmons RH, Kristoffersson U, Tuerlings JH, Ljung R, Dijhuis-Stffelsma R, Breed AS. Piebaldism in a mentally retarded girl with rare deletion of long arm of chromosome 4. Pediatr Dermatol. 1993;10:235–9. doi: 10.1111/j.1525-1470.1993.tb00367.x. [DOI] [PubMed] [Google Scholar]
- 5.Sarda P, Leffort G, Fryns JP, Humeau C, Rieu R. Interstitial deletion of long arm of chromosome 4. J Med Genet. 1992;29:259–61. doi: 10.1136/jmg.29.4.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Michaela MI, Campos PJ. Terminal deletion of 4q in a severely retarded boy. Am J Med Genet. 1989;32:228–30. doi: 10.1002/ajmg.1320330217. [DOI] [PubMed] [Google Scholar]
- 7.Godwin WS, Subha VR, Feroz KM. Po radiation due to cigarette smoking. Current Science. 2010;98:681–6. [Google Scholar]
- 8.Stern H, Elek SD, Booth JC, Fleck DG. Microbial causes of mental retardation. The role of prenatal infection with CMV, Rubella, and Toxoplasma. Lancet. 1969;2:443–8. doi: 10.1016/s0140-6736(69)90162-7. [DOI] [PubMed] [Google Scholar]
- 9.Borochowitz Z, Shalev SA, Yehudai I, Bar-el H, Dar H, Tirosh E. Deletion (4) (q33-qter): A case report and review of literature. J Child Neurol. 1977;12:335. doi: 10.1177/088307389701200510. [DOI] [PubMed] [Google Scholar]
- 10.Curtis MA, Smith RA, Silbert J, Hughes HE. Interstitial deletion, del (4) (q33-q35.1) in a mother and two children. J Med Genet. 1989;26:652–4. doi: 10.1136/jmg.26.10.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sills ES, Burns MJ, Parker LD, Carroll LP, Kepart LL, Dyer CS, et al. Further phenotypic delineation of subtelomeric (terminal) 4q deletion with emphasis on intra cranial and reporductive anatomy. Orphanet J Rare Dis. 2007;2:1172–9. doi: 10.1186/1750-1172-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherwood JB, Shivapurkar N, Asfaq R, Miller DS, Gazdar AF, Muller CY. Chromosome 4 deletions are frequent in invasive cervical cancer and differ between histologic variants. Gynecol Oncol. 2000;79:90–6. doi: 10.1006/gyno.2000.5922. [DOI] [PubMed] [Google Scholar]
- 13.Miura K, Kumagai T, Matsumoto A, Iriyama E, Watenabe K, Goto K, et al. Two cases of chromosome 4q35 deletion linked onset of facio-scapulo-humeral dystrophy with mental retardation and epilepsy. Neuro pediatrics. 1998;29:239–41. doi: 10.1055/s-2007-973568. [DOI] [PubMed] [Google Scholar]
- 14.Lin AE, Garver KL, Diggans G, Clemens M, Wenger SL, Jones MC, et al. Interstitial and terminal deletion of the long arm of chromosome 4: further delineation of phenotypes. Am J Med Genet. 1988;31:533–48. doi: 10.1002/ajmg.1320310308. [DOI] [PubMed] [Google Scholar]
- 15.Calabrese G, Giannoti A, Mingarelli R, Di Gilio MC, Piemontese MR, Palka G. Two new borns with chromosome 4 imbalances; Deletion 4q33-q35 and ring r(4) (q35.2 qter) Clin Genet. 1997;51:264–7. [PubMed] [Google Scholar]