

Abstract
(N)-Methanocarba adenosine 5′-methyluronamides containing known A3 AR (adenosine receptor)-enhancing modifications, i.e. 2-(arylethynyl)adenine and N6-methyl or N6-(3-substituted-benzyl), were nanomolar full agonists of human (h) A3AR and highly selective (Ki ~0.6 nM, N6-methyl 2-(halophenylethynyl) analogues 13, 14). Combined 2-arylethynyl-N6-3-chlorobenzyl substitutions preserved A3AR affinity/selectivity in the (N)-methanocarba series (e.g. 3,4-difluoro full agonist MRS5698 31, Ki 3 nM, human and mouse A3) better than for ribosides. Polyaromatic 2-ethynyl N6-3-chlorobenzyl analogues, such as potent linearly extended 2-p-biphenylethynyl MRS5679 34 (Ki hA3 3.1 nM; A1, A2A: inactive) and fluorescent 1-pyrene adduct MRS5704 35 (Ki hA3 68.3 nM) were conformationally rigid; receptor docking identified a large, mainly hydrophobic binding region. The vicinity of receptor-bound C2 groups was probed by homology modeling based on recent X-ray structure of an agonist-bound A2AAR, with a predicted helical rearrangement requiring an agonist-specific outward displacement of TM2 resembling opsin. Thus, X-ray structure of related A2AAR is useful in guiding design of new A3AR agonists.
Keywords: G protein-coupled receptor, purines, molecular modeling, structure activity relationship, radioligand binding, adenylate cyclase
Introduction
The structure activity relationship (SAR) of nucleoside agonists at the A3 adenosine receptor (AR), a G protein-coupled receptor (GPCR) of the rhodopsin-like family A, has been extensively explored.1 Several A3AR-selective agonists are currently in advanced clinical trials for the treatment of hepatocellular carcinoma, autoimmune inflammatory diseases,2–4 such as rheumatoid arthritis, psoriasis, and dry eye disease, and other conditions.5–8 Other potential applications of such agonists could be osteoarthritis, Crohn’s disease, ischemia, and other inflammatory disorders. As applied to cancer models, there is evidence that multiple modes of benefit from A3AR agonists are obtained: myeloprotection and reduction of neuropathic pain, in addition to the antiproliferative effects on tumors in vivo. A3AR-selective antagonists are also of interest for therapeutic applications in asthma, glaucoma, septic shock and other conditions.9–12 Thus, this subtype of AR (others are A1AR, A2AAR, and A2BAR) has provided numerous opportunities for translation to therapeutics.
In order to achieve selectivity of adenosine derivatives for the A3AR, modifications are typically introduced on the N6 position (often m-substituted benzyl groups) and on the ribose moiety (often 5′-N-methyluronamide), such as occur on prototypical agonists 1 and 2 (Chart 1).13 Replacement of the conformationally flexible ribose tetrahydrofuryl group with a rigid bicyclo[3.1.0]hexane (methanocarba) ring system in an isomeric form that enforces a North (N) envelope conformation increases both the binding affinity and selectivity of adenosine derivatives as A3AR agonists.14–16 For example, analogues that combined N6-(3-halobenzyl) and (N)-methanocarba modifications, such as 3 and 4, display anti-inflammatory activity, protection in a model of lung injury, and protection against chemotherapy-induced neuropathic pain.17–19 Cristalli and coworkers have explored the SAR leading to enhancement of A2AAR and A3AR affinity by alkyn-2-yl groups, such as hexyn-2-yl, at the adenine C2 position of adenine 9-ribosides.20 The enhancement of human (h) A3AR affinity by such groups was also observed in the (N)-methanocarba series.21 Because ligand affinity at the A3AR is often dependent on species, it was particularly important to establish the effect in species other than human. The combination of N6-(3-halobenzyl), 2-alkyn-2-yl, and 5′-N-methyluronamide modifications was found to be particularly well suited for application to agonists displaying A3AR selectivity across species.
Chart 1.

Derivatives of adenosine as A3AR-selective agonists in the ribose (1, 2) and (N)-methanocarba (3, 4) series.
There is a precedent for the lack of compatibility in AR recognition of multiple modifications of nucleoside derivatives at the N6 (i.e. 3-halobenzyl) and C2 positions (i.e. aryl or large hydrophobic groups), each of which may promote affinity at A3AR and other subtypes.13,22 There also is indication that N6-3-(substituted benzyl) and 2-arylethynyl (introduced by Cristalli and colleagues23) modifications are not additive in their effect on A3AR selectivity of 9-ribosides.24 The present study establishes that these two substitutions are fully additive and conducive to high selectivity when applied to (N)-methanocarba adenosine derivatives. We have also used molecular modeling based on a recent X-ray structure of an A2AAR complex with a 5′,2,N6 trisubstituted agonist25.26 and with insights from other GPCRs26 to probe the vicinity of the 2-arylethynyl groups when docked to the A3AR. Thus, the crystallographic structure of one AR subtype is used to guide the design of new analogues at another AR subtype following necessary customization for this ligand set.
Results
Chemical synthesis
We have explored 2-arylethynyl substitution of the adenine ring of previously reported (N)-methanocarba 5′-N-methyluronamide nucleoside derivatives that act as selective A3AR agonists.14,15,21 Two series, depending on the N6 substitution (N6-methyl: 8 – 26 and N6-3-chlorobenzyl: 27 – 36), that are closely patterned on known potent agonists are included (Scheme 1). N6-Methyladenosines are typically significantly more potent at the hA3AR than at rat (r) A3AR, while N6-(3-halobenzyl)adenosines tend to display greater species-independent selectivity.
Scheme 1.

Synthesis of (N)-methanocarba nucleoside analogues. R1 = Me or 3-Cl-benzyl. Reagents: (i) MeNH2 or 3-Cl-benzylamine, Et3N, MeOH, rt; (ii) 40% MeNH2, MeOH, rt; (iii) HC≡CAr, Pd(PPh3)2Cl2, CuI, Et3N, DMF, rt; (iv) 10% TFA, MeOH, 70°C; (v) 1-ethynylpyrene or 4-ethynylpyrene 51, Pd(PPh3)2Cl2, CuI, Et3N, DMF, rt; (vi) HCl, 1 N, dioxane, 60°C.
The synthetic route used to prepare (N)-methanocarba 5′-N-methyluronamido derivatives containing a 2-arylethynyl group involved a key Sonogashira reaction27 at a 2-iodoadenine moiety of intermediate 43 or 44 (Scheme 1). L-Ribose 38 was converted as previously reported into the 2′,3′-protected intermediate 39 containing a 5-ethyl ester, which was then subjected to a Mitsunobu condensation with 2-iodo-6-chloropurine to give 40.21,28 A N6-methyl or N6-(3-chlorobenzyl) group was added by nucleophilic substitution of 6-chloro at room temperature to provide intermediates 41 and 42, respectively, followed by aminolysis of the ester at room temperature leading to 5′-N-methyluronamides 43 and 44. A Sonogashira reaction was then carried out with a variety of commercially available arylacetylenes to give protected nucleosides 45 – 46 (N6-methyl) and 47a - f (N6-3-chlorobenzyl). Finally, acid hydrolysis of the isopropylidene protecting group provided N6-methyl 9 – 26 and N6-3-chlorobenzyl 27 – 36 nucleosides. The associated synthesis of a polyaromatic ethynyl intermediate 51 is shown in Scheme 2.
Scheme 2.

Synthesis of an intermediate arylalkynyl derivative. (i) HC≡C-TMS, Pd(PPh3)2Cl2, CuI, Et3N, DMF, rt; (ii) TBAF, THF.
Pharmacological activity
Radioligand binding assays at three hAR subtypes were carried out using standard 3H (52, 53) and 125I-labeled (54) nucleosides (Table 1).29–31 The membrane preparations were obtained from Chinese hamster ovary (CHO) cells (A1 and A3) or human embryonic kidney (HEK293) cells (A2A) stably expressing a hAR subtype.28,29,32,33 A3AR binding curves generally showed a Hill coefficient of ~1. The nucleoside analogues were not screened for activity at the hA2BAR, because other (N)-methanocarba nucleosides were previously noted to be very weak or inactive at that subtype.15 For example, in cyclic AMP assays, agonist 3 displayed 30,000-fold selectivity for the hA3AR in comparison to the hA2BAR.17 Some previously reported 2-chloro and 2-alkynyl (N)-methanocarba agonists (3 – 8) were used for comparison in the biological assays.20 For exploring species differences in AR ligand recognition, binding assays were also performed on selected nucleoside derivatives at three mouse (m) ARs (Table 2) expressed in HEK293 cells.21,33b
Table 1.
Binding affinity of a series of (N)-methanocarba adenosine derivatives at three subtypes of hARs and the functional efficacy at the hA3AR.
 | ||||||
|---|---|---|---|---|---|---|
| Compd | Structure | Affinity (Ki, nM) or % inhibitiona | %Efficacyb | |||
| R1 or R3 | R2 | hA1 | hA2A | hA3 | hA3 | |
| 3c | Cl | 3-Cl-Bn | 260±60 | 2300±100 | 0.29±0.04 | 103±7 |
| 4c,d | Cl | 3-I-Bn | 136±22 | 784±97 | 1.5±0.2 | 100 |
| 5c | H | 3-I-Bn | 700±270 | 6200±100 | 2.4±0.5 | 100 |
| 6d | C≡CH | 3-Cl-Bn | 174±23 | (48%) | 1.30±0.38 | ND |
| 7d | C≡C(CH2)2CH3 | 3-Cl-Bn | 1040±83 | (80%) | 0.82±0.20 | ND |
| 8c,d | Cl | CH3 | 2100±1700 | (6%) | 2.2±0.6 | ND |
| 9 | 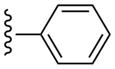 |
CH3 | (13%±6%) | (14%±7%) | 0.85±0.22 | 89.3±7.7 |
| 10 | 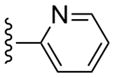 |
CH3 | (11%±4%) | (13%±4%) | 1.01±0.36 | 86.8±9.2 |
| 11 | 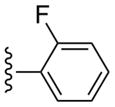 |
CH3 | (21%±4%) | (17%±2%) | 0.97±0.38 | 97.7±9.1 |
| 12 | 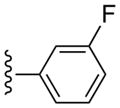 |
CH3 | (12%±2%) | (10%±5%) | 0.97±0.24 | 95.8±6.7 |
| 13 | 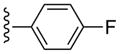 |
CH3 | (21%±11%) | (19%±3%) | 0.53±0.09 | 80.3±5.8 |
| 14 |  |
CH3 | (27%±7%) | (30%±5%) | 0.58±0.04 | 84.2±6.2 |
| 15 | 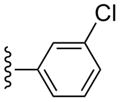 |
CH3 | (10%±2%) | 1270±300 | 1.60±0.60 | 90.9±1.9 |
| 16 | 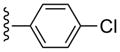 |
CH3 | (14%±1%) | (30%±1%) | 1.22±0.31 | 97.4±9.1 |
| 17 | 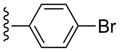 |
CH3 | (13%±7%) | (26%±1%) | 0.91±0.06 | 97.5±12.3 |
| 18 | 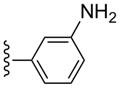 |
CH3 | (10%±5%) | (19%±14%) | 1.07±0.14 | 109±4.1 |
| 19 | 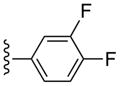 |
CH3 | (6%±3%) | (6%±6%) | 1.65±0.08 | 108±1.9 |
| 20 | 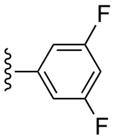 |
CH3 | (8%±3%) | (47%±4%) | 1.66±0.36 | 99.6±3.3 |
| 21 | CH3 | (13%±3%) | (38%±5%) | 3.78±1.16 | 110±4.9 | |
| 22 | CH3 | (23%±5%) | (7%±5%) | 10.1±1.9 | 78.4±6.5 | |
| 23 | CH3 | (15%±7%) | 5300±600 | 2.57±0.78 | 87.4±9.9 | |
| 24 | 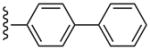 |
CH3 | (21%±6%) | (29%±8%) | 3.10±1.26 | 110±2.9 |
| 25 | 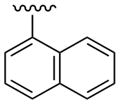 |
CH3 | (25%±1%) | (34%±8%) | 1.67±0.18 | 94.6±4.4 |
| 26 | 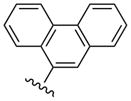 |
CH3 | (15%±5%) | (52%±1%) | 3.48±1.36 | 108±3.5 |
| 27 | 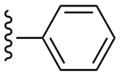 |
3-Cl-Bn | (20%±3%) | (27%±3%) | 1.34±0.30 | 101±5.9 |
| 28 | 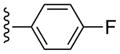 |
3-Cl-Bn | (20%±6%) | (42%±2%) | 2.16±0.34 | 102±1.4 |
| 29 | 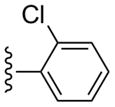 |
3-Cl-Bn | (19%±2%) | (52%±12%) | 1.92±0.57 | 103±1.5 |
| 30 | 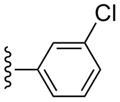 |
3-Cl-Bn | (4%±4%) | 1740±590 | 4.45±1.39 | 91.5±11.4 |
| 31 | 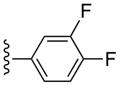 |
3-Cl-Bn | (6%±4%) | (41%±10%) | 3.49±1.84 | 95.7±6.4 |
| 32 | 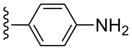 |
3-Cl-Bn | 1520±300 | (44%±4%) | 2.27±0.70 | 76.6±13.1 |
| 33 | 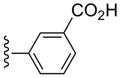 |
3-Cl-Bn | (6%±5%) | (38%±5%) | 6.75±2.78 | 86.7±5.4 |
| 34 | 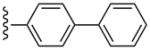 |
3-Cl-Bn | (2%±2%) | (0%±0%) | 3.06±1.35 | 89.0±4.5 |
| 35 | 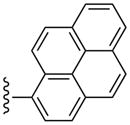 |
3-Cl-Bn | (8%±2%) | 3110±530 | 68.3±12.5 | 77.8±11.6 |
| 36 | 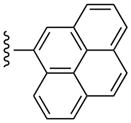 |
3-Cl-Bn | (11±5%) | (4±3%) | 660±170 | 97.1±3.3 |
All experiments were done on CHO or HEK293 (A2A only) cells stably expressing one of three subtypes of the four hARs. The binding affinity for A1, A2A and A3ARs was expressed as Ki values (n = 3–5) and was determined by using agonist radioligands ([3H]N6-R-phenylisopropyladenosine 52 (R-PIA), [3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine 53 (CGS21680), or [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyl-uronamide 54 (I-AB-MECA), respectively), unless noted.29–31 A percent in parentheses refers to inhibition of radioligand binding at 10 μM (n = 3).
Unless noted, the efficacy at the hA3AR was determined by inhibition of forskolin-stimulated cAMP production in AR-transfected CHO cells.32–34 At a concentration of 10 μM, in comparison to the maximal effect of 5′-N-ethylcarboxamidoadenosine 48 (=100%) at 10 μM. Data are expressed as mean±standard error (n = 3).
Values from Melman et al.21
ND, not determined.
Table 2.
Binding affinity of a series of (N)-methanocarba adenosine derivatives at three subtypes of mARs.
| Compd | Affinity (Ki, nM) or % inhibitiona | ||
|---|---|---|---|
| mA1 | mA2A | mA3 | |
| 3b | 15.3±5.8 | 10,400±1,700 | 1.49±0.46 |
| 4b | 7.32±1.5 | 5,350±860 | 0.80±0.14 |
| 6b | 45.6±7.9 | (41%)i | 0.85±0.08 |
| 7b | 1390±430 | (42%)i | 6.06±1.21 |
| 8b | 55.3±6.0 | 20,400±3,200 | 49.0±3.9 |
| 13 | (29±2%) | (0%) | 37.7±1.1 |
| 14 | (55±5%) | (2±1%) | 37.2±2.0 |
| 27 | (50±5%) | (2±1%) | 1.23±0.14 |
| 28 | (65±3%) | (7±2%) | 2.38±0.04 |
| 29 | (51±12%) | (19±3%) | 2.64±0.22 |
| 30 | (35±3%) | (55%) | 2.39±0.38 |
| 31 | (14±3%) | (27±2%) | 3.08±0.23 |
| 32 | 261±19 | (5±2%) | 0.82±0.06 |
| 33 | (18±3%) | (22%) | 3.66±0.25 |
| 34 | (41±6%) | (6±1%) | 10.8±0.93 |
| 35 | (8±2%) | (64%) | 47.6±4.6 |
Competition radioligand binding assays using [125I]54 (A1 and A3ARs) and [3H]53 (A2AAR) were conducted with membranes prepared from HEK293 cells expressing recombinant mA1, A2A, or A3ARs. The data (n = 3–4) are expressed as Ki values. A percent in parentheses refers to inhibition of radioligand binding at 10 μM.
Values from Melman et al.21
The simplest N6-methyl 2-phenylethynyl analogue 9 displayed a subnanomolar Ki value at the hA3AR and was nearly inactive at the hA1AR and hA2AAR, with <20% inhibition of binding at 10 μM. Therefore, the degree of A3AR selectivity of 9 was estimated to be >10,000-fold. The effects of phenyl modification of the C2-arylethynyl group at the C2 position were explored initially in the N6-methyl 5′-N-methyluronamide series. These derivatives displayed great freedom of substitution of the arylethynyl moiety, i.e. aza substitution of a CH of phenyl (10), monohalo (11 – 17) and dihalo (19, 20) or other (18, 21 – 23) substituent groups on the phenyl ring, and the presence of additional aryl rings (24 – 26). The 3,4-difluorophenyl analogue 19 was particularly A3AR-selective with insignificant inhibition at A1 and A2AARs. Planar polyaromatic groups, such as α-naphthyl 25 and the larger phenanthrene 26, in the N6-methyl series did not interfere with the binding to the hA3AR. The presence of a branched p-t-Bu group in 22 reduced A3AR binding affinity by 12-fold compared to the H analogue 9. The most potent A3AR ligands in the N6-methyl series were halo-substituted compounds 13 and 14 with Ki values of 0.5 – 0.6 nM. A p-acetyl substitution in 23 was lower in hA3AR affinity and higher in hA2AAR affinity than most other ring substitutions, unlike in the riboside series of Cristalli and coworkers,23a in which a 2(-p-acetylphenylethynyl) group favored A3AR affinity and selectivity.
The variability of binding affinity upon substitution of the arylethynyl moiety was greater at the hA2AAR than at the hA1AR. For example, the hA2AAR affinity increased substantially to a Ki value of 1.3 μM upon replacement of 3-F 12 with 3-Cl in 15. In contrast, the same change slightly decreased hA3AR affinity. At the hA1AR, only 10 to 30% of binding inhibition was typically seen at 10 μM for a variety of substitutions.
The SAR was extended by the analysis of eleven analogues prepared in the N6-(3-chlorobenzyl) series (27 – 36). The effect in 27 of adding a phenyl group to the ethynyl substituent of the simpler acetylene derivative 6 was complete retention of the affinity at hA3AR and a reduction of the hA1AR affinity from Ki = 174 nM to >10 μM, thus providing high selectivity. Furthermore, with only marginal binding of 27 at the hA2AAR, the hA3AR selectivity was roughly 10,000-fold in comparison to both hA1AR and hA2AAR. For comparison, by adding a n-propyl chain in 7 rather than a phenyl ring, the hA1AR affinity was reduced only 6-fold in comparison to 6, and the hA2AAR affinity appeared to be increased. Thus, a flat aryl ring rather than a flexible alkyl chain provides the appropriate geometry for hA3AR selectivity. A potent A3AR ligand 34 in the N6-(3-chlorobenzyl) series containing a rigid, linearly extended 2-p-biphenylethynyl group served as a model ligand for receptor docking due to its steric and conformational constraints. It displayed a Ki value at the hA3AR of 3.06 nM and exceptionally high selectivity in comparison to the hA1 and hA2AARs (no inhibition observed). The hA3AR affinities of 34 and the corresponding N6-methyl analogue 24 were equal. Thus, the combination of 2-arylethynyl, 5′-N-methyluronamide, and N6-3-chlorobenzyl substitution preserved the hA3AR selectivity of (N)-methanocarba nucleosides, even for large 2-arylethynyl moieties.
Species differences in the binding affinity of the nucleoside derivatives were explored. Nanomolar A3AR affinity was maintained in a murine species only for the N6-(substituted benzyl) series (Figure 1). The N6-methyl-2-arylethynyl derivatives 13 and 14 were determined to be ~70-fold weaker at the mA3AR than at the hA3AR, consistent with the previously reported reduced affinity at mA3AR of N6-methyl-2-Cl derivative 8.21 The mA3AR/mA1AR selectivity in the N6-methyl series was enhanced with the elongation and rigidification of the C2 substituent. However, with N6-3-chlorobenzyl substitution, elongation at C2 did not significantly reduce affinity at the mA3AR compared to hA3AR (except for a 3-fold reduction for the elongated biaryl derivative 34). In the N6-3-chlorobenzyl series, particularly high selectivity compared to mA1AR and mA2AAR was generally present. There was greater variability in the degree of binding inhibition at mA1AR than at mA2AAR. Elongation of a 2-ethynyl group in 6 with a straight alkyl chain in 7 reduced mA3AR affinity by 7-fold, but elongation with a phenyl ring in 27 only slightly reduced mA3AR affinity. The most potent derivative at the mA3AR was the p-aminophenyl analogue 32 with a Ki value of 0.82 nM. Curiously, although this derivative remained selective, its A1AR affinity was enhanced over other members of the series at both in mouse (Ki 261 nM) and human (Ki 1520 nM).
Figure 1.

Inhibition of binding of the radioligand [125I]54 (0.3 nM) at the mA3AR by compounds 13, 31, 32 and 34. Ki values are found in Table 2.
Functional data were determined in an assay consisting of hA3AR-induced inhibition of the production of adenosine 3′,5′-cyclic phosphate (cAMP) in membranes of CHO cells expressing the hA3AR (Table 1).34 Inhibition by 10 μM NECA (48, 5′-N-ethylcarboxamidoadenosine) was set at 100% relative efficacy. The novel (N)-methanocarba 5′-N-methyluronamide derivatives at 10 μM were predominantly full agonists at the A3AR, with a few analogues showing relative efficacy of 80% or less (p-fluoro 13, t-butyl-phenyl 22, and 1-pyrene 35 derivatives). A full concentration response curve in hA3AR-mediated inhibition of adenylate cyclase for 3,4-difluoro analogue 31 provided an EC50 value of 1.2±0.7 nM (Figure 2), i.e. slightly more potent than its binding affinity.
Figure 2.
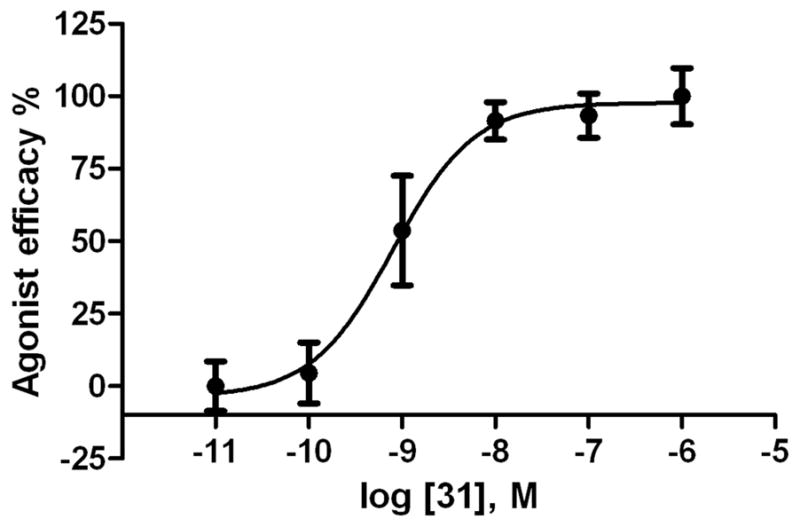
Functional agonism by the tested in an assay of adenylate cyclase in membranes of CHO cells expressing the hA3AR. Activity of the 2-(3,4-difluorophenylethynyl)-N6-3-chlorobenzyl-5′-N-methyluronamide-(N)-methanocarba analogue 31 (EC50 value 1.2±0.7 nM). The full agonist 5′-N-ethyluronamidoadenosine 48 was included for comparison (representing 100% efficacy). The experiment was repeated three times, and the average is shown.
Molecular modeling
A homology model of the hA3AR based on an agonist-bound hA2AAR X-ray structure (PDB code 3QAK)25 was used to study the putative interactions between the C2-substituted agonist 34 and the A3AR. The binding mode of 34 obtained after Induced Fit docking (IFD)35 revealed the following binding interactions of the ligand (Figure 3). The key residues embedding the adenosine moiety of the agonists in the receptor binding site are mostly conserved among AR subtypes. The 3′- and 2′-hydroxyl groups were located in proximity to Ser271 (7.42) and His272 (7.43), respectively, and could form H-bonds with these residues. The NH group of the 5′-N-methylcarboxamido moiety was involved in H-bonding with the side chain hydroxyl group of Thr94 (3.36). Both the 6-amino group and the N7 atom of the adenine ring H-bonds with Asn250 (6.55). A π–π interaction was observed between the adenine ring of 34 and Phe168 (EL2), while the side chains of Leu246 (6.51) and Ile268 (7.39) offered CH-π interactions to the adenine moiety of 34. The ligand-receptor interactions observed in the present model were in good agreement with the data of site-directed mutagenesis and with our previously published models of ARs, including the studies of AR agonists docked to the A2AAR crystal structure.36–38 In the model obtained, the 3-chlorobenzyl ring of docked 34 was located in the hydrophobic pocket formed by Val169 (EL2), Met174 (5.35), Met172 (5.33), Ile253 (6.58), and Leu264 (7.35). More precisely, the N6-3-chlorobenzyl substituent of 34 was locked in the hydrophobic pocket of the A3AR among TM5, TM6, TM7, and EL2 by CH-π interactions with the side chains of Val169 (EL2) and Ile253 (6.58). Ile253 (6.58) is not conserved among the ARs: in A1, A2A and A2B the residue at position 6.58 is a smaller threonine. Moreover, favorable interactions between the backbone group of Met172 (5.33) and the chloro atom stabilized the compound in the binding site.
Figure 3.

The binding mode (two different views with A. wire helices and B. tube helices) of linearly extended N6-3-chlorobenzyl analogue 34 obtained after IFD to the A3AR homology model. The template was built based on the crystal structure of an agonist-bound A2AAR (purple helices)25 and adjusted for movement of TM2 based on other templates (β2-adrenergic receptor, brown helices; opsin, green helices).40,41 The most successful template for TM2 was the structure of opsin.
The arylethynyl substituent at the C2 position of 34 was oriented toward the extracellular part of the A3AR in close proximity to TM2. A2AAR is characterized by four constraining disulfide bridges in the extracellular domains, unlike the A3AR, which has only one disulfide bond in that region. Thus, we expected the A3AR to be more subject to reorganization of the TMs. The disulfide bond between Cys77 (3.25) and Cys166 (EL2) is conserved not only among the AR subtypes but also among the family A GPCRs, and it is crucial for the expression and the function of the receptors. This disulfide bond involving Cys166 holds in place the backbone of two neighboring EL2 residues that are important in ligand coordination, i.e. Gln167 and Phe168. Two other disulfide bridges of A2AAR are between EL1 and EL2, namely between Cys71 and Cys159 and between Cys74 and Cys146. Another disulfide bond involves the two cysteines in EL3, Cys259 and Cys262. It has been speculated that the presence within the extracellular domains of the three disulfide bridges unique to A2AAR is not crucial for the tertiary structure and the stability of the receptor, but is indeed important for ligand recognition.39 From the structural point of view, from a comparison between the A2AAR structure and other GPCR structures, the presence of the two disulfide bridges between EL1 and EL2 forced the extracellular terminal of TM2 toward the TM bundle, thus reducing the size of the pocket embedding the agonist C2-substituents.
The IFD procedure applied to these rigid, extended A3AR agonists at the A2AAR-based model of A3AR was unable to accommodate the long and straight arylethynyl substituent at the C2 position due to a steric clash with the TM2 residues. In particular, the phenyl ring of the N6-methyl 2-phenylethynyl derivative 9, a high affinity agonist at A3AR, was clearly clashing with Ser73 in TM2, suggesting that the orientations of the extracellular terminal of TM2 and the EL1 in the A2AAR-based model of A3AR were not optimal for the accessibility of these methanocarba derivatives to the A3AR binding site.
New hybrid models of the A3AR (Figure 3) were built using different templates for the homology modeling of the extracellular part of TM2, namely an agonist-bound human β2 adrenergic receptor crystallographic structure (designated A3AR-β2adr)40 and the structure of the opsin in the activated state (designated A3AR-ops).41 In the hybrid A3AR model based on both the A2AAR and the β2-adrenergic receptor structures, the extracellular extremity of TM2 moved outward by about 4 Å at the Cα atom of Ser73. This movement created a larger pocket for the C2 arylethynyl substituents of the A3AR agonists, thereby allowing derivatives such as agonist 9 to dock in the binding cavity without steric clashes with TM2. Nevertheless, the docking of the N6-methyl 2-biphenylethynyl agonist 24 to the binding site of the A3AR-β2adr model did not produce any reasonable pose due to the steric clash between the distal phenyl ring of the compound and the residues in TM2. The unfit docked pose of 24 to the A3AR-β2adr model of A3AR suggested that a larger pocket for the longer C2-substituted agonists was needed.
The outward movement of the extracellular terminal of TM2 in the hybrid A3AR-ops model was by approximately 7 Å at the Cα of Ser73, with the creation of a larger pocket for the accessibility of C2 substituents of the present rigid chain-extended A3AR agonists, such as compounds 24 and 34. The docking poses of 24 and 34 in the A3AR-ops model showed the biphenylethynyl moiety of the C2 chain pointing toward the extracellular environment. The dihedral angle between the two phenyl rings was approximately 30–40°, making the C2 chain non-coplanar with the ethynyl moiety. The biphenylethynyl side chain was stabilized by favorable hydrophobic interactions with residues in TM2, EL2, and TM7, namely Ile268 (7.39), Tyr265 (7.36), Val72 (2.64), Leu264 (7.35), Phe168 (EL2), and the carbon chain of Gln167 (EL2). The side chain of Gln167, after the IFD optimization, pointed away from the binding cavity, opening the cavity to the agonists. The docking pose of 34 showed the distal phenyl ring of the C2 chain between the carbon chain of Gln167 and the aromatic ring of Tyr265. Tyr265 is in the wall of the binding pocket, but π interactions with the ligand were not evident. An analogue 22 that deviated from planarity because of the branched t-Bu group was significantly less potent in hA3AR binding. The presence of a halogen atom at the o-, m- or p-positions of the phenyl ring in the C2 chain was studied in the binding sites of A3AR models and the A2AAR crystal structure in order to understand the gain of affinity of compounds 15 and 30 at the A2AAR. The m-Cl atoms of the C2 side chain of 15 and 30 in the predicted orientations in the A3AR-ops model were located between Tyr271 and Gln167 (EL2), locked in the pocket by interactions between the halogen atom of the compounds and the OH group of Tyr271 and the side chain CO group of Gln167. The correspondent residue of Gln167 in human A2AAR is the hydrophobic and bulky Leu167. Nevertheless, in the docked poses of compounds 15 and 30 in the binding site of A2AAR, the m-Cl atom was able to generate strong interactions with the hydroxyl groups and the aromatic moieties of Tyr271 (7.35) and Tyr9 (1.35) of A2AAR. Instead, the orientation of the o-Cl atom of the docked agonists 14 and 29 was not optimal to create interactions with the side chain of Tyr271 (7.35) of A2AAR. On the other hand, in the docked complexes of 14 and 29 with the A3AR-ops model, the o-Cl atom was located between Tyr265 (7.35) and Val169 (EL2), creating favorable interactions with the OH group of Tyr265 and the backbone NH group of Val169. The residue corresponding to Val169 in the A2AAR is Glu169, of which the side chain was oriented such that the γ-carboxyl group interacted with the backbone NH group, i.e. no longer available to interact with the o-Cl atom of 14 or 29.
Discussion
In previous studies,13 the SAR in AR binding was studied for two series of ribonucleosides that are particularly suited for selectivity at the hA3AR: N6-methyl and N6-3-halobenzyl. N6-3-Halobenzyl substitution is greatly favored for maintaining selectivity of ribosides at the A3AR in mouse and rat. Upon addition of the (N)-methanocarba modification,21 a rise in affinity was observed at the mA1AR for certain 2-chloroadenine analogues, leading to a reduction of AR selectivity in murine species. However, straight chain alkynyl groups at the C2 position, such as pentynyl 7, alleviated this problem by reducing affinity at the mA1AR. Substitution with 2-arylethynyl groups was not examined in the (N)-methanocarba series of A3AR agonists by Melman et al.,21 but we have introduced this modification in the present study.
Previously, in the ribose-5′-uronamide series, combination of 2-phenylalkynyl groups with various bulky N6 substituents was not additive in its effect on A3AR affinity. In a series of A3AR-selective N6-arylurea derivatives, a 2-phenylalkynyl group reduced the rA3AR affinity in comparison to a 2-chloro analogue containing the same bulky N6-arylurea, although selectivity was increased.42 The combination of N6-(substituted benzyl) groups and a 2-phenylethynyl modification produced much lower affinity in hA3AR binding than the same structure with H at the C2 position.24 A study of 2-pyrazolyl-N6-substituted adenosine derivatives concluded that bulky substitutions at those two positions generally did not benefit from additivity in hA3AR binding affinity.13c
We have now explored the effects of various 2-(arylethynyl) groups at the adenine C2 position on AR affinity of (N)-methanocarba 5′-N-methyluronamido nucleosides. The resulting analogues (both N6-methyl and N6-3-halobenzyl) were full agonists of the hA3AR of nanomolar affinity that were consistently highly selective (typically >1000-fold vs. hA1AR and hA2AAR). The most potent and selective N6-methyl compounds were p-F 13 and o-Cl 14 analogues. At the mA3AR, selectivity generally remained, but for N6-methyl derivatives the high affinities achievable were somewhat lower than at hA3AR. Thus, the combination of a 2-(arylethynyl) group and large N6 substitutions was better tolerated at the A3AR in the methanocarba series than in the 9-riboside series, as characterized in earlier reports. There is also a broad flexibility of substitution of the 2-(arylethynyl) moiety, with halo, hydrophobic, and hydrophilic substitutions without losing A3AR selectivity. This feature could also benefit specific pharmacological characteristics, such as pharmacokinetic properties. The 3,4-difluoro substitution of 31 might impede possible in vivo metabolic transformation of this ring (cf. P2Y12 receptor antagonist Ticagrelor1b).
The pharmacokinetic properties of these analogues have not been measured. However, they represent an increase in hydrophobicity, as well as selectivity, in comparison to most widely used A3AR agonists. For example, the cLog P values of potent 2-chlorophenylethynyl 29, 4-fluorophenylethynyl 30 and 3,4-fluorophenylethynyl 31 derivatives are 4.65, 4.08 and 4.15, respectively. The cLog P values of more polar ribosides 1 and 2 are 0.48 and 1.20, respectively, which is possibly less desirable for full bioavailability. The total polar surface area of 29 is 121.9 Å2, which is a more favorable value for one of the physical parameters that predicts drug-likeness compared to 131.1 Å2 for both 1 and 2.43
Even large, planar polyaromatic groups at C2 (separated by an acetylene moiety) were tolerated and retained nanomolar affinity. In binding to the A2AAR, a 2-(2-(naphth-1-yl)-ethyloxy) analogue bound with high affinity, and the location of the naphthyl group was predicted by docking.44 In the present study, the size of the polycyclic C2 groups, i.e. in the potent phenanthrene-ethynyl derivative 26, exceeded that of all previous AR ligands. The 1-pyrene derivative 35 and its less potent 4-pyrene isomer 36 represent different sites of attachment of the 2-ethynyl moiety to the same large polyaromatic group. In receptor binding experiments, differences were observed that could provide insight into the geometry of the binding site considering the spatial and conformational constraints of these agonists. The 4-pyrene derivative 36 (with one additional ring added) has a close analogue in the N6-methyl series, i.e. 26.
These observations were subjected to molecular modeling analysis, which also predicted useful analogues to be synthesized. The ribose moiety is tightly anchored through a H-bond network involving TM3 and TM7, and the geometry of the unnatural glycosidic bond to the nucleobase is restricted. Thus, there is little angular flexibility of the extended, rigid C2 substituents. Thus, anchoring of the adenosine moiety to conserved amino acid residues provides a framework for exploring the region surrounding the linearly extended C2 substituent. The placement of a 2-arylethynyl substituent in the hA3AR binding site was predicted by Dal Ben et al. using the inactive structure of the hA2AAR as template, and the orientation is similar to that found here.23b However, the present study predicts the docking mode with greater detail and confidence, because the homology model is based on the agonist-bound A2AAR structure.25 With the present set of rigidified analogues, we have located a very large hydophobic pocket on the receptor that depends on an outward diplacement of TM2 in order to accommodate multiple fused rings. Thus, the binding would be allowed by the plasticity of the receptor structure to attain a ligand-specific reorganization. The reduction of hA3AR affinity for the larger C2 substituents in 35 and 36 suggests that although TM2 could shift position to accommodate such groups, there is an energetic cost for these more bulky planar polyaromatic groups. As the movement of TM2 increases, some stabilizing interactions with other regions of the receptor would be progressively lost.
The A3AR homology model built based exclusively on the crystal structure of an agonist-bound A2AAR required an adjustment of the position of TM2, which was based on the orientation of TM2 in other agonist-bound or activated GPCR templates, i.e. the β2-adrenergic receptor or opsin. The most successful template for TM2 was the structure of opsin, which displaced the upper part of TM2 relative to the agonist-bound A2AAR by ~7 Å. Thus, we predict an outward displacement of TM2 similar to the opsin structure, which is specific for agonists containing rigid C2 extensions. The inability of the A2AAR to rearrangement might contribute to the A3AR selectivity of these compounds.
The displacement of one or more TMs to accommodate a sterically bulky ligand, assuming that there are no other constraints on the helix such as a proximal disulfide bond, might be a general phenomenon in GPCRs. The ability of the helices to readjust position or “breathe” to enable a larger ligand to bind has already been proposed, specifically with respect to “multi-conformational space of the antagonist-like state of the human A3 receptor”.45 The example of altered A2AAR conformation specific to binding of a sterically extended agonist 6-(2,2-diphenylethylamino)-9-((2R,3R,4S,5S)-5-(ethylcarbamoyl)-3,4-dihydroxytetrahydrofuran-2-yl)-N-(2-(3-(1-(pyridin-2-yl)piperidin-4-yl)ureido)ethyl)-9H-purine-2-carboxamide (59, UK-432097) was already documented in the X-ray structure.25 In particular, outward movements of EL3 and extracellular portion of TM7 were associated exclusively with this bulky, multifunctionalized agonist.
The implications for receptor coupling and signaling of this type of displacement, such as the agonist-dependent movement of TM2 that we predict here, are unknown. If this conformational change associated with a family of ligands is propagated to the intracellular regions, we speculate that there could differential effects on multiple signaling pathways, and might eventually provide a rational basis for the design of biased GPCR agonists.
The enhancement of A2AAR affinity in the 2-(3-chlorophenylethynyl) analogues 15 and 30 indicated a consistent interaction with a specific site on the hA2AAR. Modeling analysis suggested halogen-π interactions46 with two tyrosine residues in the C2 binding cavity of A2AAR, namely Tyr271 (7.36) and Tyr9 (1.35), to explain the activity of the 2-(3-chlorophenylethynyl) methanocarba adenosine derivatives 15 and 30 at the A2AAR.
The freedom to insert polyaromatic ring systems on the 2-ethynyl group suggests inclusion of reporter groups, such as fluorescent dye moieties at this site. In fact, the pyrene analogue 35 is highly fluorescent and could be explored as spectroscopic probes of moderate affinity for receptor binding experiments. The fluorescent properties of pyrene are sensitive to the environment. The p-amino derivative 32 has a high affinity at both the mA3AR and hA3AR, and could potentially be derivatized for radioisotope incorporation or affinity cross-linking. We are also interested in the design of radiofluorinated nucleosides for positron emission tomographic (PET) imaging of the A3AR in vivo.47 Several fluorinated species in this study could provide the basis for incorporation of 18F into a high affinity A3AR agonist.
In conclusion, we have found a new series of highly potent and selective series of A3AR agonists containing combined substitution of the (N)-methanocarba ring system and arylethynyl groups at the adenine C2 position. The binding affinities demonstrated high tolerance of steric bulk and ring substitution by extension at the C2 position, with many aryl groups providing Ki values in the nM range. Molecular modeling suggests that this reflects conformational plasticity of the A3AR. Potent agonist ligands should be useful in future pharmacological studies of the antiinflammatory, anticancer and antiischemic properties of A3AR agonists.
Experimental Section
Chemical synthesis
Materials and instrumentation
L-ribose, and other reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). Alcohol derivative 39 was prepared as reported.28 1H NMR spectra were obtained with a Brucker 400 spectrometer using CDCl3 and CD3OD as solvents. Chemical shifts are expressed in δ values (ppm) with tetramethylsilane (δ 0.00) for CDCl3 and water (δ3.30) for CD3OD. TLC analysis was carried out on glass sheets precoated with silica gel F254 (0.2 mm) from Aldrich. The purity of final nucleoside derivatives was checked using a Hewlett–Packard 1100 HPLC equipped with a Zorbax SB-Aq 5 μm analytical column (50 × 4.6 mm; Agilent Technologies Inc, Palo Alto, CA). Mobile phase: linear gradient solvent system: 5 mM TBAP (tetrabutylammonium dihydrogenphosphate)-CH3CN from 80:20 to 0:100 in 13 min; the flow rate was 0.5 mL/min. Peaks were detected by UV absorption with a diode array detector at 230, 254, and 280 nm. All derivatives tested for biological activity showed >95% purity by HPLC analysis (detection at 254 nm). Low-resolution mass spectrometry was performed with a JEOL SX102 spectrometer with 6-kV Xe atoms following desorption from a glycerol matrix or on an Agilent LC/MS 1100 MSD, with a Waters (Milford, MA) Atlantis C18 column. High resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine, unless noted. Observed mass accuracies are those expected based on known performance of the instrument as well as trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time-dependent drift in mass accuracy. Synthetic procedures for 45 – 47 are in Supporting information. LogP and total polar surface area values were calculated using ChemBioDraw Ultra (Version 12.0.3, PerkinElmer, Boston, MA).
(1S,2R,3S,4R,5S)-2,3-Dihydroxy-N-methyl-4-(6-(methylamino)-2-(phenylethynyl)-9H-purin-9-yl)bicyclo[3.1.0]hexane-1-carboxamide (9)
A solution of compound 45a (29 mg, 0.06 mmol) in methanol (2 mL) and 10% trifluoromethane sulfonic acid (2 mL) was heated at 70 °C for 5 h. Solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 25:1) to give compound 9 (21 mg, 81%) as a syrup. 1H NMR (CD3OD) δ 8.12 (s, 1H), 7.67-7.65 (m, 2H), 7.47-7.43 (m, 2H), 5.06 (d, J = 5.2 Hz, 1H), 4.08 (d, J = 6.4 Hz, 1H), 3.15 (br s, 3H), 2.84 (s, 3H), 2.13-2.10 (m, 1H), 1.88 (t, J = 5.2 Hz, 1H), 1.41-1.39 (m, 1H). HRMS calculated for C22H23N6O3 (M + H)+: 419.1832; found 419.1818.
(1S,2R,3S,4R,5S)-2,3-Dihydroxy-N-methyl-4-(6-(methylamino)-2-(pyridin-2-ylethynyl)-9H-purin-9-yl)bicyclo[3.1.0]hexane-1-carboxamide (10)
Compound 10 (80%) was prepared from compound 46 following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.65 (s, 1H), 8.01 (s, 1H), 7.91–7.97 (m, 1H), 7.87 (d, J = 8.8 Hz, 1H), 7.53-7.48 (m, 1H), 5.15 (d, J = 5.6 Hz, 1H), 4.09 (d, J = 8.4 Hz, 1H), 3.14 (br s, 3H), 2.83 (s, 3H), 2.13-2.06 (m, 1H), 1.85 (t, J = 5.2 Hz, 1H), 1.42-1.40 (m, 1H). HRMS calculated for C21H22N7O3 (M + H)+: 420.1784; found 420.1797.
(1S,2R,3S,4R,5S)-4-(2-((2-Fluorophenyl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (11)
Compound 11 (82%) was prepared from compound 45b following the same method as used for compound 9.1H NMR (CD3OD) δ 8.11 (s, 1H), 7.49-7.39 (m, 3H), 7.25-7.20 (m, 1H), 5.06 (d, J = 5.2 Hz, 1H), 4.9 (s, 1H), 4.02 (d, J = 6.8 Hz, 1H), 3.15 (br s, 1H), 2.84 (s, 3H), 2.13-2.10 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.41-1.38 (m, 1H). HRMS calculated for C22H22FN6O3 (M + H)+: 437.1737; found 437.1753.
(1S,2R,3S,4R,5S)-4-(2-((3-Fluorophenyl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (12)
Compound 12 (78%) was prepared from compound 45c following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.11 (s, 1H), 7.67 (t, J = 6.0 Hz, 1H), 7.52-7.47 (m, 1H), 7.28-7.22 (m, 2H), 5.07 (d, J = 6.8 Hz, 1H), 4.03 (d, J = 6.8 Hz, 1H), 3.13 (br s, 1H), 2.84 (s, 3H), 2.13-2.10 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.41-1.40 (m, 1H). HRMS calculated for C22H22FN6O3 (M + H)+: 437.1737; found 437.1718.
(1S,2R,3S,4R,5S)-4-(2-((4-Fluorophenyl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (13)
Compound 13 (81%) was prepared from compound 45d following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.11 (s, 1H), 7.75-7.68 (m, 2H), 7.23-7.18 (m, 2H), 5.05 (d, J = 6.0 Hz, 1H), 4.02 (d, J = 6.4 Hz), 1H), 3.14 (br s, 3H), 2.84 (s, 3H), 2.13-2.10 (m, 1H), 1.89 (t, J = 4.8 Hz, 1H), 1.41-1.39 (m, 1H). HRMS calculated for C22H22FN6O3 (M + H)+: 437.1737; found 437.1722.
(1S,2R,3S,4R,5S)-4-(2-((2-Chlorophenyl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (14)
Compound 14 (85%) was prepared from compound 45e following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.11 (s, 1H), 7.72 (d, J = 7.2 Hz, 1H), 7.54 (d, J = 7.2 Hz, 1H), 7.46-7.36 (m, 2H), 5.10 (d, J = 6.4 Hz, 1H), 4.04 (d, J = 6.8 Hz, 1H), 3.15 (br s, 3H), 2.83 (s, 3H), 2.12-2.08 (m, 1H), 1.86 (t, J = 4.8 Hz, 1H), 1.41-1.39 (m, 1H). HRMS calculated for C22H22ClN6O3 (M + H)+: 453.1442; found 453.1449.
(1S,2R,3S,4R,5S)-4-(2-((3-Chlorophenyl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (15)
Compound 15 (82%) was prepared from compound 45f following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.11 (s, 1H), 7.67 (s, 1H), 7.59 (d, J = 7.2 Hz, 1H), 7.49-7.42 (m, 2H), 5.06 (d, J = 6.4 Hz, 1H), 4.02 (d, J = 6.8 Hz, 1H), 3.14 (br s, 3H), 2.84 (s, 3H), 2.13-2.10 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.41-1.39 (m, 1H). HRMS calculated for C22H22ClN6O3 (M + H)+: 453.1442; found 453.1442.
(1S,2R,3S,4R,5S)-4-(2-((4-Chlorophenyl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (16)
Compound 16 (84%) was prepared from compound 45g following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.10 (s, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 8.4 Hz, 2H), 5.06 (d, J = 5.6 Hz, 1H), 4.88 (s, 1H), 4.02 (d, J = 6.4 Hz, 1H), 3.15 (br s, 3H), 2.84 (s, 3H), 2.13-2.09 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.41-1.38 (m, 1H). HRMS calculated for C22H22ClN6O3 (M + H)+: 453.1460; found 453.1454.
(1S,2R,3S,4R,5S)-4-(2-((4-Bromophenyl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (17)
Compound 17 (76%) was prepared from compound 45h following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.10 (s, 1H), 7.62 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 8.4 Hz, 2H), 5.05 (d, J = 6.8 Hz, 1H), 4.88 (s, 1H), 4.02 (d, J = 6.4 Hz, 1H), 3.14 (br s, 3H), 2.84 (s, 3H), 2.13-2.09 (m, 1H), 1.88 (t, J = 5.2 Hz, 1H), 1.41-1.38 (m, 1H). HRMS calculated for C22H22BrN6O3 (M + H)+: 497.0937; found 497.0948.
(1S,2R,3S,4R,5S)-4-(2-((3-Aminophenyl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (18)
Compound 18 (67%) was prepared from compound 45i following the same method as used for compound 9.1H NMR (CD3OD) δ 8.09 (s, 1H), 7.14 (t, J = 8.0 Hz, 1H), 6.98-6.94 (m, 2H), 6.80-6.78 (m, 1H), 5.06 (d, J = 5.6 Hz, 1H), 4.02 (d, J = 6.4 Hz, 1H), 3.15 (br s, 3H), 2.85 (s, 3H), 2.12-2.09 (m, 1H), 1.87 (t, J = 4.8 Hz, 1H), 1.41-1.39 (m, 1H). HRMS calculated for C22H22N7O3 (M + H)+: 432.1784; found 432.1799.
(1S,2R,3S,4R,5S)-4-(2-((3,4-Difluorophenyl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (19)
Compound 19 (83%) was prepared from compound 45j following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.12 (s, 1H), 7.58-7.50 (m, 1H), 7.50-7.48 (m, 1H), 7.40-7.34 (m, 1H), 5.06 (d, J = 6.4 Hz, 1H), 4.02 (d, J = 6.4 Hz, 1H), 3.14 (br s, 3H), 2.84 (s, 3H), 2.13-2.09 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.41-1.40 (m, 1H). HRMS calculated for C22H21F2N6O3 (M + H)+: 455.1643; found 455.1639.
(1S,2R,3S,4R,5S)-4-(2-((3,5-Difluorophenyl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (20)
Compound 20 (84%) was prepared from compound 45k following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.12 (s, 1H), 7.31-7.26 (s, 2H), 7.14-7.09 (m, 1H), 7.49-7.42 (m, 2H), 5.06 (d, J = 5.2 Hz, 1H), 4.89 (s, 1H), 4.03 (d, J = 6.4 Hz, 1H), 3.14 (br s, 3H), 2.84 (s, 3H), 2.12-2.09 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.41-1.39 (m, 1H). HRMS calculated for C22H21F2N6O3 (M + H)+: 455.1643; found 455.1630.
(1S,2R,3S,4R,5S)-4-(2-((4-Ethylphenyl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (21)
Compound 21 (79%) was prepared from compound 45l following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.09 (s, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 5.06 (d, J = 5.2 Hz, 1H), 4.91 (s, 1H), 4.02 (d, J = 6.4 Hz, 1H), 3.15 (br s, 3H), 2.84 (s, 3H), 2.74-2.68 (m, 2H), 2.13-2.09 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.41-1.38 (m, 1H), 1.30 (t, J=7.6 Hz, 3H). HRMS calculated for C24H27N6O3 (M + H)+: 447.2145; found 447.2130.
(1S,2R,3S,4R,5S)-4-(2-((4-tert-Butylphenyl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (22)
Compound 22 (83%) was prepared from compound 45m following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.10 (s, 1H), 7.59 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 5.06 (d, J = 6.4 Hz, 1H), 4.02(d, J = 6.4 Hz, 1H), 3.15 (br s, 3H), 2.85 (s, 3H), 2.13-2.10 (m, 1H), 188(t, J = 4.8 Hz, 1H), 1.41-1.39 (m, 10H). HRMS calculated for C26H31N6O3 (M + H)+: 475.2458; found 475.2450.
(1S,2R,3S,4R,5S)-4-(2-((4-Acetylphenyl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (23)
Compound 23 (80%) was prepared from compound 45n following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.11 (s, 1H), 8.06 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.0 Hz, 2H), 5.06 (d, J = 6.4 Hz, 1H), 4.03 (d, J = 6.4 Hz, 1H), 3.15 (br s, 3H), 2.84 (s, 3H), 2.64 (s, 3H), 2.14-2.10 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.41-1.39 (m, 1H). HRMS calculated for C24H25N6O4 (M + H)+: 461.1937; found 461.1937.
(1S,2R,3S,4R,5S)-4-(2-(Biphenyl-4-ylethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (24)
Compound 24 (74%) was prepared from compound 45o following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.11 (s, 1H), 7.75-7.67 (m, 6H), 7.47 (t, J = 7.6 Hz, 2H), 7.38 (t, J = 7.2Hz, 1H), 5.06 (d, J = 6.0 Hz, 1H), 4.89 (s, 1H), 4.03 (d, J = 6.4 Hz, 1H), 3.16 (br s, 3H), 2.84 (s, 3H), 2.13-2.10 (m, 1H), 189 (t, J = 4.8 Hz, 1H), 1.42-1.39 (m, 1H). HRMS calculated for C28H27N6O3 (M + H)+: 495.2145; found 495.2141.
(1S,2R,3S,4R,5S)-2,3-Dihydroxy-N-methyl-4-(6-(methylamino)-2-(naphthalen-1-ylethynyl)-9H-purin-9-yl)bicyclo[3.1.0]hexane-1-carboxamide (25)
Compound 25 (73%) was prepared from compound 45p following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.56 (d, J = 7.6 Hz, 1H), 8.11 (s, 1H), 8.00-7.90 (m, 3H), 7.68 (t, J = 5.6 Hz, 1H), 7.62-7.53 (m, 2H), 5.10 (d, J = 5.2 Hz, 1H), 4.93 (s, 1H), 4.06 (d, J = 6.4 Hz, 1H), 3.19 (br s, 3H), 2.79 (s, 3H), 2.15-2.12 (m, 1H), 189 (t, J = 4.8 Hz, 1H), 1.41-1.38 (m, 1H). HRMS calculated for C26H25N6O3 (M + H)+: 469.1988; found 469.2005.
(1S,2R,3S,4R,5S)-2,3-Dihydroxy-N-methyl-4-(6-(methylamino)-2-(phenanthren-9-ylethynyl)-9H-purin-9-yl)bicyclo[3.1.0]hexane-1-carboxamide (26)
Compound 26 (65%) was prepared from compound 45q following the same method as used for compound 9. 1H NMR (CD3OD) δ 8.84-8.77 (m, 2H), 8.66-8.63 (m, 1H), 8.26 (s, 1H), 8.12 (s, 1H), 7.97 (d, J = 7.6 Hz, 1H), 7.80-7.73 (m, 3H), 7.66 (t, J = 7.2 Hz, 1H), 5.10 (d, J = 6.0 Hz, 1H), 4.92 (s, 1H), 4.07 (d, J = 6.4 Hz, 1H), 3.20 (br s, 3H), 2.81 (s, 3H), 2.15-2.12 (m, 1H), 1.90 (t, J = 4.8 Hz, 1H), 1.42-1.38 (m, 1H). HRMS calculated for C30H27N6O3 (M + H)+: 519.2145; found 519.2137.
(1S,2R,3S,4R,5S)-4-(6-(3-Chlorobenzylamino)-2-(phenylethynyl)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (27)
PdCl2(PPh3)2 (6.13 mg, 0.008 mmol), CuI (1.2 mg, 0.004 mmol), phenylacetylene (30 μL, 0.26 mmol) and triethylamine (60 μL, 0.4 mmol) was added to a solution of compound 44 (26 mg, 0.04 mmol) in anhydrous DMF (1 mL), and stirred at room temperature overnight. Solvent was evaporated under vacuum, and the residue was roughly purified on flash silica gel column chromatography. The resulting compound was dissolved in methanol (2 mL) and 10% trifluoromethane sulfonic acid (2 mL) and heated at 70 °C for 5 h. Solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 25:1) to give compound 27 (17 mg, 76%) as a syrup. 1H NMR (CD3OD) δ 8.13 (s, 1H), 7.66-7.63 (m, 2H), 7.46-7.42 (m, 4H), 7.37-7.26 (m, 3H), 5.06 (d, J = 5.6 Hz, 1H), 4.9 (br s, 2H) 4.04 (d, J = 6.4 Hz, 1H), 2.84 (s, 3H), 2.14-2.11 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.41-1.37 (m, 1H). HRMS calculated for C28H26ClN6O3 (M + H)+: 529.1755; found 529.1740.
(1S,2R,3S,4R,5S)-4-(6-(3-Chlorobenzylamino)-2-((4-fluorophenyl)ethynyl)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (28)
Compound 28 (68%) was prepared from compound 44 following the same method as used for compound 27. 1H NMR (CD3OD) δ 8.13 (s, 1H), 7.70-7.63 (m, 2H), 7.59-7.55 (m, 1H), 7.45 (s, 1H), 7.37-7.21 (m, 3H), 7.19-7.16 (m, 1H), 5.06 (d, J = 5.6 Hz, 1H), 4.9 (s, 1H), 4.58 (br s, 2H), 4.04 (d, J = 7.6 Hz, 1H), 2.84 (s, 3H), 2.17-2.10 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.41-1.38 (m, 1H). HRMS calculated for C28H25ClFN6O3 (M + H)+: 547.1661; found 547.1652.
(1S,2R,3S,4R,5S)-4-(6-(3-Chlorobenzylamino)-2-((2-chlorophenyl)ethynyl)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (29)
Compound 29 (65%) was prepared from compound 44 following the same method as used for compound 27. 1H NMR (CD3OD) δ 8.14 (s, 1H), 7.72-7.70 (m, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.47-7.25 (m, 6H), 5.11 (d, J = 6.8 Hz, 1H), 4.90 (s, 1H), 4.05 (d, J = 6.8 Hz, 1H), 2.82 (s, 3H), 2.12-2.09 (m, 1H), 1.86 (t, J = 4.8 Hz, 1H), 1.40-1.38 (m, 1H). HRMS calculated for C28H24Cl2N6O3Na (M + Na)+: 585.1185; found 585.1167.
(1S,2R,3S,4R,5S)-4-(6-(3-Chlorobenzylamino)-2-((3-chlorophenyl)ethynyl)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (30)
Compound 30 (66%) was prepared from compound 44 following the same method as used for compound 27. 1H NMR (CD3OD) δ 8.15 (s, 1H), 7.66-7.57 (m, 2H), 7.48-7.26 (m, 6H), 5.07 (d, J = 6.4 Hz, 1H), 4.85 (s, 1H), 4.04 (d, J = 6.8 Hz, 1H), 2.84 (s, 3H), 2.14-2.10 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.41-1.39 (m, 1H). HRMS calculated for C28H25Cl2N6O3 (M + H)+: 563.1365; found 563.1359.
(1S,2R,3S,4R,5S)-4-(6-(3-Chlorobenzylamino)-2-((3,4-difluorophenyl)ethynyl)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (31)
Compound 31 (63%) was prepared from compound 44 following the same method as used for compound 27. 1H NMR (CD3OD) δ 8.14 (s, 1H), 7.58 (t, J =8.8 Hz, 1H), 7.47-7.44 (m, 2H), 7.39-7.25 (m, 4H), 5.06 (d, J = 6.4 Hz, 1H), 4.89 (s, 1H), 4.04 (d, J = 6.4 Hz, 1H), 2.84 (s, 3H), 2.13-2.10 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.41-1.38 (m, 1H). HRMS calculated for C28H24F2ClN6O3 (M + H)+: 565.1566; found 565.1559.
(1S,2R,3S,4R,5S)-4-(2-((4-Aminophenyl)ethynyl)-6-(3-chlorobenzylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (32)
Compound 32 (59%) was prepared from compound 44 following the same method as used for compound 27. 1H NMR (CD3OD) δ 8.02 (s, 1H), 7.84 (d, J =8.8 Hz, 2H), 7.33-7.23 (m, 4H), 6.60 (d, J = 8.4 Hz, 2H), 5.17 (d, J = 6.4 Hz, 1H), 4.85 (s, 1H), 4.73 (br s, 2H), 4.02 (d, J = 6.8 Hz, 1H), 2.83 (s, 3H), 2.08-2.05 (m, 1H), 1.81 (t, J = 4.8 Hz, 1H), 1.41-1.37 (m, 1H). HRMS calculated for C28H27ClN7O3 (M + H)+: 545.1041; found 545.1045.
3-((6-(3-Chlorobenzylamino)-9-((1S,2R,3S,4R,5S)-3,4-dihydroxy-5-(methyl-carbamoyl)bicyclo[3.1.0]hexan-2-yl)-9H-purin-2-yl)ethynyl)benzoic acid (33)
PdCl2(PPh3)2 (3.0 mg, 0.004 mmol), CuI (1.0 mg, 0.004 mmol), phenylacetylene (18.7 mg, 0.12 mmol) and triethylamine (20 μL, 0.2 mmol) was added to a solution of compound 44 (12.68 mg, 0.02 mmol) in anhydrous DMF (1 mL), and stirred at room temperature overnight. Solvent was evaporated under vacuum, and the residue was roughly purified on flash silica gel column chromatography. The resulting compound was dissolved in dioxane (2 mL) and 1N HCl (1.5 mL) and heated at 60 °C for 2 h. After completion of starting material, solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH:TFA = 25:1:0.1) to give compound 33 (7 mg, 61%) as a syrup. 1H NMR (CD3OD) δ 8.28 (s, 1H), 8.18-8.16 (m, 1H), 8.11-8.07 (m, 1H), 7.87 (d, J =7.6 Hz, 1H), 7.59-7.54 (m, 2H), 7.47 (s, 1H), 7.41-7.26 (m, 2H), 5.09 (d, J = 6.4 Hz, 1H), 4.91 (s, 1H), 4.05 (d, J = 6.4 Hz, 1H), 2.85 (s, 3H), 2.14-2.11 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.42-1.38 (m, 1H). HRMS calculated for C29H26ClN6O5 (M + H)+: 573.1653; found 573.1646.
(1S,2R,3S,4R,5S)-4-(2-(Biphenyl-4-ylethynyl)-6-(3-chlorobenzylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (34)
Compound 34 (68%) was prepared from compound 44 following the same method as used for compound 27. 1H NMR (CD3OD) δ 8.13 (s, 1H), 7.74-7.66 (m, 7H), 7.49-7.45 (m, 2H), 7.40-7.26 (m, 4H), 5.07 (d, J = 5.6 Hz, 1H), 4.9 (s, 1H), 4.60 (br s, 2H), 4.05 (d, J = 6.4 Hz, 1H), 2.85 (s, 3H), 2.14-2.11 (m, 1H), 1.89 (t, J = 4.8 Hz, 1H), 1.42-1.40 (m, 1H). HRMS calculated for C34H30ClN6O3 (M + H)+: 605.2068; found 605.2083.
(1S,2R,3S,4R,5S)-4-(6-(3-Chlorobenzylamino)-2-(pyren-1-ylethynyl)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (35)
Compound 35 (91%) was prepared from compound 44 following the same method as used for compound 33. 1H NMR (CD3OD) δ 8.71 (d, J = 9.2 Hz, 1H), 8.26-8.23 (m, 4H), 8.16-8.13 (m, 2H), 8.08-8.03 (m, 3H), 7.54 (s, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.34 (t, J = 8.0 Hz, 1H), 7.27 (d, J = 8.0 Hz, 1H), 5.06 (d, J = 6.4 Hz, 1H), 4.83 (s, 1H), 4.04 (d, J = 6.4 Hz, 1H), 2.81 (s, 3H), 2.10-2.07 (m, 1H), 1.89 (t, J = 4.8 Hz, 1H), 1.41-1.37 (m, 1H). HRMS calculated for C38H30ClN6O3 (M + H)+: 653.2068; found 653.2078.
(1S,2R,3S,4R,5S)-4-(6-(3-Chlorobenzylamino)-2-(pyren-4-ylethynyl)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (36)
Compound 36 (69%) was prepared from compound 44 following the same method as used for compound 33. 1H NMR (CD3OD) δ 8.82 (d, J = 7.6 Hz, 1H), 8.51 (s, 1H), 8.29-8.21 (m, 3H), 8.16-8.12 (m, 4H), 8.04 (t, J = 7.6 Hz, 1H), 7.54 (s, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.35 (t, J = 8.0 Hz, 1H), 7.28 (d, J = 8.4 Hz, 1H), 5.1 (d, J = 6.0 Hz, 1H), 4.06 (d, J = 6.0 Hz, 1H), 2.80 (s, 3H), 2.18-2.09 (m, 1H), 1.89 (t, J = 4.8 Hz, 1H), 1.42-1.39 (m, 1H). HRMS calculated for C38H30ClN6O3 (M + H)+: 653.2068; found 653.2056.
(1S,2R,3S,4R,5S)-Ethyl-(2,3-O-isopropylidene)-4-(2-iodo-6-(methylamino)-9H-purin-9-yl)bicyclo[3.1.0]hexane-1-carboxylate (41)
Methylamine hydrochloride (0.353g, 5.23 mmol) and triethylamine (1.4 mL, 16.6 mmol) was added to a solution of compound 40 (0.528g, 1.04 mmol) in anhydrous methanol (15 mL) and stirred at room temperature overnight. Solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (hexane:ethylacetate=1:1) to give compound 41 (0.470 g, 94%) as a foamy solid. 1H NMR (CD3OD) δ 7.94 (s, 1H), 5.83 (d, J = 7.2 Hz, 1H), 4.94 (s, 1H), 4.80 (d, J = 6.0 Hz, 1H), 4.33-4.27 (m, 2H), 3.05 (br s, 3H), 2.25-2.21 (m, 1H), 1.65-1.61 (m, 1H), 1.53-1.49 (m, 4H), 1.34 (t, J = 7.2 Hz, 3H), 1.29 (s, 3H). HRMS calculated for C18H23IN5O4 (M + H)+: 500.1072; found 500.1075.
(1S,2R,3S,4R,5S)-(2,3-O-Isopropylidene)-4-(2-iodo-6-(methylamino)-9H-purin-9-yl)-N-methylbicyclo[3.1.0]hexane-1-carboxamide (43)
40% Methylamine solution (10 mL) was added to a solution of compound 41 (0.470 g, 0.94 mmol) in methanol (15 mL) and stirred at room temperature for 48 h. Solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH=40:1) to give compound 43 (0.360 g, 79%) as a syrup. 1H NMR (CD3OD) δ 7.95 (s, 1H), 5.72 (d, J = 7.2 Hz, 1H), 4.93 (s, 1H), 4.84 (d, J = 7.2 Hz, 1H), 3.05 (br s, 3H), 2.90 (s, 3H), 2.17-2.11 (m, 1H), 1.54-1.49 (m, 4H), 1.39 (t, J = 5.2 Hz, 1H), 1.30 (s, 3H). HRMS calculated for C17H22IN6O3 (M + H)+: 485.0798; found 485.0803.
Pharmacological characterization
The nucleoside derivatives, dissolved as stock solutions in DMSO (5 mM) and stored frozen, were evaluated in binding29–31 and a functional assay34 at the A3AR, binding assays at the A1AR and A2AAR, and a functional assay at the hA3AR (details in Supporting Information). Use of heterologously expressed mouse ARs was as reported.33b,49 Protein content was determined48 and IC50 values in binding inhibition transformed to Ki values51 as reported.
Molecular modeling
The Homology Model module of MOE was utilized to build a new molecular model of the hA3AR (details in Supporting Information). The recently reported model of the complex of the nonselective AR agonist 59 docked to the crystal structure of the A2AAR was used as a template for modeling of the A3AR.25,51 The sequence alignment of the A2A and A3ARs was performed with MOE, taking into account positions of highly conserved amino acid residues. The position of full agonist 59 inside the A2AAR was taken into account during the modeling. In the resulting initial homology model of the A3AR (prior to movement of TM2), the complex 5′,2,N6 trisubstituted agonist 59 used to crystallize the A2AAR was removed and the simpler 5′-uronamide 58 docked inside the receptor. The Glide program of the Schrodinger package52 was used to dock the other agonists to the A3AR model obtained. The receptor grid generation was performed for the box with a center in the centroid of 58 in its initial position. The size of the box was determined automatically. The extra precision mode (XP) of Glide was used for the docking. The binding site was defined as 58 and all amino acid residues located within 5Å from 58. All A3AR residues located within 2Å from the binding site were used as a shell. The following parameters of energy minimization were used: OPLS2005 force field, water was used as an implicit solvent, a maximum of 5000 iterations of the Polak-Ribier conjugate gradient minimization method was used with a convergence threshold of 0.01 kJ·mol−1·Å−1. Another reference nucleoside 58 was also docked in this hA3AR homology model.
Hybrid A3AR models involving movement of TM2, as explained above, were utilized to study the binding mode of analogue 34 and all other novel analogues in Table 1 using InducedFit docking implemented in the Schrödinger package. The grid generation was performed for a cubic box with sides of 26 Å and having a center in the centroid of the agonist molecules. The default values were used for other parameters.
Supplementary Material
Acknowledgments
We thank Dr. John Lloyd and Dr. Noel Whittaker (NIDDK) for mass spectral determinations. This research was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases and by NIH R01 HL077707.
Abbreviations
- AR
adenosine receptor
- cAMP
adenosine 3′,5′-cyclic phosphate
- CHO
Chinese hamster ovary
- Cl-IB-MECA
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine
- DIPEA
diisopropylethylamine
- DCM
dichloromethane
- DMF
N,N-dimethylformamide
- DMEM
Dulbecco’s modified Eagle’s medium
- EDTA
ethylenediaminetetraacetic acid
- EL
extracellular loop
- GPCR
G protein-coupled receptor
- HEK
human embryonic kidney
- I-AB-MECA
N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyl-uronamide
- IFD
Induced Fit docking
- NECA
5′-N-ethylcarboxamidoadenosine
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HRMS
high resolution mass spectroscopy
- NMR
nuclear magnetic resonance
- R-PIA
N6-R-phenylisopropyladenosine
- TEA
triethylamine
- TLC
thin layer chromatography
- TM
transmembrane domain
Footnotes
Supporting information available:
Docking figures for compounds 14, 15 and 34, procedures for synthesis of intermediates, detailed molecular modeling procedures, and biological assays for novel nucleoside derivatives, and coordinates of complexes with 13, 31 and 35. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a) Cheong SL, Federico S, Venkatesan G, Mandel AL, Shao YM, Moro S, Spalluto G, Pastorin G. The A3 adenosine receptor as multifaceted therapeutic target: pharmacology, medicinal chemistry, and in silico approaches. Med Res Rev. 2012 doi: 10.1002/med.20254. in press. [DOI] [PubMed] [Google Scholar]; b) Jacobson KA, Balasubramanian R, Deflorian F, Gao ZG. G protein-coupled adenosine (P1) and P2Y receptors. Purinergic Signal. 2012 doi: 10.1007/s11302-012-9294-7.. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fishman P, Jacobson KA, Ochaion A, Cohen S, Bar-Yehuda S. The anti-cancer effect of A3 adenosine receptor agonists: A novel, targeted therapy. Immun Endoc Metab Agents in Med Chem. 2007;7:298–303. doi: 10.2174/187152207781369878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, Rosner I, Rozenbaum M, Mader R, Adawi M, Caspi D, Tishler M, Langevitz P, Rubinow A, Friedman J, Green L, Tanay A, Ochaion A, Cohen S, Kerns WD, Cohn I, Fishman-Furman S, Farbstein M, Bar-Yehuda S, Fishman P. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: Data from a phase II clinical trial. J Rheumatol. 2008;35:41–48. [PubMed] [Google Scholar]
- 4.Gessi S, Merighi S, Varani K, Cattabriga E, Benini A, Mirandola P, Leung E, MacLennan S, Feo C, Baraldi S, Borea PA. Adenosine receptors in colon carcinoma tissues colon tumoral cell lines: focus on the A3 adenosine subtype. J Cell Physiol. 2007;211:826–836. doi: 10.1002/jcp.20994. [DOI] [PubMed] [Google Scholar]
- 5.Zheng J, Wang R, Zambraski E, Wu D, Jacobson KA, Liang BT. A novel protective action of adenosine A3 receptors: Attenuation of skeletal muscle ischemia and reperfusion injury. Am J Physiol, Heart and Circ Physiol. 2007;293:3685–3691. doi: 10.1152/ajpheart.00819.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wan TC, Ge ZD, Tampo A, Mio Y, Bienengraeber MW, Tracey WR, Gross GJ, Kwok WM, Auchampach JA. The A3 adenosine receptor agonist CP-532,903 [N6-(2,5-dichlorobenzyl)-3′-aminoadenosine-5′-N-methylcarboxamide] protects against myocardial ischemia/reperfusion injury via the sarcolemmal ATP-sensitive potassium channel. J Pharmacol Exp Ther. 2008;324:234–243. doi: 10.1124/jpet.107.127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guzman J, Yu JG, Suntres Z, Bozarov A, Cooke H, Javed N, Auer H, Palatini J, Hassanain HH, Cardounel AJ, Javed A, Grants I, Wunderlich JE, Christofi FL. ADOA3R as a therapeutic target in experimental colitis: Proof by validated high-density oligonucleotide microarray analysis. Inflamm Bowel Dis. 2006;12:766–789. doi: 10.1097/00054725-200608000-00014. [DOI] [PubMed] [Google Scholar]
- 8.(a) Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA. Pharmacological and therapeutic effects of A3 adenosine receptor (A3AR) agonists. Drug Disc Today. 2012;17:359–366. doi: 10.1016/j.drudis.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gessi S, Merighi S, Varani K, Leung E, MacLennan S, Borea PA. The A3 adenosine receptor: An enigmatic player in cell biology. Pharmacology & Therapeutics. 2008;117:123–140. doi: 10.1016/j.pharmthera.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Yamano K, Inoue M, Masaki S, Saki M, Ichimura M, Satoh M. Generation of adenosine A3 receptor functionally humanized mice for the evaluation of the human antagonists. Biochem Pharmacol. 2006;71:294–306. doi: 10.1016/j.bcp.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 10.Yang H, Avila MY, Peterson-Yantorno K, Coca-Prados M, Stone RA, Jacobson KA, Civan MM. The cross-species A3 adenosine-receptor antagonist MRS 1292 inhibits adenosine-triggered human nonpigmented ciliary epithelial cell fluid release and reduces mouse intraocular pressure. Current Eye Res. 2005;30:747–754. doi: 10.1080/02713680590953147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hua X, Chason KD, Fredholm BB, Deshpande DA, Penn RB, Tilley SL. Adenosine induces airway hyperresponsiveness through activation of A3 receptors on mast cells. J Allergy Clin Immunol. 2008;122:107–113. doi: 10.1016/j.jaci.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bulger EM, Tower CM, Warner KJ, Garland T, Cuschieri J, Rizoli S, Rhind S, Junger WG. Increased neutrophil adenosine A3 receptor expression is associated with hemorrhagic shock and injury severity in trauma patients. Shock. 2011;36:435–439. doi: 10.1097/SHK.0b013e318231ee2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Gao ZG, Kim SK, Biadatti T, Chen W, Lee K, Barak D, Kim SG, Johnson CR, Jacobson KA. Structural determinants of A3 adenosine receptor activation: Nucleoside ligands at the agonist/antagonist boundary. J Med Chem. 2002;45:4471–4484. doi: 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jeong LS, Lee HW, Jacobson KA, Kim HO, Shin DH, Lee JA, Gao ZG, Lu C, Duong HT, Gunaga P, Lee SK, Jin DZ, Chun MW, Moon HR. Structure-activity relationships of 2-chloro-N6-substituted-4′-thioadenosine-5′-uronamides as highly potent and selective agonists at the human A3 adenosine receptor. J Med Chem. 2006;49:273–281. doi: 10.1021/jm050595e. [DOI] [PubMed] [Google Scholar]; (c) Elzein E, Palle V, Wu Y, Maa T, Zeng D, Zablocki J. 2-Pyrazolyl-N6-substituted adenosine derivatives as high affinity and selective adenosine A3 receptor agonists. J Med Chem. 2004;47:4766–4773. doi: 10.1021/jm049682h. [DOI] [PubMed] [Google Scholar]; (d) DeNinno MP, Masamune H, Chenard LK, DiRico KJ, Eller C, Etienne JB, Tickner JE, Kennedy SP, Knight DR, Kong J, Oleynek JJ, Tracey WR, Hill RJ. The synthesis of highly potent, selective, and water-soluble agonists at the human adenosine A3 receptor. Bioorg Med Chem Lett. 2006;16:2525–2527. doi: 10.1016/j.bmcl.2006.01.088. [DOI] [PubMed] [Google Scholar]; (e) Cosyn L, Palaniappan KK, Kim SK, Duong HT, Gao ZG, Jacobson KA, Van Calenbergh S. 2-Triazole-substituted adenosines: A new class of selective A3 adenosine receptor agonists, partial agonists, and antagonists. J Med Chem. 2006;49:7373–7383. doi: 10.1021/jm0608208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobson KA, Ji X-d, Li AH, Melman N, Siddiqui MA, Shin KJ, Marquez VE, Ravi RG. Methanocarba analogues of purine nucleosides as potent and selective adenosine receptor agonists. J Med Chem. 2000;43:2196–2203. doi: 10.1021/jm9905965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee K, Ravi RG, Ji X-d, Marquez VE, Jacobson KA. Ring-constrained (N)methanocarba-nucleosides as adenosine receptor agonists: Independent 5′-uronamide and 2′-deoxy modifications. Bioorg Med Chem Lett. 2001;11:1333–1337. doi: 10.1016/s0960-894x(01)00213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tchilibon S, Joshi BV, Kim SK, Duong HT, Gao ZG, Jacobson KA. Methanocarba 2,N6-disubstituted adenine nucleosides as highly potent and selective A3 adenosine receptor agonists. J Med Chem. 2005;48:1745–1758. doi: 10.1021/jm049580r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ochaion A, Bar-Yehuda S, Cohen S, Amital H, Jacobson KA, Joshi BV, Gao ZG, Barer F, Patoka R, Del Valle L, Perez-Liz G, Fishman P. The A3 adenosine receptor agonist CF502 inhibits the PI3K, PKB/Akt and NF-κB signaling pathway in synoviocytes from rheumatoid arthritis patients and in adjuvant induced arthritis rats. Biochem Pharmacol. 2008;76:482–494. doi: 10.1016/j.bcp.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matot I, Weininger CF, Zeira E, Galun E, Joshi BV, Jacobson KA. A3 Adenosine receptors and mitogen activated protein kinases in lung injury following in-vivo reperfusion. Critical Care. 2006;10:R65. doi: 10.1186/cc4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Z, Janes K, Chen C, Doyle T, Tosh DK, Jacobson KA, Salvemini D. Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J. 2012 doi: 10.1096/fj.11-201541. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Volpini R, Dal Ben D, Lambertucci C, Taffi S, Vittori S, Klotz KN, Cristalli GJ. N6-Methoxy-2-alkynyladenosine derivatives as highly potent and selective ligands at the human A3 adenosine receptor. J Med Chem. 2007;50:1222–1230. doi: 10.1021/jm060963u. [DOI] [PubMed] [Google Scholar]; (b) Cristalli G, Lambertucci C, Marucci G, Volpini R, Dal Ben D. A2A Adenosine receptor and its modulators: Overview on a druggable GPCR and on structure-activity relationship analysis and binding requirements of agonists and antagonists. Curr Pharm Des. 2008;14:1525–1552. doi: 10.2174/138161208784480081. [DOI] [PubMed] [Google Scholar]
- 21.Melman A, Gao ZG, Kumar D, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Design of (N)-methanocarba adenosine 5′-uronamides as species-independent A3 receptor-selective agonists. Bioorg Med Chem Lett. 2008;18:2813–2819. doi: 10.1016/j.bmcl.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dooley MJ, Quinn RJ. The three binding domain model of adenosine receptors: molecular modeling aspects. J Med Chem. 1992;35:211–217. doi: 10.1021/jm00080a002. [DOI] [PubMed] [Google Scholar]
- 23.(a) Volpini R, Buccioni M, Dal Ben D, Lambertucci C, Lammi C, Marucci G, Ramadori AT, Klotz KN, Cristalli G. Synthesis and biological evaluation of 2-alkynyl-N6-methyl-5′-N-methylcarboxamidoadenosine derivatives as potent and highly selective agonists for the human adenosine A3 receptor. J Med Chem. 2009;52:7897–7900. doi: 10.1021/jm900754g. [DOI] [PubMed] [Google Scholar]; (b) Dal Ben D, Lambertucci C, Lammi C, Marucci G, Thomas A, Volpini R, Cristalli G. Molecular modeling study on potent and selective adenosine A3 receptor agonists. Bioorg Med Chem. 2010;18:7923–7930. doi: 10.1016/j.bmc.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 24.Sevillano LG, McGuigan C, Davies RH. Compounds useful as A3 adenosine receptor agonists. U.S. Patent 7,414,036 B2. 2008
- 25.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Congreve M, Langmead CJ, Mason JS, Marshall FH. Progress in structure based drug design for G protein-coupled receptors. J Med Chem. 2011;54:4283–4311. doi: 10.1021/jm200371q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chinchilla R, Nájera C. The Sonogashira reaction: A booming methodology in synthetic organic chemistry. Chem Rev. 2007;107:874–922. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 28.Tosh DK, Chinn M, Ivanov AA, Klutz AM, Gao ZG, Jacobson KA. Functionalized congeners of A3 adenosine receptor-selective nucleosides containing a bicyclo[3.1.0]hexane ring system. J Med Chem. 2009;52:7580–7592. doi: 10.1021/jm900426g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwabe U, Trost T. Characterization of adenosine receptors in rat brain by (−)-[3H]N6-phenylisopropyladenosine. Naunyn Schmiedebergs Arch Pharmacol. 1989;313:179–187. doi: 10.1007/BF00505731. [DOI] [PubMed] [Google Scholar]
- 30.Jarvis MF, Schutz R, Hutchison AJ, Do E, Sills MA, Williams M. [3H]CGS 21680 an A2 selective adenosine receptor agonist directly labels A2 receptors in rat brain tissue. J Pharmacol Exp Ther. 1989;251:888–893. [PubMed] [Google Scholar]
- 31.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. 125I-4-Aminobenzyl-5′-N-methylcarboxamidoadenosine, a high affinity radioligand for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:978–982. [PMC free article] [PubMed] [Google Scholar]
- 32.Englert M, Quitterer U, Klotz KN. Effector coupling of stably transfected human A3 adenosine receptors in CHO cells. Biochem Pharmacol. 2002;64:61–65. doi: 10.1016/s0006-2952(02)01071-7. [DOI] [PubMed] [Google Scholar]
- 33.(a) Jacobson KA, Park KS, Jiang J-l, Kim YC, Olah ME, Stiles GL, Ji Xd. Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology. 1997;36:1157–1165. doi: 10.1016/s0028-3908(97)00104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ge ZD, Peart JN, Kreckler LM, Wan TC, Jacobson MA, Gross GJ, Auchampach JA. Cl-IB-MECA [2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J Pharmacol Exp Ther. 2006;319:1200–1210. doi: 10.1124/jpet.106.111351. [DOI] [PubMed] [Google Scholar]
- 34.Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 35.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 36.Ivanov AA, Barak D, Jacobson KA. Evaluation of homology modeling of GPCRs in light of the A2A adenosine receptor crystallographic structure. J Med Chem. 2009;52:3284–3292. doi: 10.1021/jm801533x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaakola VP, Lane JR, Lin JY, Katritch V, IJzerman AP, Stevens RC. Ligand binding and subtype selectivity of the human A2A adenosine receptor: identification and characterization of essential amino acid residues. J Biol Chem. 2010;285:13032–13044. doi: 10.1074/jbc.M109.096974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Costanzi S, Ivanov AA, Tikhonova IG, Jacobson KA. Structure and function of G protein-coupled receptors studied using sequence analysis, molecular modeling and receptor engineering: adenosine receptors. Front Drug Des Discov. 2007;3:63–79. [Google Scholar]
- 39.O’Malley MA, Naranjo AN, Lazarova T, Robinson AS. Analysis of adenosine A2A receptor stability: Effects of ligands and disulfide bonds. Biochemistry. 2010;49:9181–9189. doi: 10.1021/bi101155r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunihara RK, Kobilka BK. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 42.Baraldi PG, Cacciari B, Pineda de las Infantas MJ, Romagnoli R, Spalluto G, Volpini R, Costanzi S, Vittori S, Cristalli G, Melman N, Park KS, Ji X-d, Jacobson KA. Synthesis and biological activity of a new series of N6-arylcarbamoyl-, 2-(ar)alkynyl-N6-arylcarbamoyl, and N6-carboxamido- derivatives of adenosine-5′-N-ethyluronamide (NECA) as A1 and A3 adenosine receptor agonists. J Med Chem. 1998;41:3174–3185. doi: 10.1021/jm980147p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nature Chem. 2012;4:90–98. doi: 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deflorian F, Kumar TS, Phan K, Gao ZG, Xu F, Wu H, Katritch V, Stevens RC, Jacobson KA. Evaluation of molecular modeling of agonist binding in light of the crystallographic structure of the agonist-bound A2A adenosine receptor. J Med Chem. 2012;55:538–552. doi: 10.1021/jm201461q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moro S, Deflorian F, Bacilieri M, Spalluto G. Ligand-based homology modeling as attractive tool to inspect GPCR structural plasticity. Curr Pharm Des. 2006;12:2175–2185. doi: 10.2174/138161206777585265. [DOI] [PubMed] [Google Scholar]
- 46.Hardegger LA, Kuhn B, Spinnler B, Anselm L, Ecabert R, Stihle M, Gsell B, Thoma R, Diez J, Benz J, Plancher JM, Hartmann G, Banner DW, Haap W, Diederich F. Systematic investigation of halogen bonding in protein-ligand interactions. Angew Chem Int Engl Ed. 2011;50:314–318. doi: 10.1002/anie.201006781. [DOI] [PubMed] [Google Scholar]
- 47.Kiesewetter DO, Lang L, Ma Y, Bhattacharjee AK, Gao ZG, Joshi BV, Melman A, Castro S, Jacobson KA. Synthesis and characterization of [76Br]-labeled high affinity A3 adenosine receptor ligands for positron emission tomography. Nucl Med Biol. 2009;36:3–10. doi: 10.1016/j.nucmedbio.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 49.Kreckler LM, Wan TC, Ge ZD, Auchampach JA. Adenosine inhibits tumor necrosis factor-α release from mouse peritoneal macrophages via A2A and A2B but not the A3 adenosine receptors. J Pharmacol Exp Ther. 2006;317:172–180. doi: 10.1124/jpet.105.096016. [DOI] [PubMed] [Google Scholar]
- 50.Cheng YC, Prusoff WH. Relationship between inhibition constant (K1) and concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic-reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 51.Tosh DK, Phan K, Deflorian F, Wei Q, Gao ZG, Jacobson KA. Truncated (N)-Methanocarba Nucleosides as A1 Adenosine Receptor Agonists and Partial Agonists: Overcoming Lack of a Recognition Element. ACS Med Chem Lett. 2011;2:626–631. doi: 10.1021/ml200114q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mohamadi FN, Richards GJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. Macromodel an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J Comput Chem. 1990;11:440–467. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.