

Abstract
We have characterized the properties of immunopurified transcription complexes arrested at a specifically located cyclobutane pyrimidine dimer (CPD) using enzymatic probes and an in vitro transcription system with purified RNA polymerase II (RNAP II) and initiation factors. To help understand how RNAP II distinguishes between a natural impediment and a lesion in the DNA to initiate a repair event, we have compared the conformation of RNAP II complexes arrested at a CPD with complexes arrested at a naturally occurring elongation impediment. The footprint of RNAP II arrested at a CPD, using exonuclease III and T4 DNA polymerase’s 3′→5′ exonuclease, covers ~35 base pairs and is asymmetrically located around the dimer. A similar footprint is observed when RNAP II is arrested at the human histone H3.3 arrest site. Addition of elongation factor SII to RNAP II arrested at a CPD produced shortened transcripts of discrete lengths up to 25 nucleotides shorter than those seen without SII. After addition of photolyase and exposure to visible light, some of the transcripts could be reelongated beyond the dimer, suggesting that SII-mediated transcript cleavage accompanied significant RNAP II backup, thereby providing access of the repair enzyme to the arresting CPD.
Transcription-coupled repair (TCR)1 is a subpathway of nucleotide excision repair that removes lesions from the transcribed strand of actively transcribed genes (1). TCR has been shown to occur in mammalian cells (2), in Escherichia coli (3), and in Saccharomyces cerevisiae (4-6). Several lines of evidence have suggested that an active RNA polymerase elongation complex is necessary for preferential repair of the transcribed strand (7). A model for TCR has been proposed in which the RNA polymerase stalled at a lesion directs repair enzymes to the transcribed strand of an active gene (2). This model assumes that the polymerase must be removed from the site of the lesion to provide access for the repair complex to the lesion site and to allow reannealing of the DNA strands to form a proper substrate for repair.
An essential question about the mechanism of TCR is how the repair proteins recognize an RNAP II elongation complex arrested at a lesion and distinguish it from a transcription complex arrested at a natural arrest site. The process of transcriptional arrest can provide some clues to this question. RNAP II arrest sites have been identified and characterized (8). Arrest can occur at a bend in the helix axis of template DNA (9). It can also be induced by nucleotide depletion (10), DNA-binding drugs (11), and sequence-specific DNA-binding proteins (12). RNAP II can bypass arrest sites by activation of a cryptic endonuclease function that resides in the polymerase, a process mediated by the transcription elongation factor SII. SII-mediated transcript cleavage removes short oligonucleotides of discrete lengths from the 3′ end of the nascent RNA. Transcript shortening is thought to restore the association of the 3′ end of the transcript with the catalytic site in the polymerase after arrest. Footprinting analysis of elongation complexes arrested at a natural arrest site located in the first intron of the human H3.3 gene has shown that the footprint covers ~35 base pairs (bp) and is characterized by a shorter distance of the 3′ end of the transcript to the leading edge than to the rear edge of the polymerase (13-17). After addition of SII the boundaries of the footprint do not change significantly (13), but the 3′ end of the transcript is positioned near the catalytic site of the polymerase so that transcription can continue past the arrest site.
We have speculated that a change in RNA polymerase conformation similar to that resulting from natural arrest sites may also occur at the site of a lesion and that this may be the signal required to recruit repair proteins to the damage site. This hypothesis was supported by our observation that complexes arrested at the site of a CPD are substrates for SII-induced transcript shortening and that the shortened transcripts can be reelongated up to the point of blockage (18). Importantly, this observation demonstrates that the transcript is not released from the CPD-arrested complex. To determine the likely conformation of RNAP II arrested at a CPD we have carried out footprinting experiments using enzymatic probes and immunopurified ternary complexes arrested at a specifically located CPD. We have determined the conformation of RNAP II arrested at a CPD before and after SII-mediated transcript cleavage and compared this conformation with RNAP II arrested at the H3.3 arrest site. Furthermore, we show conditions in which SII-mediated transcript cleavage is accompanied by the actual displacement of RNAP II from the CPD site. This was indicated by the ability of Anacystis nidulans photolyase to access and repair the CPD site. After SII-mediated transcript cleavage and photoreversal, some of the transcripts could be reelongated beyond the resulting thymines.
EXPERIMENTAL PROCEDURES
Proteins and Reagents
RNAP II, transcription initiation factors, and SII were purified from rat liver or recombinant sources as described previously (19, 20). T4 polynucleotide kinase, T4 DNA ligase, and E. coli exonuclease III were from Roche Molecular Biochemicals. Photolyase from A. nidulans was a gift from Dr. Anders Eker (Erasmus University, Rotterdam, The Netherlands). E. coli strain MV1184 was a gift of Dr. Joachim Messing (Rutgers University, Piscataway, NJ). D44 IgG anti-RNA antibodies (21) were purified from ascites fluid as described previously (19). Highly purified NTPs were purchased from Amersham Pharmacia Biotech, formalin-fixed Staphylococcus aureus was from Calbiochem, and T4 DNA polymerase was from New England Biolabs. Radiolabeled nucleotides were purchased from Amersham Pharmacia Biotech.
Plasmids and Templates
Plasmid pAdH1 contains a 42-bp fragment of the human histone H3.3 arrest site. Construction of pAdH1 has been described (22). Plasmid pUC118 was from Worthington. Plasmid pUCH2 was produced by replacing a 51-bp EcoRI-HindIII fragment of pUC118 with a 470-bp EcoRI-HindIII fragment of pAdH1. This resulted in cloning the H3.3 arrest sequence in opposite orientation with respect to pAdH1. pUCH2 was transformed into the F′ E. coli strain MV1184 to produce single-stranded DNA for primer extension (see below).
DNA templates used for transcription reactions consisted of plasmid DNA linearized with HindIII. DNA templates used in footprinting experiments consisted of EcoRI-HindIII fragments labeled at the 5′ end of either the template or the non-template strand (see Figs. 1 and 2). They were obtained by linearizing plasmid DNA with HindIII or EcoRI, labeling the 5′ end with [γ-32P]ATP, and digesting with EcoRI and PvuII or HindIII and PvuII, respectively. This digestion produced a DNA fragment labeled at the 5′ end of the template or the non-template strand, including the promoter and the H3.3 sequence or the CPD, a small end-labeled EcoRI-PvuII or HindIII-PvuII fragment, and a long unlabeled portion of the plasmid DNA. DNA was purified by phenol/chloroform extraction and ethanol precipitation before use in footprinting experiments.
Fig. 1. DNA templates used in foot-printing experiments.

EcoRI-HindIII DNA fragments (470 bp) containing the H3.3 arrest site (H3.3) or a site-specific CPD in the transcribed strand (H-T^T-16) downstream of AdMLP was generated as described under “Experimental Procedures.” Numbers in parentheses indicate nucleotide positions on the plasmid DNA sequence. Runoff RNA (RO) and RNA resulting from transcription arrest at the H3.3 site (H3.3) or at a CPD (T^T) are marked with dashed lines together with their expected sizes. The transcription start site (+1) is represented with a bent arrow. TATA, TATA binding region.
Fig. 2. Schematic representation of ExoIII mapping of RNAP II complexes arrested by a CPD in the transcribed strand.

Immunopurified complexes arrested at a CPD were assembled on a DNA fragment labeled at the 5′ end of the template strand to map the up-stream boundary (A) or the non-template strand to map the downstream boundary (B). The bent arrow represents the transcription start site. RNA polymerase II arrested at a CPD is shown as a gray oval. TATA, TATA binding region; T^T, cyclobutane thymine dimer; *, 5′ end-labeled strand.
Insertion of Adducted Oligonucleotides into Plasmids
Sixteen base oligonucleotides of the sequence 5′-AAAGAGGGACGTTTTT-3′ containing a site-specific CPD at positions 14 and 15 (H-T^T-16) were obtained from Dr. John-Stephen Taylor (Washington University, St. Louis, MO). Covalently closed circular DNA containing a single CPD adduct on the transcribed strand was produced by priming 10 μg of the plus strand of pUCH2 with a 5-fold molar excess of CPD-containing oligonucleotide phosphorylated at the 5′ end in a 300-μl reaction mixture containing 10 mm Tris-HCl, pH 7.9, 50 mm NaCl, 10 mm MgCl2, 1 mm dithiothreitol, 600 μm each of dATP, dCTP, dTTP, and dGTP, 1 mm ATP, 30 units of T4 DNA polymerase, and 5 units of T4 DNA ligase. Covalently closed circular molecules were purified from an agarose gel containing 0.3 μg/ml ethidium bromide. Under these conditions, covalently closed circular DNA migrates as supercoiled DNA and can be resolved from single-stranded closed circular and nicked double-stranded plasmids. Treatment of closed circular DNA molecules with T4 endonuclease V indicated that ~80% contained a CPD (data not shown).
Transcription Reactions
DNA templates were incubated for 30 min at 28 °C with rat liver protein fraction D (2 μg, containing TFIID and TFIIH) and rat liver RNAP II (0.5 μg) in a 20-μl mixture containing 20 mm HEPES-NaOH, pH 7.9, 20 mm Tris-HCl, pH 7.9, 2.2 mm polyvinylalcohol, 212 units RNasin, 0.5 mg/ml acetylated bovine serum albumin, 150 mm KCl, 2 mm dithiothreitol, and 3% glycerol. After incubation, 33 μl of a solution containing fraction B′ (1 μg, containing TFIIF and TFIIE) and recombinant rat TFIIB (3 ng) in the same buffer as fraction D but without KCl were added, and incubation continued for 20 min at 28 °C to form preinitiation complexes. 7 mm MgCl2, 20 μm ATP, 20 μm UTP, and 40 μCi of [α-32P]CTP or unlabeled CTP (20 μm) were added, and incubation continued for 20 min. Elongation proceeds to nucleotide 15, at which the first GTP is required for incorporation. Heparin was added to prevent further initiation and then 800 μm of each NTP was added to allow elongation to continue, typically for 15 min. Arrested complexes were immunoprecipitated with D44 anti-RNA antibodies and formalin-fixed S. aureus and then washed three times in reaction buffer containing 20 mm Tris-HCl, pH 7.9, 3 mm HEPES-NaOH, pH 7.9, 60 mm KCl, 0.5 mm EDTA, 2 mm dithiothreitol, 0.2 mg/ml acetylated bovine serum albumin, and 2.2% (w/v) polyvinyl alcohol. Washed complexes were resuspended in 60 μl of reaction buffer for further treatment. For SII-mediated transcript cleavage, arrested complexes were incubated with SII for 1 h at 28 °C in 60 μl of reaction buffer containing 7 mm MgCl2. In experiments with photolyase the reaction buffer was adjusted to contain 5 mm dithiotreitol. Reactions were stopped with SDS and proteinase K, and nucleic acids were precipitated with ethanol. Samples were resuspended in formamide loading dye, heat denatured, and electrophoresed through a 6% polyacrylamide gel in TBE (89 mm Tris, 89 mm boric acid, 1 mm EDTA, pH 8) with 8.3 m urea. Gels were dried and autoradiographed using intensifying screens.
Exonuclease III and T4 DNA Polymerase 3′→5′ Exonuclease Mapping
Washed complexes arrested at a CPD or at the H3.3 arrest site were incubated with 7 mm MgCl2 and 65 units of E. coli exonuclease III (ExoIII) in reaction buffer at 37 °C for 3 min or with 12 units of T4 DNA polymerase 3′→5′ exonuclease (T4 exo) at 37 °C for 30 min. For the analysis of the upstream boundary, washed elongation complexes were first digested with FspI for 15 min at 37 °C in reaction buffer containing 7 mm MgCl2. Reactions were stopped by addition of SDS and proteinase K. Nucleic acids were precipitated with ethanol and dried.
RESULTS
Mapping of RNAP II Complexes Arrested at a CPD
To characterize the structural properties of transcription complexes arrested at a CPD we have used an in vitro transcription system with DNA templates in which a thymine-thymine CPD is situated at a specific site downstream of the major late promoter of adenovirus (AdMLP) (Fig. 1), partially purified rat liver RNAP II, and initiation factors to carry out transcription, as described previously (18). Oligonucleotides containing site-specific CPDs were inserted at a long distance (>150 bp) down-stream of the major late promoter of adenovirus to make sure that RNA polymerase elongation complexes had completed any transitions out of the initiating state before encountering the CPD (23). Transcription proceeds until RNAP II reaches a site-specific CPD in the transcribed strand of the DNA template (18). Transcription complexes arrested at a CPD were immunoprecipitated with D44 anti-RNA antibodies to remove most of the DNA template that does not participate in the transcription reaction. This step is essential to obtain a homogeneous population of ternary complexes arrested at lesions.
To map the boundaries of arrested complexes we used ExoIII and T4 exo as footprinting agents. ExoIII is a processive 3′→5′, double strand-dependent nuclease used commonly to detect the boundaries of RNA polymerase on DNA (13, 17, 24, 25). Tightly bound proteins block the nuclease, resulting in a resistant core that delineates the protein’s boundary (Fig. 2). This footprinting method has the advantage of a rather sharp transition from the protected to the unprotected area on the DNA with the borders more clearly defined than in footprints obtained with other probes. Furthermore, the digestion with ExoIII in contrast to DNase I is sequence-independent. Also, we can compare our data with previous ExoIII analyses of RNA polymerase II-arrested and -halted complexes (13, 16, 17). T4 exo was used as an alternative footprinting probe to confirm the results obtained with Exo III.
To map the upstream boundary, RNAP II complexes arrested at a CPD or at the H3.3 arrest site were assembled on linear templates labeled with 32P at the 5′ end of the template strand (Figs. 1 and 2). The extent of RNA synthesis was verified by labeling transcripts with [α-32P]CTP. Transcription of templates containing the H3.3 sequence produced transcripts arrested at the H3.3 site and full-length runoff transcripts resulting from read-through of this sequence (Fig. 3, lane 1). Transcription of templates containing a site-specific CPD in the transcribed strand produced mostly shortened transcripts arrested at the CPD and a small portion of full-length runoff transcripts, resulting from transcription of undamaged templates present in the preparation (Fig. 3, lane 6). Arrested complexes were immunopurified with anti-RNA antibodies. Identical complexes containing unlabeled transcripts were prepared for ExoIII or T4 exo mapping. Before ExoIII or T4 exo treatment, we separated the promoter region from the labeled fragment in arrested complexes by digesting with restriction enzyme FspI (Fig. 2A). This step was required to remove blockage of ExoIII or T4 exo in the AdMLP promoter by factors bound to the TATA box binding region (13) that prevented identification of the upstream boundary (Fig. 2A). ExoIII digestion of complexes arrested at the H3.3 arrest site from this new FspI-generated end revealed one prominent product (Fig. 3, lane 3) that mapped 25 bp upstream from the position of the transcript 3′ end. A similar result was obtained with T4 exo (Fig. 3, lane 5). Complexes arrested at a CPD produced a fragment that mapped 22 bp from the T^T dimer (Fig. 3, lane 8; see Fig. 6, lane 4) and a shorter product also present when untranscribed DNA was digested (Fig. 3, lane 7). This product resulted from the blockage of digestion by ExoIII or T4 exo molecules when they encountered the dimer.
Fig. 3. Mapping of the upstream boundaries of RNAP II-arrested complexes.

Complexes arrested at the H3.3 site (lanes 1 and 3–5) or at a CPD (lanes 6 and 8–10) containing 32P-labeled (lanes 1 and 7) or unlabeled (lanes 3–5 and 8–10) RNA were assembled on a DNA fragment labeled at the 5′ end of the template strand (lanes 1 and 6, band longer than runoff (RO) RNA). Untranscribed DNA (lanes 2, 4, 7, and 9) or ternary complexes digested with ExoIII (lanes 3 and 8) or T4 exo (lanes 5 and 10) are shown. DNA fragments protected from ExoIII or T4 exo digestion are indicated with arrows. L, 10-bp ladder; GTFs, general transcription factors.
Fig. 6. Mapping of the upstream boundaries of RNAP II complexes arrested at a CPD after SII-mediated transcript cleavage.

Complexes arrested at a CPD containing 32P-labeled (lanes 1–3) or unlabeled (lanes 4–9) RNA were assembled on a DNA fragment labeled at the 5′ end of the template strand. After incubation with or without elongation factor SII (1 ng) and MgCl2 for 1 h at 28 °C, arrested complexes were digested with ExoIII (lanes 4–6) or T4 exo (lanes 7–9). DNA fragments protected from Ex-oIII or T4 exo digestion are indicated with a white or a black arrow. The sequence around the CPD is shown at the top of the figure with the positions of the upstream boundaries detected with ExoIII (white arrow) or T4 exo (black arrow). L, 10-bp ladder; GTFs, general transcription factors.
To define the downstream boundary of these arrested complexes an identical experiment was performed with the duplex DNA labeled at the 5′ end of the non-template strand (Fig. 2B). ExoIII digestion of complexes arrested at the H3.3 arrest site produced three fragments (Fig. 4, lane 3), positioning the front edge of the polymerase 2-12 nt downstream from the 3′ end of the transcript. Mapping with T4 exo (Fig. 4, lane 5) positioned the front edge 2-8 nt from the 3′ end of the transcript. Complexes arrested by a T^T dimer produced a major product with ExoIII (Fig. 4, lane 8; see Fig. 7, lane 4) and a broader set of bands with T4 exo (Fig. 4, lane 10, see Fig. 7, lane 7) corresponding to a downstream boundary located 10-15 nt from the CPD. These results indicated that RNAP II arrested at a CPD covers about 35 bp. Furthermore, the 3′ end of the transcript is closer to the front edge than to the rear edge of the polymerase, similar to complexes arrested at the H3.3 site.
Fig. 4. Mapping of the downstream boundaries of RNAP II-arrested complexes.

Complexes arrested at the H3.3 site (lanes 1 and 3–5) or at a CPD (lanes 6 and 8–10) containing 32P-labeled (lanes 1 and 6) or unlabeled (lanes 2–5 and 7–10) RNA were assembled on a DNA fragment labeled at the 5′ end of the non-template strand (lanes 1 and 6, band longer than runoff RNA). Untranscribed DNA (lanes 2, 4, 7, and 9) or ternary complexes digested with ExoIII (lanes 3 and 8) or T4 exo (lanes 5 and 10) are shown. DNA fragments protected from ExoIII or T4 exo digestion are indicated with brackets. L, 10-bp ladder; GTFs, general transcription factors.
Fig. 7. Mapping of the downstream boundaries of elongation complexes arrested at a CPD after SII-mediated transcript cleavage.

Complexes arrested at a CPD containing 32P-labeled (lanes 1–3) or unlabeled (lanes 4–9) RNA were assembled on a DNA fragment labeled at the 5′ end of the non-template strand. After incubation with or without elongation factor SII (1 ng) and MgCl2 for 1 h at 28 °C, arrested complexes were digested with ExoIII (lanes 4–6) or T4 exo (lanes 7–9). The sequence around the CPD is shown at the top of the figure with the positions of the downstream boundaries detected with ExoIII (white arrow) or T4 exo (black arrow). L, 10-bp ladder; GTFs; general transcription factors.
RNAP II Arrested at a CPD Can Resume Elongation Past the Dimer Site after SII-mediated Transcript Cleavage and Photo-reactivation
Previously we have shown that RNAP II complexes arrested at a CPD are substrates of SII-mediated transcript cleavage and that the cleaved transcripts can be reelongated up to the CPD (18). Here we have compared the SII cleavage products of transcripts arrested at the H3.3 arrest site with products resulting from cleavage of transcripts arrested at a CPD. Cleavage of transcripts arrested at the H3.3 sequence produced RNAs of discrete length, shortened from 5 to 20 nt, with identifiable intermediates found in steps of 5 nt (Fig. 5, lanes 2–4). Surprisingly, SII-mediated cleavage of transcripts arrested at a CPD produced mostly transcripts that were only about 5 nt shorter (Fig. 5, lane 6). The upstream boundary of these complexes moved 2–3 nt upstream from the position occupied before cleavage (Fig. 6). The downstream boundary remained unchanged (Fig. 7). However, a 5-fold increase in SII concentration produced a few transcripts shortened as much as 25 nt (Fig. 5, lane 8). If these shortened transcripts corresponded to RNAP II molecules backed up from the lesion, then photolyase might be able to access the CPD and reverse it, allowing transcription to proceed past the resulting thymines. Complexes arrested at the CPD were treated with A. nidulans photolyase and light after transcript cleavage with SII, and then NTPs were added to allow transcription to proceed on any repaired templates. To distinguish between the full-length transcripts normally observed in these reactions (Fig. 3, lane 6) and RNAs deriving from read-through after photoreactivation, template DNA was shortened by digestion with restriction enzyme HhaI prior to NTP addition. As expected, most transcripts were reelongated only up to the dimer site (Fig. 8, lane 7), but some were elongated to the HhaI site. To verify that the photolyase was active under the conditions of the transcription assay, elongation complexes were arrested at position 14 on the template by omission of GTP, treated with photolyase to repair the CPD, and finally incubated with NTPs to allow elongation to resume. All the transcripts observed under these conditions were full-length, indicating that the photolyase was active (data not shown).
Fig. 5. SII-mediated transcript cleavage of RNAP II-arrested complexes.
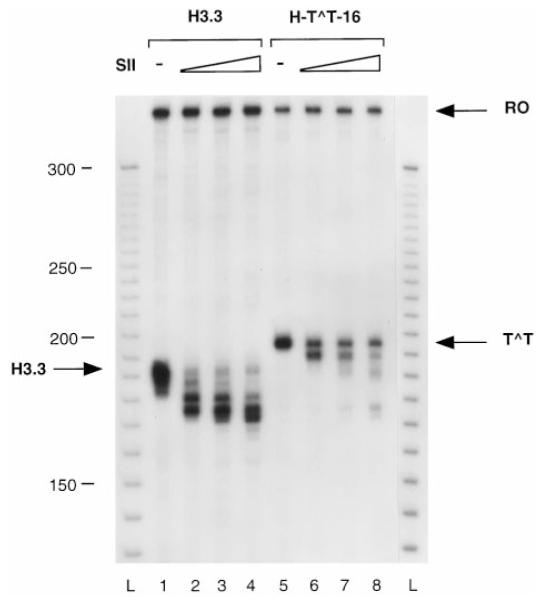
Complexes arrested at the H3.3 arrest site (lanes 1–4) or at a CPD (lanes 5–8) were incubated with increasing amounts of elongation factor SII (1 ng, lanes 2 and 6; 2.5 ng, lanes 3 and 7; 5 ng, lanes 4 and 8) and MgCl2 for 1 h at 28 °C. Runoff RNA (RO) and RNA resulting from transcription arrest at the H3.3 site (H3.3) and at a CPD (T^T) are marked with an arrow. L, 10-bp ladder.
Fig. 8. RNAP II elongation past a dimer site after SII-mediated transcript cleavage and photoreactivation.
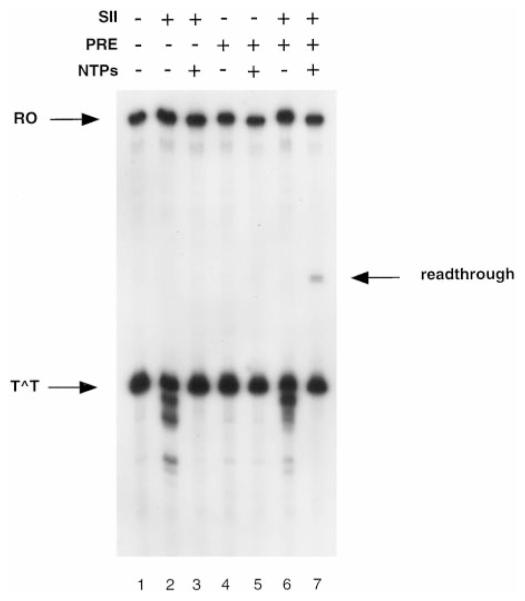
Lanes 1–3, complexes arrested at a CPD were incubated without (lane 1) or with (lanes 2 and 3) elongation factor SII (5 ng) and NTPs as indicated; lanes 4 and 5, complexes arrested at a CPD were treated with photolyase (PRE) and NTPs as indicated; lanes 6 and 7, complexes arrested at a CPD were treated with SII for 1 h before photolyase and NTP treatment as indicated. RO, runoff transcript; T^T, transcript arrested at a CPD; read-through, product resulting from transcription of repaired template.
DISCUSSION
Using ExoIII and T4 exo as footprinting agents and immunopurified RNAP II complexes arrested by a specifically located CPD in the transcribed strand as substrates, we have shown that the footprint of RNAP II arrested by a CPD in the transcribed strand covers ~35 bp, in agreement with Whitson2 and with Selby et al. (26) using T4 exo and λ exonuclease. However, we have found that the footprint is asymmetric around the dimer rather than symmetrical. This conformation is similar to that of complexes arrested at the natural arrest site contained in the human histone H3.3 gene (Refs. 13, 17, and this study). Furthermore, addition of elongation factor SII produced a population of shortened transcripts of discrete lengths up to 25 nt shorter than the transcripts arrested at a CPD. After photoreversal, a fraction of shortened transcripts could be reelongated past the dimer site, suggesting that SII-mediated transcript cleavage resulted in RNAP II backup, thereby providing access of photolyase to the arresting CPD.
The similarity between the footprint at the H3.3 site and at a CPD suggests that arrest at a CPD may occur with a similar mechanism. Recent models of transcription arrest postulate that arrest occurs at certain template locations at which RNAP II fails to continue translocation, resulting in a configuration characterized by a decreased distance between the 3′ end of the transcript and the leading edge of the polymerase and misalignment of the transcript 3′ end from the catalytic site (8). Similarly, complexes arrested at a CPD are characterized by a shorter distance between the position of the CPD and the leading edge of the polymerase.
A stable RNA·DNA hybrid in the elongation complex is necessary to maintain the RNA 3′ terminus engaged with the active site of RNA polymerase (25, 27, 28). This is consistent with arrest occurring at T-rich sequences in the non-transcribed strand where the dA·dT hybrid at the leading edge of the transcription bubble is energetically favored over the dA·rU hybrid (29). Furthermore, this process involves kinetic competition between incorporation of the next nucleotide and decay into the inactive state (30). Formation of a CPD causes a small deformation of the double helix consisting of unwinding by ~15° (31) and bending of at least 7° relative to the B form (32). The neighboring pyrimidines must rotate from their usual B form DNA alignment with overlapping of the 5,6 bonds. This small deformation decreases duplex stability and Watson-Crick hydrogen bonding interaction (33). It is likely that the presence of a CPD in the transcribed strand affects formation of the RNA·DNA hybrid, and this in turn may shift the equilibrium from nucleotide addition toward arrest. Conversely, a CPD in the non-transcribed strand may facilitate read-through of the H3.3 site by shifting the equilibrium toward the RNA·DNA hybrid rather than the DNA·DNA hybrid (22). The lower stability of CPD-containing duplexes has also been proposed to facilitate binding of T4 DNA endonuclease V to a thymine dimer-containing duplex by destacking the base pair flanking the 5′ side of the dimer and “flipping out” the base opposite the 5′ T of the dimer (34).
RNAP II complexes arrested at the H3.3 site can resume elongation after removal of a short transcript from the 3′ end of the nascent RNA by an endonucleolytic activity that resides in the polymerase and is activated by the transcription factor SII (35). After SII-activated transcript cleavage, the polymerase boundaries do not change significantly. However, the transcript 3′ end is repositioned near the catalytic site, and elongation can resume from this new 3′ end (13). A similar conformation is observed after transcript cleavage of RNAP II complexes arrested at a CPD (Figs. 6 and 7). However, unlike the case of RNAP II molecules arrested at the H3.3 site, this conformation allows elongation to proceed up to, but not past, the lesion site (18). This may be explained by the observation that a CPD in the transcribed strand is a complete block to transcription elongation, whereas the H3.3 site causes arrest of only a portion of RNAP II molecules. SII-mediated transcript cleavage of complexes arrested at the H3.3 site is supposed to increase the number of chances a polymerase molecule has to read through the block.
If SII-mediated transcript cleavage were to be of importance in the TCR mechanism, the RNAP II backup would have to be sufficient to permit access of repair enzymes to the lesion. Previously, we have shown that after SII-mediated transcript cleavage RNAP II arrested at a CPD prevented access of photolyase to the lesion site (18). In the present study we have found conditions in which higher concentrations of SII were used that resulted in transcripts shortened as much as 25 nt. Under these conditions we have observed read-through after photoreversal of the CPD. This suggests that these transcription complexes had been displaced from the lesion site a distance sufficient to allow access of photolyase to the CPD. Furthermore, it is consistent with our previous finding that a polymerase arrested at a CPD shields the dimer from photoreactivation (18). These results support the original TCR model in which RNAP II must be removed from the site of the lesion to provide access for repair enzymes to the lesion site (2). They do not exclude the possibility that a fraction of RNAP II molecules covering the lesion may be released from DNA, as suggested by recent evidence indicating that UV irradiation induces ubiquitination of RNAP II followed by proteasomal degradation (36). It is likely that multiple biochemical scenarios may ensue when RNAP II is arrested at a lesion. Depending upon the nature of the lesion and its sequence context, the polymerase might either reverse translocate or be released from the DNA to permit access of repair enzymes.
Acknowledgments
We thank Ann K. Ganesan and C. Allen Smith for helpful discussion and critical reading of this manuscript. We thank A. Eker for a generous gift of A. nidulans photolyase and for helpful advice on the photolyase experiment. We are indebted to Joyce Hunt and John Mote, Jr. for expert technical assistance. We are most grateful to John-Stephen Taylor for providing the oligomer with site-specific CPD.
Footnotes
The abbreviations used are: TCR, transcription-coupled repair; RNAP II, RNA polymerase II; CPD, cyclobutane pyrimidine dimer; bp, base pair(s); TFIID, transcription factor IID; ExoIII, Escherichia coli exonuclease III; T4 exo, T4 DNA polymerase 3′→5′ exonuclease; AdMLP, adenovirus major late promoter; nt, nucleotide.
H. Whitson, unpublished observations.
This work was supported by Grant CA-77712 from the National Cancer Institute, United States Department of Health and Human Services. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Mellon I, Spivak G, Hanawalt PC. 1987;51:241–249. doi: 10.1016/0092-8674(87)90151-6. [DOI] [PubMed] [Google Scholar]
- 2.Mellon I, Bohr VA, Smith CA, Hanawalt PC. Proc. Natl. Acad. Sci. U. S. A. 1986;83:8878–8882. doi: 10.1073/pnas.83.23.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mellon I, Hanawalt PC. Nature. 1989;342:95–98. doi: 10.1038/342095a0. [DOI] [PubMed] [Google Scholar]
- 4.Leadon SA, Lawrence DA. J. Biol. Chem. 1992;267:23175–23182. [PubMed] [Google Scholar]
- 5.Sweder KS, Hanawalt PC. Proc. Natl. Acad. Sci. U. S. A. 1992;89:10696–10700. doi: 10.1073/pnas.89.22.10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smerdon MJ, Thoma F. Cell. 1990;61:675–684. doi: 10.1016/0092-8674(90)90479-x. [DOI] [PubMed] [Google Scholar]
- 7.Tornaletti S, Hanawalt PC. Biochimie (Paris) 1999;81:139–146. doi: 10.1016/s0300-9084(99)80046-7. [DOI] [PubMed] [Google Scholar]
- 8.Uptain SM, Kane CM, Chamberlin MJ. Annu. Rev. Biochem. 1997;66:117–172. doi: 10.1146/annurev.biochem.66.1.117. [DOI] [PubMed] [Google Scholar]
- 9.Kerppola TK, Kane CM. Biochemistry. 1990;29:269–278. doi: 10.1021/bi00453a037. [DOI] [PubMed] [Google Scholar]
- 10.Rice GA, Kane CM, Chamberlin MJ. Proc. Natl. Acad. Sci. U. S. A. 1991;88:4245–4249. doi: 10.1073/pnas.88.10.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mote JJ, Ghanouni P, Reines D. J. Mol. Biol. 1994;236:725–737. doi: 10.1006/jmbi.1994.1185. [DOI] [PubMed] [Google Scholar]
- 12.Reines D. In: Transcription. Conaway R, Conaway JW, editors. Raven Press Ltd.; New York: 1994. pp. 263–278. [Google Scholar]
- 13.Gu W, Powell W, Mote J, Jr., Reines D. J. Biol. Chem. 1993;268:25604–25616. [PMC free article] [PubMed] [Google Scholar]
- 14.Chamberlin MJ. Harvey Lect. 1995;88:1–21. [PubMed] [Google Scholar]
- 15.Nudler E, Goldfarb A, Kashlev M. Science. 1994;265:793–796. doi: 10.1126/science.8047884. [DOI] [PubMed] [Google Scholar]
- 16.Wang D, Meier TI, Chan CL, Feng G, Lee DN, Landick R. Cell. 1995;81:341–350. doi: 10.1016/0092-8674(95)90387-9. [DOI] [PubMed] [Google Scholar]
- 17.Samkurashvili I, Luse DS. J. Biol. Chem. 1996;271:23495–23505. doi: 10.1074/jbc.271.38.23495. [DOI] [PubMed] [Google Scholar]
- 18.Donahue BA, Yin S, Taylor J-S, Reines D, Hanawalt P. Proc. Natl. Acad. Sci. U. S. A. 1994;91:8502–8506. doi: 10.1073/pnas.91.18.8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reines D. J. Biol. Chem. 1991;266:10510–10517. [PMC free article] [PubMed] [Google Scholar]
- 20.Gu W, Reines D. J. Biol. Chem. 1995;270:11238–11244. doi: 10.1074/jbc.270.19.11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eilat D, Hochberg M, Fischel R, Laskov R. Proc. Natl. Acad. Sci. U. S. A. 1982;79:3818–3822. doi: 10.1073/pnas.79.12.3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tornaletti S, Donahue BA, Reines D, Hanawalt PC. J. Biol. Chem. 1997;272:31719–31724. doi: 10.1074/jbc.272.50.31719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samkurashvili I, Luse DS. Mol. Cell. Biol. 1998;9:5343–5354. doi: 10.1128/mcb.18.9.5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaychikov E, Denissova L, Heumann H. Proc. Natl. Acad. Sci. U. S. A. 1995;92:1739–1743. doi: 10.1073/pnas.92.5.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nudler E, Mustaev A, Lukhtanov E, Goldfarb A. Cell. 1997;89:33–41. doi: 10.1016/s0092-8674(00)80180-4. [DOI] [PubMed] [Google Scholar]
- 26.Selby CP, Drapkin R, Reinberg D, Sancar A. Nucleic Acids Res. 1997;25:787–793. doi: 10.1093/nar/25.4.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landick R. Cell. 1997;88:741–744. doi: 10.1016/s0092-8674(00)81919-4. [DOI] [PubMed] [Google Scholar]
- 28.Sidorenkov I, Komissarova N, Kashlev M. Mol. Cell. 1998;2:55–64. doi: 10.1016/s1097-2765(00)80113-6. [DOI] [PubMed] [Google Scholar]
- 29.Yager TD, von Hippel PH. Biochemistry. 1991;30:1097–1118. doi: 10.1021/bi00218a032. [DOI] [PubMed] [Google Scholar]
- 30.von Hippel PH. Science. 1998;281:660–665. doi: 10.1126/science.281.5377.660. [DOI] [PubMed] [Google Scholar]
- 31.Ciarrocchi G, Pedrini AM. J. Mol. Biol. 1982;155:177–183. doi: 10.1016/0022-2836(82)90445-4. [DOI] [PubMed] [Google Scholar]
- 32.Wang C-I, Taylor J-S. Proc. Natl. Acad. Sci. U. S. A. 1991;88:9072–9076. doi: 10.1073/pnas.88.20.9072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jing Y, Kao JF, Taylor J-S. Nucleic Acids Res. 1998;26:3845–3853. doi: 10.1093/nar/26.16.3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vassylyev DG, Kashiwagi T, Mikami Y, Ariyoshi M, Iwai S, Ohtsuka E, Morikawa K. Cell. 1995;83:773–782. doi: 10.1016/0092-8674(95)90190-6. [DOI] [PubMed] [Google Scholar]
- 35.Reines D, Mote JJ. Proc. Natl. Acad. Sci. U. S. A. 1993;90:1917–1921. doi: 10.1073/pnas.90.5.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ratner JN, Balasubramanian B, Corden J, Warren SL, Bregman DB. J. Biol. Chem. 1998;273:5184–5189. doi: 10.1074/jbc.273.9.5184. [DOI] [PubMed] [Google Scholar]