

Abstract
Intracellular generation of reactive oxygen species (ROS) via thiol-mediated reduction of copper(II) to copper(I) has been assumed as the major mechanism underlying the anticancer activity of copper(II) complexes. The aim of this study was to compare the anticancer potential of copper(II) complexes of Triapine (3-amino-pyridine-2-carboxaldehyde thiosemicarbazone; currently in phase II clinical trials) and its terminally dimethylated derivative with that of 2-formylpyridine thiosemicarbazone and that of 2,2′-bipyridyl-6-carbothioamide. Experiments on generation of oxidative stress and the influence of biologically relevant reductants (glutathione, ascorbic acid) on the anticancer activity of the copper complexes revealed that reductant-dependent redox cycling occurred mainly outside the cells, leading to generation and dismutation of superoxide radicals resulting in cytotoxic amounts of H2O2. However, without extracellular reductants only weak intracellular ROS generation was observed at IC50 levels, suggesting that cellular thiols are not involved in copper-complex-induced oxidative stress. Taken together, thiol-induced intracellular ROS generation might contribute to the anticancer activity of copper thiosemicarbazone complexes but is not the determining factor.
Keywords: Thiosemicarbazone, Copper, Anticancer, Reactive oxygen species, Glutathione
Introduction
Cancer cells are known to differ distinctly from healthy tissues in their redox metabolism [1-3]. Besides enhanced levels of intracellular reactive oxygen species (ROS), the specific milieu of the solid tumor is often characterized by high metabolic activity, hypoxia, and, in general, reductive conditions [1, 2]. Consequently, interference with the cellular redox homeostasis of cancer cells seems to be an attractive and promising approach for cancer therapy [4, 5]. Copper complexes are known for their redox-active properties under physiological conditions, which led to the synthesis and biological characterization of a number of copper-containing anticancer drugs and radiopharmaceuticals [6-8]. Current knowledge of anticancer copper compounds is primarily based on investigations using complexes of α-N-heterocyclic thiosemicarbazones (TSC) [9, 10], 2,2′-bipyridyl-6-carbothioamide (BPYTA) [11, 12], and 1,10-phenanthroline [13, 14]. Thiosemicarbazones and BPYTA are well-known tridentate chelators and have been used for the synthesis of a wide range of metal complexes including, besides copper, also iron, cobalt, zinc, nickel, and gallium complexes [12, 15-18]. BPYTA [12, 19] and thiosemicarbazones [17, 20] are known for their ribonucleotide reductase (RR) inhibitory potential. One of these, 3-amino-pyridine-2-carboxaldehyde thiosemicarbazone (Triapine), has already reached clinical phase I/II evaluation, where it showed promising activity against advanced leukemia [21-24]. With regard to copper complexes, Cu-BPYTA [11] and Cu-TSC [25] were also shown to inhibit the RR tyrosyl radical. Interestingly, co-incubation of copper(II) with Triapine significantly increased its RR inhibitory potential [26]. However, no copper complex of Triapine has been synthesized so far. In addition to the RR inhibition by Cu-TSC, generation of ROS is assumed to be a major mechanism of anticancer activity of many copper complexes in general [9, 25, 27-33]. Crucial for this redox reactivity is the presence of a reducing agent such as dithiothreitol, mercaptoethanol, or the biologically relevant glutathione (GSH), which leads to the reduction of copper(II) to copper(I) [9, 25, 27-30]. The reoxidation of copper(I) with molecular oxygen results in the formation of superoxide radicals (O2·−), which has been confirmed in cell-free systems by EPR spin-trapping experiments [30]. The observation of plasmid DNA cleavage provides further evidence that reaction of Cu-TSC complexes with reducing agents generates ROS [34, 35]. Consequently, it is currently widely suggested that these complexes exert their biological activity by intracellular ROS formation after reaction with cellular thiols such as GSH. In accordance, depletion of the intracellular thiol content was reported after treatment of Ehrlich ascites cells with a pyridoxal thiosemicarbazone copper complex [9]. The aim of this study was, on the one hand, to compare the electrochemical properties as well as anticancer activity of the first copper(II) complexes of Triapine and its terminally dimethylated derivative (3-aminopyridine-2-carboxaldehyde 4N,4N-dimethylthiosemicarbazone, APTSC) with those of the already known complexes of 2-formylpyridine thiosemicarbazone (FTSC) and BPYTA. On the other hand, the study focuses on the similarities/differences in their interaction with biologically relevant antioxidants (GSH and ascorbic acid, AA) as well as the role of ROS formation in their anticancer activity against tumor cells.
Materials and methods
All solvents and reagents were obtained from commercial suppliers and used without further purification. FTSC, Triapine, and APTSC·HCl were synthesized according to the methods described in our previous publication [18]. Elemental analyses were conducted with a Carlo Erba microanalyzer at the Microanalytical Laboratory of the University of Vienna and are within ±0.4%, confirming 95% or greater purity. Electrospray ionization mass spectrometry (ESI–MS) was performed with a Bruker Esquire 3000 instrument (Bruker Daltonic, Bremen, Germany). Expected and experimental isotope distributions were compared. UV–vis spectra were recorded with a PerkinElmer Lambda 650 UV–vis spectrophotometer using samples dissolved in methanol (900–200 nm). Thermogravimetric analysis and differential thermal analysis measurements were performed simultaneously using a Mettler Toledo TGA/SDTA851e apparatus, with a 3 °C/min heating rate under an air atmosphere. For all cell- and molecular biological experiments, the compounds were dissolved in DMSO and diluted into the culture media (or buffer) at the concentrations indicated (DMSO concentrations were always below 0.1%).
Synthesis of metal complexes
Pyridine-2-carboxaldehydethiosemicarbazonato-N,N,S-dichloridocopper(II), [Cu(FTSC)Cl2]
The compound was synthesized following a slightly modified published procedure [36]. FTSC (300 mg, 1.66 mmol) was slowly added to a mixture of copper(II)chloride dihydrate (284 mg, 1.66 mmol) and concentrated HCl (278 μL) in methanol (20 mL) at 50 °C. Subsequently, the mixture was stirred for 1 h at room temperature. The green precipitate was filtered off, washed with methanol and diethyl ether, and dried in vacuo. Yield: 380 mg (73%). Anal. Calcd. for C7H8Cl2CuN4S (Mr = 314.68): C, 26.72; H, 2.56; N, 17.80. Found: C, 26.83; H, 2.25; N, 17.43. ESI–MS in methanol (positive): m/z 242, [M–H–2Cl]+; 521, [2M–3Cl–2H]+. UV–vis (MeOH), λmax, nm (ε, M−1 cm−1): 292 (14,200), 326 (9,519)sh, 406 (8,430), 636 (160). EPR [dimethyl sulfoxide (DMSO)/dimethylformamide (DMF) (1:3 v/v); −196 °C]: g = 2.029, g|| = 2.170; hyperfine coupling constants ACu = 26.8 G, A||Cu = 179.7 G; superhyperfine coupling constants AN1 = 20.1 G, A||N1 = 16.8 G; AN2 = 7.0 G, A||N2 = 22.8 G.
= 2.029, g|| = 2.170; hyperfine coupling constants ACu = 26.8 G, A||Cu = 179.7 G; superhyperfine coupling constants AN1 = 20.1 G, A||N1 = 16.8 G; AN2 = 7.0 G, A||N2 = 22.8 G.
3-Aminopyridine-2-carboxaldehyde thiosemicarbazonato-N,N,S-dichloridocopper(II) monohydrate, [Cu(Triapine)Cl2]·H2O
Triapine (150 mg, 0.77 mmol) dissolved in hot methanol (40 mL) was slowly added to a mixture of copper(II)chloride dihydrate (132 mg, 0.77 mmol) and concentrated HCl (130 μL) in methanol (15 mL) at 60 °C [37]. Subsequently, the mixture was stirred for 1.5 h at room temperature. The green precipitate was filtered off, washed with methanol and diethyl ether, and dried in vacuo. Yield: 181 mg (68%). Anal. Calcd. for C7H9Cl2CuN5S·H2O (Mr = 347.71): C, 24.18; H, 3.19; N, 20.14; S, 9.22. Found: C, 24.46; H, 3.09; N, 19.89; S, 9.22. ESI–MS in methanol (negative): m/z 291, [M–2H–Cl]−. UV–vis (MeOH), λmax, nm (ε, M−1 cm−1): 291 (13,450), 367 (3,290), 443 (9,650), 616 (180). EPR [DMSO/DMF (1:3 v/v); −196 °C]: g = 2.032, g|| = 2.180; hyperfine coupling constants ACu = 22.4 G, A||Cu = 181.7 G; superhyperfine coupling constants AN1 = 11.4 G, A||N1 = 0.0 G; AN2 = 12.0 G, A||N2 = 34.6 G. Crystals suitable for X-ray data collection were obtained by slow diffusion of acetone into an aqueous solution of the complex.
3-Aminopyridine-2-carboxaldehyde N,N-dimethylthiosemicarbazonato-N,N,S-dichloridocopper(II), [Cu(APTSC)Cl2]
APTSC·HCl (60 mg, 0.23 mmol) dissolved in hot methanol (12 mL) was slowly added to a mixture of copper(II)chloride dihydrate (40 mg, 0.23 mmol) and concentrated HCl (38 μL) in methanol (5 mL). The mixture was stirred for 1 h at room temperature. The green precipitate was filtered off, washed with methanol and diethyl ether, and dried in vacuo. Yield: 73 mg (87%). Anal. Calcd. for C9H13Cl2CuN5S (Mr = 357.75): C, 30.22; H, 3.66; N, 19.58; S, 8.96. Found: C, 30.30; H, 3.76; N, 19.27; S, 8.95. ESI–MS in methanol (negative): m/z 319, [M–2H–Cl]−. UV–vis (MeOH), λmax, nm (ε, M−1 cm−1): 300 (15,300), 371 (4,146), 458 (15,790), 626 (190). EPR [DMSO/DMF (1:3 v/v); −196 °C]: g = 2.028, g|| = 2.168; hyperfine coupling constants ACu = 23.2 G, A||Cu = 185.4 G; superhyperfine coupling constants AN1 = 17.0 G, A||N1 = 7.2 G; AN2 = 20.7 G, A||N2 = 24.8 G.
Acetato(3-aminopyridine-2-carboxaldehyde thiosemicarbazonato-N,N,S)copper(II)
Triapine (100 mg, 0.51 mmol) dissolved in hot methanol (25 mL) was slowly added to copper(II)acetate monohydrate (104 mg, 0.52 mmol) in methanol (10 mL) at 50 °C. Subsequently, the mixture was stirred for 2 h at room temperature. The brown precipitate was filtered off, washed with methanol and diethyl ether, and dried in vacuo. Yield: 125 mg (77%). Anal. Calcd. for C9H11Cu-N5O2S (Mr = 316.83): C, 34.12; H, 3.50; N, 22.10; S, 10.12. Found: C, 34.27; H, 3.54; N, 21.93; S, 9.83. ESI–MS in methanol (positive): m/z 257, [M–OAc]+; 515, [2M–2OAc–H]+, 573, [2M–OAc]+. UV-vis (MeOH), λmax, nm (ε, M−1 cm−1): 291 (17,040), 368 (4,130), 443 (12,220), 616 (130). EPR [DMSO/DMF (1:3 v/v); −196 °C]: g = 2.031, g|| = 2.191; hyperfine coupling constants ACu = 26.0 G, A||Cu = 183.2 G; superhyperfine coupling constants AN1 = 9.0 G, A||N1 = 27.2 G; AN2 = 14.6 G, A||N2 = 16.3 G.
2,2′-Bipyridyl-6-carbothioamido-N,N,S-dichloridocopper(II), [Cu(BPYTA)Cl2]
The compound was synthesized following a published procedure [19]. Anal. Calcd. for C11H9Cl2CuN3S·0.5H2O (Mr = 358.73): C, 36.83; H, 2.81; N, 11.71; S, 8.94. Found: C, 36.76; H, 2.74; N, 11.49; S, 8.97. ESI–MS in methanol (negative): m/z 347, [M–H]−; 659, [2M–Cl–2H]−. EPR [DMSO/DMF (1:3 v/v); −196 °C]: g = 2.033, g|| = 2.189; hyperfine coupling constants ACu = 18.9 G, A||Cu = 180.4 G; superhyperfine coupling constants AN1 = 10.3 G, A||N1 = 30.6 G; AN2 = 15.3 G, A||N2 = 14.6 G.
Electrochemistry
Cyclic voltammograms were measured in a three-electrode cell using a 2.0-mm-diameter glassy carbon working electrode, a platinum auxiliary electrode, and an Ag|AgCl reference electrode containing 3 M NaCl. Measurements were performed at room temperature using an EG & G PARC 273A potentiostat/galvanostat. Deaeration of solutions was accomplished by passing a stream of argon through the solution for 5 min prior to the measurement and then maintaining a blanket atmosphere of argon over the solution during the measurement. The potentials were measured in DMF/phosphate-buffered saline (PBS) pH 7.4 (2:1 v/v) containing 0.10 M [n-Bu4N][BF4], and are quoted relative to the normal hydrogen electrode (NHE).
Crystallographic structure determination
X-ray diffraction measurements were performed with a Bruker X8 APEXII CCD diffractometer. A twin crystal was positioned at 35 mm from the detector, and 1,033 frames were measured, each for 70 s over 1° scan width. The orientation matrices for two components in the crystal were found by using CELL NOW and were verified by RLATT [38]. The two-component integration was performed using SAINT-Plus [39]. The data were processed using TWINABS [40]. Structure solution and initial refinement were performed in the usual way by direct methods using the HKLF 4 format file. Final refinement including twin fractions (BASF) was accomplished using the HKLF 5 format file. Crystal data, data collection parameters, and structure refinement details are given in Table S1. Non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were inserted in calculated positions and refined with a riding model. Structure solution was achieved with SHELXS-97, refinement was achieved with SHELXL-97 [41], and graphics were produced with ORTEP-3 [42]. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre (CCDC) with number CCDC-810211. Copies of the data can be obtained free of charge from the CCDC (12 Union Road, Cambridge CB2 1EZ, UK; deposit@ccdc.com.ac.uk).
EPR spectroscopy
The copper(II) complexes (1 mM) were dissolved in DMSO/DMF (1:3 v/v) and transferred in an EPR quartz tube (outer diameter 4 mm). Oxygen was removed by passing a stream of argon through the solution for 30 min. Subsequently, the samples were frozen in liquid nitrogen and transferred into a quartz finger dewar for EPR analysis at −196 °C. EPR spectra of the complexes were recorded with a Bruker EMX instrument and a TE102 cavity using the following instrument settings: 9.4-GHz microwave frequency, 5-mW microwave power, 3,134-G center field, 1,080-G sweep, 2-G modulation amplitude, 100-kHz modulation frequency, 1 × 105 receiver gain, 96 G/min scan rate, 0.327-s time constant, and three scans.
Spectral simulations of the copper complexes were performed using the program EasySpin [43]. A coupling of two nonequivalent 14N nuclei with the copper metal center (electron spin of ½) was assumed. Simulations were performed using the Esfit module and the Pepper function of EasySpin. Stepwise g factors, coupling constants, and line widths were optimized using genetic/simplex algorithms, and baseline distortions were corrected by the lsq2 parameter.
Cell culture
The following human cell lines were used in this study: the colon carcinoma cell line SW480, the promyelocytic leukemia cell line HL60, the ovarian carcinoma cell line A2780, and its cisplatin-resistant subline A2780cis. SW480 and HL60 were purchased from American Type Culture Collection (Manassas, VA, USA), and the A2780 lines were obtained from Sigma-Aldrich. All cells were grown in RPMI-1640 supplemented with 10% fetal bovine serum, with the exception of SW480 cells, which were cultivated in minimal essential medium with 10% fetal bovine serum. Cultures were regularly checked for Mycoplasma contamination.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays
To determine cell viability, cells were plated (2 × 103 cells in 100 μl per well) in 96-well plates and allowed to recover for 24 h. Drugs were added in another 100 μl growth medium and cells were exposed for the time periods indicated. For N-acetylcysteine (NAC) and AA experiments, cells were preincubated with 50 μl NAC solution (1 and 2 mM in growth medium) for 30 min. Then, drugs were added in another 50 μl. For washout experiments, NAC-containing medium was replaced with fresh (drug-free) culture medium after the preincubation before Cu-TSC treatment. After 72 h drug treatment, the proportion of viable cells was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay following the manufacturer’s recommendations (EZ4U, Biomedica, Vienna, Austria).
Hoechst 33258/propidium iodide staining
To determine early and late apoptosis as well as necrosis, Hoechst 33258 and propidium iodide (PI) (both from Sigma) were added directly to the culture medium of SW480 cells to final concentrations of 5 and 2 mg/mL, respectively, after 24 h drug incubation. After an incubation period of 1 h at 37 °C, the cells were examined with a Zeiss Axiovert 35 fluorescence microscope with 4′,6-diamidino-2-phenylindole filters. Cells were photographed on Kodak Ektachrome P1600 film (Eastman Kodak, Rochester, NY, USA). Viable cells, apoptotic cells, and necrotic cells were counted. The Hoechst dye stains the nuclei of live and dead cells; therefore, nuclear changes associated with apoptosis, such as chromatin condensation and nuclear fragmentation, can be monitored [44]. On the other hand, PI is excluded from viable and early apoptotic cells. Consequently, PI uptake indicates the loss of membrane integrity characteristic of necrotic and late apoptotic cells. In combination with fluorescence microscopy, selective uptake of the two dyes allows to monitor the induction of apoptosis in intact cultures and to distinguish it from nonapoptotic cell death (necrosis). Necrosis is characterized in this system by nuclear PI uptake without chromatin condensation or nuclear fragmentation. Duplicate slides were prepared for each cell type/treatment group and more than 500 cells were counted for each sample.
Measurement of intracellular oxidants
2′,7′-Dichlorofluorescein diacetate (DCF-DA) was used to detect the production of ROS [45]. DCF-DA stock solutions (33.4 mM) in DMSO were stored at −20 °C. HL60 cells (2.5 × 105 cells per sample in phenol-free Hanks balanced salt solution) were incubated with DCF-DA for 30 min. Subsequently, the copper complexes were added in the indicated concentrations. After incubation for another 30 min, the mean fluorescence intensity was measured by flow cytometry using an FACSCalibur instrument (Becton Dickinson, Palo Alto, CA, USA). A concentration of 200 μM H2O2 was used as the control. The resulting histograms were quantified using ModeFit (BD). In the case of experiments with NAC and AA, the reducing agents were added to the samples 15 min prior to addition of the copper complexes.
Generation of superoxide radicals
To examine the cell-free production of superoxide radicals, the reduction of nitroblue tetrazolium (NBT) was analyzed. Briefly, 0.6 mM NBT was incubated with 25 μM copper complexes with or without 2 mM NAC or 50 μM AA. The experiments were performed in 0.1 M PBS (pH 7.8). The extent of NBT reduction was determined spectrophotometrically by measuring the absorbance at 560 nm after 10 min of incubation.
Determination of superoxide dismutase activity by xanthine oxidase assay
Superoxide dismutase (SOD) activity of the complexes was investigated by using their ability to inhibit the reduction of NBT by xanthine/xanthine oxidase generated superoxide radicals [46]. The reaction system contained 0.2 mM xanthine, 0.6 mM NBT, increasing concentrations of the copper compounds, and 70 mU/mL xanthine oxidase to start the reaction in 0.1 M PBS at pH 7.8. The extent of NBT reduction was followed spectrophotometrically by measuring the absorbance at 560 nm. Each experiment was performed in duplicate, and the SOD activity was defined as the concentration of the copper complex tested necessary to inhibit 50% of the NBT reduction (IC50 value) caused by superoxide.
Quantification of H2O2 production
To measure cell-free production of H2O2, the PerOXOquant assay (Pierce, Rockford, IL, USA) was used according to the manufacturer’s recommendations. The detection of peroxides by this assay is based on oxidation of ferrous to ferric ions in the presence of xylenol orange at acidic pH. The Fe3+ ions are coordinated by xylenol orange, yielding a purple product with an absorbance maximum at 560 nm. Peroxide levels in test samples were determined using H2O2 standards.
Results and discussion
Synthesis and characterization
Triapine and APTSC were synthesized in a three-step procedure according to methods described in the literature [18]. FTSC was synthesized by condensation of 2-formylpyridine and thiosemicarbazide in boiling EtOH/H2O.
Owing to the generally low aqueous solubility of 1:2 complexes of the type [Cu(L)2] (where L is a monodeprotonated TSC) and their tendency to partially dissociate into the corresponding 1:1 complexes [10], all copper(II) complexes were synthesized in a 1:1 metal-to-ligand ratio (Fig. 1). The reaction of CuCl2·2H2O with the thiosemicarbazone ligands in methanol in the presence of 2 equiv of concentrated HCl resulted in green complexes of the general formula [Cu(HL)Cl2] [HL is FTSC (Cu-FTSC), Triapine (Cu-Triapine), APTSC (Cu-APTSC)] in 66–87% yield. In the case of Cu-Triapine, according to the elemental analysis one molecule of water co-crystallized, which was confirmed by thermogravimetric measurements. In addition, reaction of Triapine and Cu(OAc)2·H2O in methanol resulted in deprotonation of the ligand and isolation of the brown product [Cu(L)OAc] [HL is Triapine; Cu(OAc)-Triapine] in 77% yield. [Cu(BPYTA)Cl2] (Cu-BPYTA) was synthesized according to a literature procedure [19].
Fig. 1.

Overview of compounds investigated
The negative ESI mass spectra of the chlorido complexes Cu-Triapine and Cu-APTSC showed exclusively a strong peak due to [M–2H–Cl]− ions at m/z 291 and 319, respectively. In the case of the acetato complex of Triapine, only the positive ion mode displayed strong peaks due to [M–OAc]+ at m/z 257 and additionally two dimeric fragments at m/z 515 and 573 attributed to [2M–2OAc–H]+ and [2M–OAc]+, respectively. The UV–vis spectra of the copper(II) thiosemicarbazone complexes in methanol showed the characteristic very weak d–d absorption band in the range 616–636 nm. The strong metal-to-ligand charge transfer absorption is redshifted from 406 nm (Cu-FTSC) to 443 nm (Cu-Triapine) and 458 nm (Cu-APTSC) in accordance with the electron-donating properties of the amino and methyl groups.
X-ray crystallography
X-ray diffraction quality single crystals of Cu-Triapine were obtained by slow diffusion of acetone into an aqueous solution of Cu-Triapine. The result of the X-ray diffraction study is shown in Fig. 2. Selected bond distances and angles are quoted in the legend to Fig. 2. The coordination polyhedron around the copper(II) center approaches a square-planar geometry, with a chlorido ligand and the monodeprotonated tridentate Triapine coordinated to the metal ion. In addition, an H3O+ molecule and a chlorido counterion are present in the asymmetric unit. The deprotonation in aqueous solution is in agreement with the concentration distribution curves of the CuII-Triapine system indicating the presence of the [CuL]+ species (HL is Triapine) in a wide pH range [47]. In the solid state the square-planar molecules are linked via intermolecular Cu–S bonds [2.986(4) Å], forming a chain structure (see Fig. S1).
Fig. 2.
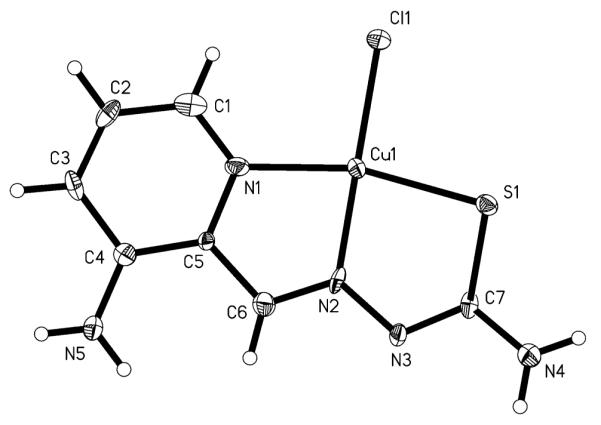
ORTEP plot of Cu-Triapine with thermal ellipsoids depicted at the 50% probability level (the H3O+Cl− fragment is omitted). Selected bond distances (Å) and angles (°): Cu–Cl, 2.2493(5); Cu–S, 2.281(3); Cu–N2, 1.937(9); Cu–N1, 2.031(8); S1–C7, 1.723(10); C7–N3, 1.311(13); N3–N2, 1.383(11); N2–C6, 1.317(13); N1–Cu–S, 164.5(2); N2–Cu–Cl, 176.9(3); N1–Cu–N2, 81.1(3); N2–Cu–S, 83.6(3)
EPR spectroscopy
The EPR spectra of the five copper(II) complexes in DMSO/DMF solutions (1:3 v/v) at −196 °C show the typical four hyperfine lines due to the d9 configuration (S = ½) and a nuclear spin I = 3/2 of the main isotopes 63Cu and 65Cu as well as a superhyperfine splitting of the high-field lines due to the interaction of copper with the 14N nuclei (I = 1) (Fig. 3, S2–S5). All complexes show g|| and g parameters typical of axial symmetry (see “Synthesis of metal complexes”), and g|| > g > 2.0023 is consistent with a dx2–y2 ground state in a square-planar or square-pyramidal geometry [48]. The spectra were simulated using EasySpin [43] assuming the coupling of two nonequivalent 14N nuclei with the copper(II) center and are in good agreement with the experimental data (Fig. 3, S2–S5). Except for the superhyperfine coupling, in the EPR spectra of the copper(II) complexes only small changes could be observed, which is obvious considering the existence of the NNS coordination pattern in all compounds. Comparison of the EPR spectra of Cu-Triapine and Cu(OAc)-Triapine in DMSO/DMF (Fig. S6a) reveals significant differences in the superhyperfine coupling, whereas the same complexes in DMSO/PBS pH 7.4 (1:1 v/v; Fig. S6, plot b) exhibit almost the same spectrum. This further supports the assumption that Cu-Triapine releases 1 equiv of HCl (see above) so that both copper(II) Triapine complexes form a square-planar [CuL(H2O)]+ species (HL is Triapine) in aqueous solution. The formation of [CuL(H2O)]+ is also supported by the spectra of the already square-planar complex Cu(OAc)-Triapine, which distinctly differs upon changing the solvent from DMSO/DMF to DMSO/PBS (Fig. S6).
Fig. 3.
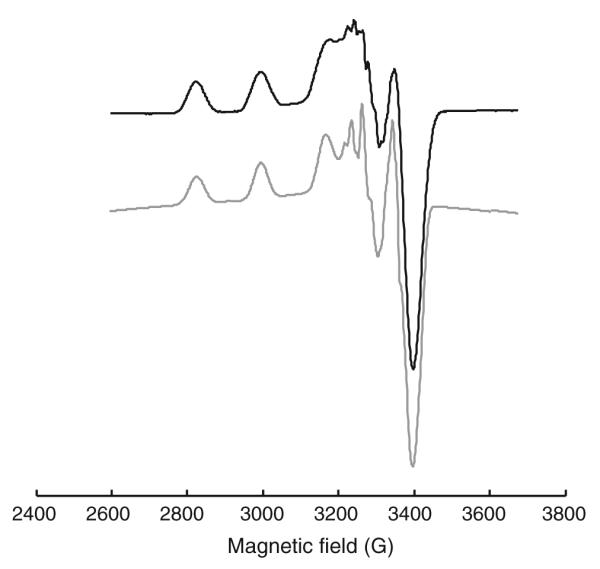
EPR spectrum of 1 mM Cu-Triapine in dimethyl sulfoxide (DMSO)/dimethyl formamide (DMF) (1:3 v/v) (black) and simulated spectrum (gray). Experimental conditions: X-band; temperature −196 °C, microwave frequency 9.4 GHz; microwave power 5 mW; modulation amplitude 2 G
Cyclic voltammetry
The electrochemical properties of the five copper(II) complexes were investigated by cyclic voltammetry in DMF/PBS pH 7.4 (2:1 v/v) containing 0.10 M [n-Bu4N] [BF4] as the supporting electrolyte. All complexes display an irreversible copper(II)/copper(I) reduction peak between −0.23 and +0.10 V versus NHE at 200 mV/s scan rate (Table 1). The decrease of the redox potentials in the order Cu-FTSC > Cu-Triapine > Cu-APTSC is in line with the electron donor properties of the NH2 and CH3 groups. Owing to the low aqueous solubility of the complexes, the measurements were performed in DMF/PBS mixtures. However, this makes the direct comparison with the literature redox potentials of AA (+0.06 V vs. NHE) [49], NAC (−0.18 V vs. NHE) [50], and GSH (−0.24 V vs. NHE) [51] determined in pure aqueous solutions at pH 7.0 difficult. Nevertheless, the redox potentials of the thiosemicarbazone complexes investigated suggest that AA is not able to reduce them, whereas the redox potentials of NAC and GSH are in the same potential range as the copper(II) to copper(I) reduction.
Table 1. Electrochemical data.
| Ep/CuII/I | |
|---|---|
| Cu-FTSC | −0.14 |
| Cu-Triapine | −0.19 |
| Cu(OAc)-Triapine | −0.21 |
| Cu-APTSC | −0.23 |
| Cu-BPYTA | +0.10 |
See Fig. 1 for an explanation of the compounds. Potentials in volts ± 0.02 versus the normal hydrogen electrode in 2:1 dimethylformamide/0.2 M phosphate-buffered saline pH 7.4 containing 0.10 M [n-Bu4N][BF4]
Anticancer activity and intracellular ROS generation
The antiproliferative activity of the five copper complexes was compared in MTT viability assays using SW480 colon cancer and leukemic HL60 cells. In general, both cell lines displayed widely similar sensitivity to the test compounds, with IC50 values in the low micromolar range (Table 2). The most active compound in our test panel was Cu-Triapine, with an IC50 value of approximately 1.2 μM, whereas Cu-BPYTA was least active, with an IC50 value of approximately 8.5 μM. On the basis of the rather similar anticancer activity of Cu-Triapine and Cu(OAc)-Triapine and the results obtained by the physicochemical characterization, all further experiments were performed only with Cu-Triapine. The antiproliferative activity of the copper(II) complexes was also compared with that of the corresponding metal-free ligands (Table 2). All copper(II) complexes were found to be slightly less effective than the respective metal-free ligands, with exception of Cu-FTSC, where in SW480 cells an about threefold increased activity compared with metal-free FTSC was observed. However, in HL60 cells these two compounds were comparably active. These results are in accordance with the observations described in previous publications, where complexation of FTSC and copper(II) was reported to result in increasing or decreasing activity depending on the cell type investigated [15, 52, 53].
Table 2.
Cytotoxic/cytostatic activity against two human cancer cell lines after 72 h treatment
| IC50 (μM) ± SD |
||
|---|---|---|
| SW480 | HL60 | |
| Cu-FTSC | 3.3 ± 1.2 | 3.4 ± 0.4 |
| Cu-Triapine | 1.3 ± 0.3 | 1.1 ± 0.1 |
| Cu(OAc)-Triapine | 2.1 ± 0.2 | n.d. |
| Cu-APTSC | 5.5 ± 0.7 | 3.1 ± 0.6 |
| Cu-BPYTA | 8.4 ± 0.4 | 8.7 ± 0.6 |
| FTSC | 10.6 ± 0.1 | 3.3 ± 0.5 |
| Triapine | 0.4 ± 0.05 | 0.4 ± 0.1 |
| APTSC | 0.4 ± 0.01 | 0.2 ± 0.02 |
| BPYTA | 5.8 ± 3.0 | 7.1 ± 0.5 |
SD standard deviation
To investigate whether the reduction in cell viability observed by the MTT assays is based on cytostatic or cytotoxic effects, Hoechst 33258/PI stainings were performed [44]. As shown in Fig. 4, the apoptosis-inducing potential of the copper complexes strongly differed. In the case of Cu-Triapine, virtually no increase in the number of apoptotic cells was observed, indicating that this compound acts solely as a cytostatic agent. In contrast, apoptosis induction by Cu-FTSC correlated with the results obtained in MTT assay (at IC50 levels, 45.2% of the cells were apoptotic), identifying this compound as mainly a cytotoxic drug. In the case of Cu-APTSC and Cu-BPYTA, no induction of apoptosis was found at IC50 levels. However, at higher drug concentrations a profound increase in the number of apoptotic cells up to 100% was detected. Thus, these drugs have both cytostatic and cytotoxic properties.
Fig. 4.
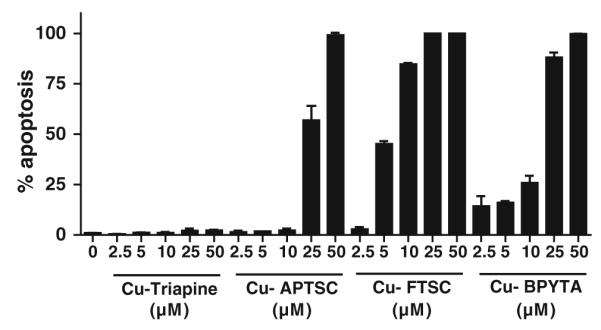
Cell death induction. The proportion of apoptotic cells (early and late apoptosis) was determined by Hoechst 33258/propidium iodide staining [44] after 24 h treatment with the indicated concentrations of the copper complexes tested. More than 500 cells from at least two samples for each concentration were analyzed and the percentages of early and late apoptotic cells at the indicated concentrations were determined by counting. One representative experiment of two giving comparable results is shown
The generation of ROS is assumed to be one of the main mechanisms underlying the anticancer activity of copper(II) complexes [31-33]. To evaluate the impact of the copper complexes on the intracellular levels of ROS, the cell-permeable, fluorescent dye DCF–DA was used. An enzymatic hydrolysis by intracellular esterases is necessary to gain the ROS-detecting DCF–H. This assay is frequently applied for the detection of intracellular H2O2 and ROS such as OH. and ROO . [45]. Figure 5 shows the induction of ROS after treatment with increasing concentrations of the copper complexes. In accordance with previous reports [9, 25], treatment with Cu-FTSC and Cu-BPYTA led to a significant (in the case of Cu-FTSC dose-dependent) increase of DCF–DA fluorescence (up to approximately fivefold after treatment with 50 μM Cu-FTSC and approximately fourfold after treatment with 25 and 50 μM Cu-BPYTA). In contrast, Cu-Triapine and Cu-APTSC only slightly enhanced intracellular ROS levels (about 1.25-fold). Unexpectedly, treatment with 50 μM Cu-APTSC even strongly decreased the DCF-DA fluorescence by 0.2-fold. None of the metal-free ligands displayed any significant effect on DCF-DA fluorescence (data not shown).
Fig. 5.
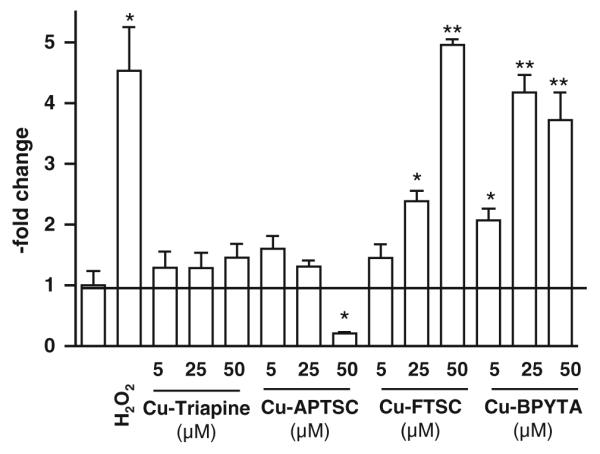
Intracellular reactive oxygen species (ROS) generation. Intracellular production of ROS in HL60 cells by the indicated concentrations of the copper complexes was determined after 30 min incubation using the ROS indicator 2′,7′-dichlorofluorescein diacetate (DCF-DA). Fluorescence was measured by flow cytometry. One representative experiment of three giving comparable results is shown. Significant differences were calculated by Student’s t test: single asterisk p < 0.05, two asterisks p < 0.01
In general, the ROS generation data are in good agreement with the apoptosis-inducing potential of the test compounds, indicating that (in agreement with the literature) the apoptosis induction at higher drug concentrations is mainly based on intracellular ROS. However, it has to be noted that at IC50 levels (5 μM) the increase in intracellular ROS levels induced by all the copper complexes is rather low, with Cu-BPYTA displaying the strongest effect (twofold increase). Together with the lack of enhanced anticancer activity of the copper complexes in comparison with their metal-free ligands observed in MTT assay, the data question the relevance of intracellular ROS generation for the anticancer activity of copper thiosemicarbazone complexes in general and especially for Cu-APTSC and Cu-Triapine. Notably, preliminary uptake experiments of the copper complexes using inductively coupled plasma mass spectrometry indicate that the lack of intracellular ROS generation is not based on insufficient cellular drug uptake. Even distinctly higher cellular copper levels were detected in the case of Cu-APTSC and Cu-FTSC as compared with equimolar concentrations of CuSO4 (data not shown).
Impact of reductants on the anticancer activity of the copper complexes
It is well established that reduction of copper complexes (e.g., by thiols such as GSH) leads to redox cycling and generation of ROS [9, 25, 27-30]. Thus, the effect of co-incubation with the GSH precursor NAC on the substance panel was investigated. As expected, drastically enhanced anticancer activity of all copper complexes in SW480 cells (Fig. 6, left panel) and HL60 cells (Fig. S7, left panel) was observed in the presence of NAC (1.0 and 2.0 mM). Notably, in the case of Cu-APTSC, addition of NAC had the weakest effect on the activity. In contrast to the NAC experiments, co-incubation with the antioxidant AA (25 and 50 μM) only weakly enhanced the activity of Cu-BPYTA, whereas it had no significant effects on Cu-FTSC and Cu-APTSC (Fig. 6, S7, right panel). In the case of Cu-Triapine, cotreatment with 50 μM AA even slightly decreased the anticancer activity 1.4-fold. These observations are in good agreement with the reduction potential of the copper(II) complexes obtained by cyclic voltammetry (see Table 1), with the highest redox potential for Cu-BPYTA and the lowest for Cu-APTSC.
Fig. 6.

Impact of reductants on the anticancer activity of the copper complexes. To evaluate the effects of reductants, the glutathione (GSH) precursor N-acetylcysteine (NAC) and the antioxidant ascorbic acid (AA) were used. Briefly, after 30 min preincubation with NAC (1 and 2 mM) or AA (25 and 50 μM), SW480 cells were treated for 72 h with the indicated concentrations of the copper complexes. Viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The values given are the mean ± the standard deviation of three determinations from three experiments
To evaluate whether these changes in anticancer activity are based on enhanced intracellular ROS levels, DCF-DA stainings were performed after cotreatment with NAC or AA. Figures 7 and S8 show that combination of the copper complexes with 2 mM NAC induced a strong increase of the intracellular ROS levels. In experiments using 25 μM copper complexes (Fig. 7), the strongest effects were observed with Cu-FTSC (eightfold increase) and the weakest effects were observed with Cu-APTSC (fivefold increase). At lower drug concentrations (5 μM), similar effects, however to a lesser extent, were found (Fig. S8). In contrast to the results of the 72-h viability assays (where no or only a minor influence of AA was observed), co-incubation with 50 μM AA significantly enhanced intracellular ROS levels after treatment with the copper complexes, however, in general, to a weaker extent than co-incubation with NAC. The strongest effects of AA on intracellular ROS levels were observed in the case of Cu-FTSC and Cu-BPYTA (fivefold increase).
Fig. 7.
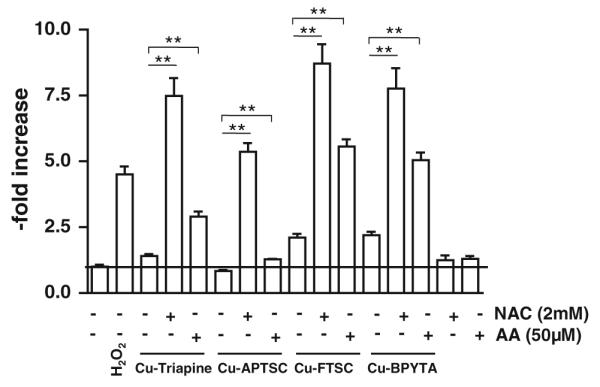
Reductant-induced ROS generation by the copper complexes. The influence of pretreatment with 2 mM NAC or 50 μM AA on the intracellular ROS levels in HL60 cells after incubation with the copper complexes (25 μM) was determined using the ROS indicator DCF-DA. Fluorescence was measured by flow cytometry. One representative experiment of three giving comparable results is shown. Significant differences were calculated by Student’s t test: two asterisks p < 0.01
To test whether cells with increased amounts of GSH are more susceptible to copper compounds, the ovarian cancer cell model A2780 and its cisplatin-resistant subline A2780cis containing higher intracellular GSH levels [54] were used. In general, parental A2780 cells displayed drug sensitivity (Table 3) very similar to the HL60 cells (see Table 2). On the basis of the higher GSH levels in A2780cis cells, enhanced thiol-induced ROS generation leading to increased activity of the copper(II) complexes was expected. In contrast, A2780cis cells were significantly resistant (up to 22-fold in the case of Cu-Triapine) to all the copper complexes tested.
Table 3.
Cytotoxic/cytostatic activity in A2780 and A2780cis cell lines at 72 h treatment
| IC50 (μM) ± SD |
|||
|---|---|---|---|
| A2780 | A2780cis | Resistance | |
| Cu-FTSC | 3.0 ± 1.0 | 8.8 ± 2.2 | 2.9-fold |
| Cu-Triapine | 1.3 ± 0.1 | 29 ± 3 | 22-fold |
| Cu-APTSC | 5.6 ± 0.1 | 8.3 ± 0.6 | 1.5-fold |
| Cu-BPYTA | 7.3 ± 1.8 | 25 ± 70 | 3.4-fold |
To gain more insight into the impact of the intracellular GSH levels on the anticancer activity and ROS-inducing capability of the copper compounds tested, the intracellular GSH pools of SW480 cells were boosted by preloading with NAC, which is then quickly metabolized to GSH inside the cell. After 30 min preincubation, remaining extracellular NAC was removed and the copper compounds were added to the cells in fresh culture medium. As shown in Fig. 8a, in contrast to the drastic increase in cytotoxic/cytostatic activity caused by NAC co-incubation observed in previous experiments (Fig. 6), enhanced intracellular GSH pools did not increase the activity of the copper complexes tested. Notably, in case of Cu-Triapine, NAC preloading even had protective effects (twofold and fourfold increase in the IC50 value caused by preincubation with 1 and 2 mM NAC, respectively). In accordance, no enhanced ROS generation was detected in NAC-preloaded cells (after washout) via DCF–DA stainings (Fig. 8b, right panel). This indicates extracellular generation of ROS, which then react after membrane diffusion with DCF–H inside the cell. As H2O2 is the only diffusible ROS species, we suggested that thiol-induced redox cycling of the copper complexes leads to extracellular production of H2O2.
Fig. 8.

Impact of enhanced intracellular GSH levels on the anticancer activity of the copper complexes. a To evaluate the impact of elevated intracellular GSH levels on the copper complexes tested, SW480 cells were incubated with the GSH precursor NAC. After 30 min pretreatment, NAC-containing medium was replaced by fresh culture medium. Then the copper complexes were added at the indicated concentrations. After 72 h incubation, viability was determined using MTT assay. The values given are the mean ± the standard deviation of three determinations from three experiments. b Left DCF-DA-loaded HL60 cells were incubated for 15 min with 2 mM NAC, then the test compounds (50 μM) were added in the presence of NAC. After 30 min incubation, fluorescence was measured by flow cytometry. Right after preincubation, the NAC-containing buffer was replaced by fresh Hanks balanced salt solution and the test compounds (50 μM) were added. After 30 min incubation, fluorescence was measured by flow cytometry. One representative experiment of three giving comparable results is shown
Extracellular H2O2 and superoxide radical formation by cotreatment with biologically relevant reductants
To test whether coincubation of copper compounds with NAC and AA leads to generation of H2O2 under cell-free conditions, H2O2 levels were determined using the xylenol orange-based PerOXOquant assay. Figure 9 shows that without addition of reductants, no H2O2 production by the copper complexes was detectable. Co-incubation with 2 mM NAC distinctly induced H2O2 formation in the case of all four copper complexes (50 μM). For Cu-APTSC and Cu-BPYTA, 2–3 equiv of H2O2 per copper complex molecule was produced, whereas Cu-Triapine and Cu-FTSC were less effective, inducing production of only 1 equiv of H2O2. Thus, redox cycling was only observed in the case of Cu-APTSC and Cu-BPYTA. Notably, also after AA cotreatment, minor levels of H2O2 were detected. Overall, the results obtained in these cell-free settings are in good agreement with the findings of the intracellular DCF-DA staining experiments (see Fig. 7).
Fig. 9.
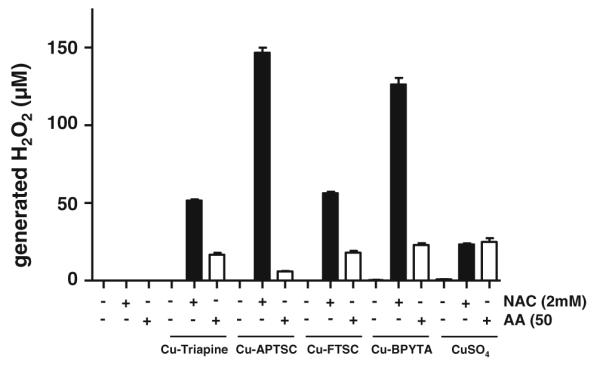
NAC- and AA-induced H2O2 production by the copper complexes. The dependence of the level of copper-complex-generated H2O2 on NAC (2 mM) and AA (50 μM) was determined using the xylenol orange-based PerOXOquant assay. The copper complexes were used at concentrations of 50 μM. The values given are the mean ± the standard deviation of three determinations
To prove that H2O2 is the ROS responsible for the DCF-DA fluorescence reported in Fig. 7, the respective experiments were repeated in the presence of the H2O2-metabolizing enzyme catalase (Figs. 10a, S9). In the case of all four copper complexes, addition of extracellular catalase significantly reduced intracellular NAC-induced ROS levels. Application of SOD together with catalase further reduced ROS levels, especially in the case of Cu-FTSC and Cu-APTSC, suggesting that the extracellular H2O2 formation is O2·−-dependent (Fig. S9). The protective effects of catalase against the NAC-induced ROS formation were further confirmed for Cu-Triapine using MTT assays (Fig. 10b).
Fig. 10.

Effect of extracellular superoxide dismutase (SOD) and catalase (CAT) on thiol-induced ROS generation. a Influence of SOD and CAT cotreatment (100 U/mL) on the NAC-induced (2 mM) ROS formation by Cu-Triapine (25 μM) in HL60 cells was determined using DCF-DA. Fluorescence was measured by flow cytometry. One representative experiment of three giving comparable results is shown. Significant differences were calculated by Student’s t test: two asterisks p < 0.01, three asterisks p < 0.001. b Left impact of CAT cotreatment (100 U/mL) on the anticancer activity of Cu-Triapine in the presence and absence of NAC. Right protective effects of CAT against H2O2-induced cell death. Cell viability was determined by MTT assay after 72 h drug treatment
For several copper compounds (including CuSO4), SOD-like activity has been reported [55]. As this ability is probably the underlying mechanism of H2O2 formation shown in Fig. 9, the superoxide radical dismutation ability of our complex panel was measured using the xanthine/xanthine oxidase assay (Table 4). In these experiments, the xanthine/xanthine oxidase system was used to generate superoxide radicals, which in turn led to the reduction of NBT, resulting in blue staining. In accordance with already published data [46], the IC50 value for the SOD-like activity of CuSO4 was found to be approximately 20 μM. Also Cu-FTSC and Cu-BPYTA had potent activity, with IC50 values of 45 and 6 μM, respectively. In the case of Cu-Triapine, only a weak SOD-like activity with an IC50 value of 98 μM was detected. Up to 100 μM, Cu-APTSC had no effect on the detected O2·− levels. These effects are in strong correlation with the redox potential (see Table 1), indicating that the redox potential is important for the SOD-like activity of copper compounds. This is in good agreement with a study on copper 1,10-phenanthroline complexes, which also concluded that there is a direct correlation between the redox potential and SOD-like activity within the phenanthroline compounds studied [56].
Table 4.
Superoxide dismutase (SOD)-like activity
| IC50 (μM) | |
|---|---|
| Cu-FTSC | 45 |
| Cu-Triapine | 98 |
| Cu-APTSC | ⪢100 |
| Cu-BPYTA | 6 |
| CuSO4 | 20 |
| Native Cu/Zn-SOD | 0.04a |
SOD-like activity of the copper complexes was investigated by spectrophotometric determination of their ability to inhibit the reduction of nitroblue tetrazolium by xanthine/xanthine oxidase generated superoxide radicals
Taken from [57]
With respect to the thiol-induced redox cycling of thiosemicarbazone copper compounds, it has to be kept in mind that the xanthine/xanthine oxidase assay is performed without addition of NAC or AA [46]. Moreover, if the generated H2O2 is produced by dismutation of O2·−, the question of the source of O2·− production arises. To determine whether the copper complexes are able to produce the necessary superoxide radicals upon reduction by NAC (or AA), NBT was used as an O2·−-detecting agent (without xanthine/xanthine oxidase). As shown in Figs. 11 and S10, distinct formation of O2·− was detected in the samples containing copper complexes with NAC, and this formation was inhibited by cotreatment with natural SOD (Fig. S10). In contrast, without NAC or in the presence of AA, no significant formation of O2·− was found. Together with the H2O2 generation, this indicates that in the presence of NAC, the copper complexes tested generate O2·−, which is then further dismutated to the membrane-permeable H2O2, leading to intracellular oxidative stress.
Fig. 11.

O2·− generation ability of the copper complexes in the presence of NAC and AA. The dependence of the level of copper-complex-generated O2·− on NAC (2 mM) and AA (50 μM) was determined by measuring the reduction of nitroblue tetrazolium spectrophotometrically. The copper complexes were used at concentrations of 25 μM. The values given are the mean ± the standard deviation of three determinations. Significant differences were calculated by Student’s t test: single asterisk p < 0.05, two asterisks p < 0.01
Conclusions
Generation of ROS based on the reduction of copper(II) to copper(I) by intracellular reducing agents has so far been assumed to be the major mode of action underlying the anticancer activity of copper(II) complexes [9, 25, 27-33]. In accordance, also in our study addition of the GSH precursor NAC drastically increased the anticancer activity of the copper(II) complexes investigated. However, several of our observations indicate that reductant-induced intracellular redox cycling does not dominate the anticancer activity of copper complexes: (1) although potent ROS generation was induced by NAC co-incubation, single treatment with Cu-Triapine, Cu-APTSC, or Cu-FTSC at IC50 levels did not induce significant intracellular DCF-DA fluorescence (Fig. 5); (2) cells with significantly enhanced intracellular GSH levels (A2780cis cells) did not show increased sensitivity to treatment with the copper complexes (Table 3); and (3) the presence of extracellular catalase strongly protected cells from reductant-induced copper-complex-mediated oxidative stress and cell death (Fig. 10). Consequently, our data suggest that the frequently reported thiol-induced redox cycling of copper(II) complexes occurs predominantly in the extracellular space. The proposed reaction scheme is shown in Fig. 12. Upon copper(II) reduction by NAC, the copper(I) complex formed is able to extracellularly generate H2O2 by O2·− dismutation (accompanied by reoxidation of the copper complex). H2O2 is then able to enter the cell by diffusion, where ROS are generated, e.g., by Fenton reactions. Additionally, the copper(I) complexes are able to produce O2·− in the presence of oxygen. This, on the one hand, leads again to reoxidation of the copper complex and, on the other hand, to generation of O2·− essential for H2O2 production. After reoxidation to copper(II), again reduction by NAC can occur, leading to redox cycling of the copper center. Additionally, the distinct SOD-like activity of Cu-FTSC and Cu-BPYTA suggests that these compounds are able to be reduced by O2·−, which is in contrast to Cu-Triapine and Cu-APTSC. These differences in the redox activity might be responsible for the observed intracellular ROS generation by high concentrations of Cu-FTSC and Cu-BPYTA in single treatments (see Fig. 5). Owing to the lower redox potential, Cu-Triapine and Cu-APTSC are probably not reduced by O2·− under physiological conditions and need stronger reductants for H2O2 generation. On the basis of our experiments on living cells, we conclude that the intracellular milieu present under cell culture is not sufficient to allow significant redox cycling and ROS generation, especially in the case of Cu-Triapine and Cu-APTSC. Consequently, the assumed mode of action for copper compounds based on thiol-induced intracellular ROS generation urgently needs further investigations and probably reformulation of the proposed mechanisms. Although the similarity in IC50 values between the complex and the metal-free ligand indicates that both might share the same mode of action, preliminary data (e.g., in the case in Cu-BPYTA [11]) indicate that not ROS, but other redox-dependent mechanisms underlie the anticancer activity of the copper complexes, which is distinctly different from that of the metal-free ligands. The investigation of this issue is the subject of ongoing studies. In general, on the basis of the strong differences in the cellular redox balance between cancer and nonmalignant cells, the redox activity of copper complexes opens the opportunity for targeting cancer cells. Additionally, this might be further enhanced by targeting strategies (e.g., via albumin binding) to increase the selective accumulation of the compounds in the malignant tissue. Furthermore, the redox activity of the copper complexes can be tuned to selectively target the hypoxic tumor environment, as already shown for copper(II)-containing radiopharmaceuticals.
Fig. 12.

Proposed extracellular redox reactions underlying the Cu-TSC-induced ROS generation
Supplementary Material
Acknowledgments
We are indebted to Vera Bachinger for the skilful handling of cell cultures, Irene Herbacek for fluorescence-activated cell sorting analysis, and Christian Balcarek for competent technical assistance. We also thank Aliona Luganschi for the measurement of mass spectra, and Alexander Roller as well as Vladimir Arion for the collection/refinement of the X-ray crystal data. This work was supported by Austrian Science Fond (FWF) grants L212-B11 and P22072-B11 (to W.B.).
Abbreviations
- AA
Ascorbic acid
- APTSC
3-Aminopyridine-2-carboxaldehyde 4N,4N-dimethylthiosemicarbazone
- BPYTA
2,2′-Bipyridyl-6-carbothioamide
- CCDC
Cambridge Crystallographic Data Centre
- DCF-DA
2′,7′-Dichlorofluorescein diacetate
- DMF
Dimethylformamide
- DMSO
Dimethyl sulfoxide
- ESI-MS
Electrospray ionization mass spectrometry
- FTSC
2-Formylpyridine thiosemicarbazone
- GSH
Glutathione
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAC
N-Acetylcysteine
- NBT
Nitroblue tetrazolium
- NHE
Normal hydrogen electrode
- PBS
Phosphate-buffered saline
- PI
Propidium iodide
- ROS
Reactive oxygen species
- RR
Ribonucleotide reductase
- SOD
Superoxide dismutase
- TSC
α-N-heterocyclic thiosemicarbazone
Footnotes
Electronic supplementary material The online version of this article (doi: 10.1007/s00775-011-0864-x) contains supplementary material, which is available to authorized users.
Contributor Information
Christian R. Kowol, Institute of Inorganic Chemistry, University of Vienna, Waehringer Str. 42, 1090 Vienna, Austria; Comprehensive Cancer Center, Medical University Vienna, Borschkegasse 8a, 1090 Vienna, Austria; Research Platform “Translational Cancer Therapy Research“, University of Vienna and Medical University of Vienna, Vienna, Austria
Petra Heffeter, Comprehensive Cancer Center, Medical University Vienna, Borschkegasse 8a, 1090 Vienna, Austria; Research Platform “Translational Cancer Therapy Research“, University of Vienna and Medical University of Vienna, Vienna, Austria; Department of Medicine I, Institute of Cancer Research, Medical University of Vienna, Borschkegasse 8a, 1090 Vienna, Austria.
Walter Miklos, Department of Medicine I, Institute of Cancer Research, Medical University of Vienna, Borschkegasse 8a, 1090 Vienna, Austria.
Lars Gille, Molecular Pharmacology and Toxicology Unit, Department of Biomedical Sciences, University of Veterinary Medicine Vienna, Veterinaerplatz 1, 1210 Vienna, Austria.
Robert Trondl, Institute of Inorganic Chemistry, University of Vienna, Waehringer Str. 42, 1090 Vienna, Austria.
Loredana Cappellacci, Medicinal Chemistry Unit, School of Pharmacy, University of Camerino, Via S. Agostino 1, 62032 Camerino, Italy.
Walter Berger, Comprehensive Cancer Center, Medical University Vienna, Borschkegasse 8a, 1090 Vienna, Austria; Research Platform “Translational Cancer Therapy Research“, University of Vienna and Medical University of Vienna, Vienna, Austria; Department of Medicine I, Institute of Cancer Research, Medical University of Vienna, Borschkegasse 8a, 1090 Vienna, Austria.
Bernhard K. Keppler, Institute of Inorganic Chemistry, University of Vienna, Waehringer Str. 42, 1090 Vienna, Austria; Comprehensive Cancer Center, Medical University Vienna, Borschkegasse 8a, 1090 Vienna, Austria; Research Platform “Translational Cancer Therapy Research“, University of Vienna and Medical University of Vienna, Vienna, Austria
References
- 1.Gupte A, Mumper RJ. Cancer Treat Rev. 2009;35:32–46. doi: 10.1016/j.ctrv.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Trachootham D, Alexandre J, Huang P. Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 3.Denny WA. Future Oncol. 2010;6:419–428. doi: 10.2217/fon.10.1. doi:10.2217/fon.10.1. [DOI] [PubMed] [Google Scholar]
- 4.Hartinger CG, Zorbas-Seifried S, Jakupec MA, Kynast B, Zorbas H, Keppler BK. J Inorg Biochem. 2006;100:891–904. doi: 10.1016/j.jinorgbio.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Hall MD, Hambley TW. Coord Chem Rev. 2002;232:49–67. [Google Scholar]
- 6.Wang T, Guo Z. Curr Med Chem. 2006;13:525–537. doi: 10.2174/092986706776055742. [DOI] [PubMed] [Google Scholar]
- 7.Tisato F, Marzano C, Porchia M, Pellei M, Santini C. Med Res Rev. 2010;30:708–749. doi: 10.1002/med.20174. doi:10.1002/med.20174. [DOI] [PubMed] [Google Scholar]
- 8.Bonnitcha PD, Vavere AL, Lewis JS, Dilworth JR. J Med Chem. 2008;51:2985–2991. doi: 10.1021/jm800031x. doi:10.1021/jm800031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byrnes RW, Mohan M, Antholine WE, Xu RX, Petering DH. Biochemistry. 1990;29:7046–7053. doi: 10.1021/bi00482a014. [DOI] [PubMed] [Google Scholar]
- 10.Jansson PJ, Sharpe PC, Bernhardt PV, Richardson DR. J Med Chem. 2010;53:5759–5769. doi: 10.1021/jm100561b. doi:10.1021/jm100561b. [DOI] [PubMed] [Google Scholar]
- 11.Nocentini G, Barzi A. Biochem Pharmacol. 1996;52:65–71. doi: 10.1016/0006-2952(96)00139-6. [DOI] [PubMed] [Google Scholar]
- 12.Nocentini G, Barzi A. Gen Pharmacol. 1997;29:701–706. doi: 10.1016/s0306-3623(97)00011-6. [DOI] [PubMed] [Google Scholar]
- 13.Sigman DS, Mazumder A, Perrin DM. Chem Rev. 1993;93:2295–2316. [Google Scholar]
- 14.Jungwirth U, Kowol CR, Keppler BK, Hartinger C, Berger W, Heffeter P. Antioxid Redox Signal. 2011;15:1085–1127. doi: 10.1089/ars.2010.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saryan LA, Ankel E, Krishnamurti C, Petering DH, Elford H. J Med Chem. 1979;22:1218–1221. doi: 10.1021/jm00196a013. [DOI] [PubMed] [Google Scholar]
- 16.Sammes PG, Yahioglu G. Chem Soc Rev. 1994;23:327–334. [Google Scholar]
- 17.Yu Y, Kalinowski DS, Kovacevic Z, Siafakas AR, Jansson PJ, Stefani C, Lovejoy DB, Sharpe PC, Bernhardt PV, Richardson DR. J Med Chem. 2009;52:5271–5294. doi: 10.1021/jm900552r. [DOI] [PubMed] [Google Scholar]
- 18.Kowol CR, Trondl R, Heffeter P, Arion VB, Jakupec MA, Roller A, Galanski M, Berger W, Keppler BK. J Med Chem. 2009;52:5032–5043. doi: 10.1021/jm900528d. [DOI] [PubMed] [Google Scholar]
- 19.Nocentini G, Federici F, Franchetti P, Barzi A. Cancer Res. 1993;53:19–26. [PubMed] [Google Scholar]
- 20.Shao J, Zhou B, Di Bilio AJ, Zhu L, Wang T, Qi C, Shih J, Yen Y. Mol Cancer Ther. 2006;5:586–592. doi: 10.1158/1535-7163.MCT-05-0384. [DOI] [PubMed] [Google Scholar]
- 21.Giles FJ, Fracasso PM, Kantarjian HM, Cortes JE, Brown RA, Verstovsek S, Alvarado Y, Thomas DA, Faderl S, Garcia-Manero G, Wright LP, Samson T, Cahill A, Lambert P, Plunkett W, Sznol M, DiPersio JF, Gandhi V. Leuk Res. 2003;27:1077–1083. doi: 10.1016/s0145-2126(03)00118-8. [DOI] [PubMed] [Google Scholar]
- 22.Karp JE, Giles FJ, Gojo I, Morris L, Greer J, Johnson B, Thein M, Sznol M, Low J. Leuk Res. 2008;32:71–77. doi: 10.1016/j.leukres.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nutting CM, van Herpen CM, Miah AB, Bhide SA, Machiels JP, Buter J, Kelly C, de Raucourt D, Harrington KJ. Ann Oncol. 2009;20:1275–1279. doi: 10.1093/annonc/mdn775. doi:10.1093/annonc/mdn775. [DOI] [PubMed] [Google Scholar]
- 24.Kunos CA, Waggoner S, von Gruenigen V, Eldermire E, Pink J, Dowlati A, Kinsella TJ. Clin Cancer Res. 2010;16:1298–1306. doi: 10.1158/1078-0432.CCR-09-2469. doi:10.1158/1078-0432.CCR-09-2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Narasimhan J, Antholine WE, Chitambar CR, Petering DH. Arch Biochem Biophys. 1991;289:393–398. doi: 10.1016/0003-9861(91)90429-m. [DOI] [PubMed] [Google Scholar]
- 26.Finch RA, Liu MC, Cory AH, Cory JG, Sartorelli AC. Adv Enzyme Regul. 1999;39:3–12. doi: 10.1016/s0065-2571(98)00017-x. [DOI] [PubMed] [Google Scholar]
- 27.Chikira M, Tomizawa Y, Fukita D, Sugizaki T, Sugawara N, Yamazaki T, Sasano A, Shindo H, Palaniandavar M, Antholine WE. J Inorg Biochem. 2002;89:163–173. doi: 10.1016/s0162-0134(02)00378-1. [DOI] [PubMed] [Google Scholar]
- 28.Antholine WE, Taketa F. J Inorg Biochem. 1984;20:69–78. doi: 10.1016/0162-0134(84)80007-0. [DOI] [PubMed] [Google Scholar]
- 29.Sigman DS, Graham DR, D’Aurora V, Stern AM. J Biol Chem. 1979;254:12269–12272. [PubMed] [Google Scholar]
- 30.Saryan LA, Mailer K, Krishnamurti C, Antholine W, Petering DH. Biochem Pharmacol. 1981;30:1595–1604. doi: 10.1016/0006-2952(81)90386-5. [DOI] [PubMed] [Google Scholar]
- 31.Byrnes RW, Antholine WE, Petering DH. Free Radic Biol Med. 1992;13:469–478. doi: 10.1016/0891-5849(92)90141-3. [DOI] [PubMed] [Google Scholar]
- 32.McCann M, Geraghty M, Devereux M, O’Shea D, Mason J, O’Sullivan L. Met Based Drugs. 2000;7:185–193. doi: 10.1155/MBD.2000.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsang SY, Tam SC, Bremner I, Burkitt MJ. Biochem J. 1996;317:13–16. doi: 10.1042/bj3170013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruiz R, Garcia B, Garcia-Tojal J, Busto N, Ibeas S, Leal JM, Martins C, Gaspar J, Borras J, Gil-Garcia R, Gonzalez-Alvarez M. J Biol Inorg Chem. 2010;15:515–532. doi: 10.1007/s00775-009-0620-7. [DOI] [PubMed] [Google Scholar]
- 35.Aguirre MdC, Borras J, Castineiras A, Garcia-Monteagudo JM, Garcia-Santos I, Niclos J, West DX. Eur J Inorg Chem. 2006;6:1231–1244. [Google Scholar]
- 36.Garcia-Tojal J, Garcia-Jaca J, Cortes R, Rojo T, Urtiaga MK, Arriortua MI. Inorg Chim Acta. 1996;249:25–32. [Google Scholar]
- 37.Popović-Bijelić A, Kowol CR, Lind MES, Luo J, Himo F, Enyedy EA, Arion VB, Gräslund A. J Inorg Biochem. 2011;105:1422–1431. doi: 10.1016/j.jinorgbio.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruker-Nonius AXS. RLATT. Bruker-Nonius AXS; Madison: 2008. [Google Scholar]
- 39.Bruker-Nonius AXS. SAINT-Plus. Bruker-Nonius AXS; Madison: 2004. [Google Scholar]
- 40.Bruker-Nonius AXS. TWINABS. Bruker-Nonius AXS; Madison: 2008. [Google Scholar]
- 41.Sheldrick GM. Acta Crystallogr Sect A Found Crystallogr. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 42.Johnson GK. Oak Ridge National Laboratory. Oak Ridge: 1976. [Google Scholar]
- 43.Stoll S, Schweiger A. J Magn Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 44.Grusch M, Fritzer-Szekeres M, Fuhrmann G, Rosenberger G, Luxbacher C, Elford HL, Smid K, Peters GJ, Szekeres T, Krupitza G. Exp Hematol. 2001;29:623–632. doi: 10.1016/s0301-472x(01)00624-5. [DOI] [PubMed] [Google Scholar]
- 45.Gomes A, Fernandes E, Lima JL. J Biochem Biophys Methods. 2005;65:45–80. doi: 10.1016/j.jbbm.2005.10.003. doi: 10.1016/j.jbbm.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 46.Ghosh K, Kumar P, Tyagi N, Singh UP, Aggarwal V, Baratto MC. Eur J Med Chem. 2010;45:3770–3779. doi: 10.1016/j.ejmech.2010.05.026. doi:10.1016/j.ejmech. 2010.05.026. [DOI] [PubMed] [Google Scholar]
- 47.Enyedy EA, Nagy NV, Zsigo E, Kowol CR, Arion VB, Keppler BK, Kiss T. Eur J Inorg Chem. 2010;11:1717–1728. [Google Scholar]
- 48.Hathaway BJ, Billing DE. Coord Chem Rev. 1970;5:143–207. [Google Scholar]
- 49.Fasman GD. CRC handbook of biochemistry and molecular biology. CRC Press; Cleveland: 1976. [Google Scholar]
- 50.Noszal B, Visky D, Kraszni M. J Med Chem. 2000;43:2176–2182. doi: 10.1021/jm9909600. [DOI] [PubMed] [Google Scholar]
- 51.Schafer FQ, Buettner GR. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 52.Ainscough EW, Brodie AM, Denny WA, Finlay GJ, Ranford JD. J Inorg Biochem. 1998;70:175–185. doi: 10.1016/s0162-0134(98)10011-9. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Tojal J, Garcia-Orad A, Diaz AA, Serra JL, Urtiaga MK, Arriortua MI, Rojo T. J Inorg Biochem. 2001;84:271–278. doi: 10.1016/s0162-0134(01)00184-2. [DOI] [PubMed] [Google Scholar]
- 54.Jansen BA, Brouwer J, Reedijk J. J Inorg Biochem. 2002;89:197–202. doi: 10.1016/s0162-0134(02)00381-1. [DOI] [PubMed] [Google Scholar]
- 55.Diaz AM, Villalonga R, Cao R. J Coord Chem. 2009;62:100–107. [Google Scholar]
- 56.Bijloo GJ, van der Goot H, Bast A, Timmerman H. J Inorg Biochem. 1990;40:237–244. doi: 10.1016/0162-0134(90)80057-5. [DOI] [PubMed] [Google Scholar]
- 57.Pierre J-L, Chautemps P, Refaif S, Beguin C, El Marzouki A, Serratrice G, Saint-Aman E, Rey P. J Am Chem Soc. 1995;117:1965–1973. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.