

Abstract
We have adapted transposase-based in vitro shotgun library construction (“tagmentation”) for whole-genome bisulfite sequencing. This method, Tn5mC-seq, enables a >100-fold reduction in starting material relative to conventional protocols, such that we generate highly complex bisulfite sequencing libraries from as little as 10 ng of input DNA, and ample useful sequences from 1 ng of input DNA. We demonstrate Tn5mC-seq by sequencing the methylome of a human lymphoblastoid cell line to ∼8.6× high-quality coverage of each strand.
DNA methylation is a widespread epigenetic modification that plays a pivotal role in the regulation of the genomes of diverse organisms. The most prevalent and widely studied form of DNA methylation in mammalian genomes occurs at the five carbon position of cytosine residues, usually in the context of the CpG dinucleotide. Microarrays, and more recently massively parallel sequencing, have enabled the interrogation of cytosine methylation (5mC) on a genome-wide scale (Zilberman and Henikoff 2007). However, the in vivo study of DNA methylation and other epigenetic marks, e.g., in specific cell types or anatomical structures, is sharply limited by the relatively high amount of input material required for contemporary protocols.
Methods for genome-scale interrogation of methylation patterns include several that are preceded by the enrichment of defined subsets of the genome (Meissner et al. 2005; Down et al. 2008; Deng et al. 2009), e.g., reduced representation bisulfite sequencing (RRBS) (Meissner et al. 2005) and anti-methylcytosine DNA immunoprecipitation followed by sequencing (MeDIP-seq) (Down et al. 2008). An advantage of such methods is that they can be performed with limited quantities of starting DNA (Gu et al. 2011). However, they are constrained in that they are not truly comprehensive. For example, the digestion-based RRBS method interrogates only ∼12% of CpGs, primarily in CpG islands (Harris et al. 2010), with poor coverage of methylation in gene bodies (Ball et al. 2009) and elsewhere. Furthermore, RRBS does not target cytosines in the CHG or CHH (H = A,C,T) contexts, which have been shown to be methylated at elevated levels in the early stages of mammalian development (Lister et al. 2009). While a small proportion of non-CpG methylation sites can be observed using RRBS, they are restricted to regions within or highly proximal to CpG-islands (Ziller et al. 2011).
The most comprehensive, highest resolution method for detecting 5mC is whole-genome bisulfite sequencing (WGBS) (Cokus et al. 2008; Lister et al. 2009; Harris et al. 2010). Treatment of genomic DNA with sodium bisulfite chemically deaminates cytosines much more rapidly than 5mC, preferentially converting them to uracils (Clark et al. 1994). With massively parallel sequencing, these can be detected on a genome-wide scale at single-base-pair resolution. This approach has revealed complex and unexpected methylation patterns and variation, particularly in the CHG and CHH contexts. Furthermore, as the costs of massively parallel sequencing continue to plummet, WGBS is increasingly affordable. However, a key limitation of WGBS is that the current protocols for library construction are based on ligation chemistry and call for 5 μg of genomic DNA as input (Cokus et al. 2008; Lister et al. 2009; Li et al. 2010) which is essentially prohibitive for many samples obtained in vivo.
We recently characterized a transposase-based in vitro shotgun library construction method (“tagmentation”) that allows for construction of sequencing libraries from greatly reduced amounts of DNA (Fig. 1A; Adey et al. 2010). Briefly, the method utilizes a hyperactive derivative of the Tn5 transposase loaded with discontinuous synthetic oligonucleotides to simultaneously fragment and append adaptors to genomic DNA. The resulting products are subjected to PCR amplification followed by high-throughput sequencing. The increased efficiency of genomic DNA conversion to viable amplicons and the greatly reduced number of steps allow the construction of low-bias, highly complex libraries from <50 ng of genomic DNA.
Figure 1.

The Tn5mC-seq method and resulting methylation profiles. (A) Tagmentation-based DNA-seq library construction. Genomic DNA is attacked by transposase homodimers loaded with synthetic, discontinuous oligos (yellow, purple) that allow for fragmentation and adaptor incorporation in a single step. Subsequent PCR appends outer flowcell-compatible primers (pink, green). (B) Tn5mC-seq library construction. Loaded transposase attacks genomic DNA with a single methylated adaptor (yellow). An oligo-replacement approach anneals a second methylated adaptor (purple), which is then subject to gap-repair. Bisulfite treatment then converts unmethylated cytosine to uracil (orange) followed by PCR to append outer flowcell-compatible primers (pink, green). Methylation is represented as black lollipops. (C) Coverage of cytosine positions genome-wide. More than 96% of Cs in all three contexts are covered at least once. Slight decrease in CpG coverage is due to reduced read alignment ability at regions with a high density of methylation. (D) Normalized methylated cytosine over total cytosine positions in 10-kb windows across chromosome 12 (blue and purple, left axis), and normalized methylated CpG over total CpG in 100-kb windows across chromosome 12 (green, right axis). (E) Normalized methylated CpG over total CpG residues at annotated genic loci. Promoter is defined as 2-kb region upstream of TSS. (F) Elevated CpG methylation levels in gene body (intron, exon) compared to intergenic regions.
Here we describe a modified approach, which we call Tn5mC-seq, that retains the advantages of transposase-based library preparation in the context of WGBS. Because the target of the transposition reaction is double-stranded DNA, whereas bisulfite treatment yields single-stranded DNA, the method was extensively modified such that the tagmentation reaction could take place prior to bisulfite treatment (Fig. 1B). First, the adaptors to be incorporated were methylated at all cytosine residues to maintain cytosine identity during bisulfite treatment, with the exception of the 19-bp transposase recognition sequence (in order to minimize differential binding during transposome assembly). Second, an oligonucleotide replacement scheme (Supplemental Fig. S1B; Grunenwald et al. 2011; Gertz et al. 2012) was utilized to ensure that each strand would have adaptors covalently attached to both ends of the molecule. Specifically, this entails initial transposition with a single adaptor in which the double-stranded transposase recognition sequence is truncated to 16 bp (Tm = 36°C), thereby facilitating its post-incorporation removal by denaturation. A second adaptor is then annealed and the gap repaired, resulting in each strand being covalently flanked by both a 3′ and 5′ adaptor. The fragmented, adapted, double-stranded genomic DNA is then subjected to standard bisulfite treatment for the conversion of unmethylated cytosine to uracil. Degradation during the conversion process likely remains a primary source of loss, but the increased efficiency of the prior steps and the lack of gel-based size selection result in an overall increase in the fraction of DNA that is converted, PCR-amplified, and sequenced.
Results
Ultra-low-input transposase-based WGBS library performance
We applied Tn5mC-seq to sequence the methylome of a lymphoblastoid cell line (GM20847) using libraries constructed from 1–200 ng of input genomic DNA. Each library was barcoded during PCR amplification and subjected to either a spike-in (5%) or majority (80%–90%) of a lane of sequencing on an Illumina HiSeq2000 (paired-end 100 bp [PE100]; v2 chemistry with custom sequencing primers). These data are summarized in Table 1 and Supplemental Figure S2. In addition, several PCR conditions were investigated to optimize amplification uniformity (Supplemental Fig. S3), as well as a modified protocol (Tn5mC-seq 1.1) (Supplemental Figs. S1D, S4) that eliminates the need for custom sequencing primers and may increase library construction efficiency. Reads were aligned to an in silico converted hg19 (GRC37) to both the top (C→T) and bottom (G→A) strands using BWA (Li and Durbin 2009) followed by read trimming of unmapped reads and secondary alignment using the same parameters. Unaligned reads typically consisted of low-quality artifacts that likely arose during amplification due to the reduced base complexity of bisulfite converted amplicons.
Table 1.
Summary of Tn5mC-seq libraries and sequencing
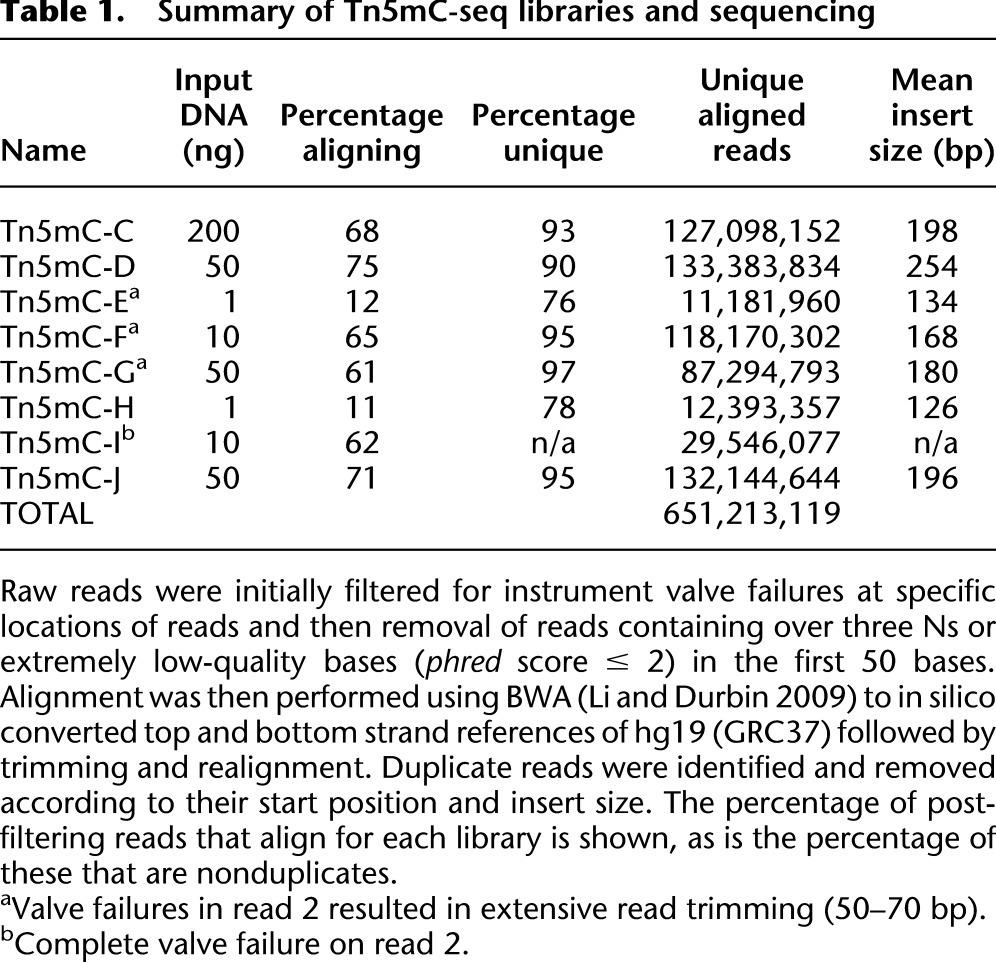
For each library constructed using ≥10 ng of genomic DNA, over 100 million aligned reads were obtained (60%–75% of total filtered reads; see Methods) of high complexity (90%–97% nonduplicates). Despite the significantly reduced performance of libraries prepared from 1 ng, ∼12 million reads were still aligned and the library was of reasonable complexity (78% nonduplicates). Post-alignment reads were merged and quality filtered for a total of 51.7 Gb of aligned, unique sequence. The average read depth was 8.6× per strand with >96% of CpG and >98% of non-CpG cytosines covered genome-wide (Fig. 1C; Supplemental Fig. S2). Because unmethylated nucleotides are incorporated during the gap-repair step (first 9 bp of the second read and last 9 bp before the adaptor as determined by insert size on the first read), the gap-repair regions must be excluded from methylation analysis. However, these bases also serve as an internal control for the conversion rate of the bisulfite treatment. We found this to be >99% for all libraries, and this was independently confirmed using unmethylated lambda DNA spike-ins to two libraries.
For comparison, ligation chemistry–based libraries were constructed using 1000, 100, and 10 ng of GM20847 DNA of the same isolation as the batch used for Tn5mC-seq. These libraries were prepared following the protocols outlined by Lister et al. (2009) with the exception of PCR, which was performed using Kapa Robust due to its higher efficiency over other polymerase choices (Supplemental Fig. S3) During amplification, the 100 and 10 ng preparations did not show significant amplification above a negative control background and were not carried through to sequencing, precluding a comparison of Tn5mC-seq and ligation chemistry–based library construction with identical inputs (a 1000 ng Tn5mC-seq preparation was also not feasible due to the dilute concentrations of the commercially available transposase, which would result in a reduced density of transposition events on a high input mass).
Post-alignment, the 1000 ng ligation chemistry–based library provided slightly more uniform coverage than Tn5mC-seq 1.1 (Supplemental Fig. S1D) libraries constructed from 10 ng, particularly at the lower CpG densities that represent the majority of the genome (Supplemental Fig. S5A). Comparable uniformity was also observed with respect to G+C content as well as for tetramer/pentamer sequence contexts (Supplemental Fig. S5B,C). We also compared the methylation levels of CpGs well-covered by sequencing of libraries corresponding to both methods, and observed good agreement at positions with 5× or greater coverage (r2 = 0.55) as well as 10× or greater coverage (r2 = 0.82) (Supplemental Fig. S5D).
Lymphoblastoid cell line methylation
We were able to detect ∼46 million 5mC positions (1% FDR; see Methods), accounting for 4.2% of total cytosines with coverage. The majority of methylation observed was in the CpG context (97.1%), and the global CpG methylation level was 69.1%. This level is similar to that of the fetal fibroblast cell line IMR90 sequenced by Lister et al. (2009; 67.7%) and is consistent with the observation that CpG methylation levels are reduced in differentiated cell types. Additionally, CHG and CHH methylation levels were substantially lower than in ES cells, at 0.36% and 0.37%, respectively, again consistent with the differentiated cell type. On the chromosome scale, the methylation density correlated with banding patterns and increasing levels were observed extending distally through subtelomeric regions (Fig. 1D). An analysis of functionally annotated genic regions revealed a sharp decrease in CpG methylation through the promoter region followed by a minor increase in the 5′ UTR and then elevated levels of methylation throughout the gene body, particularly at introns (Fig. 1E,F), consistent with previously described CpG methylation profiles (Lister et al. 2009).
Discussion
We developed Tn5mC-seq as a novel method for rapidly preparing complex, shotgun bisulfite sequencing libraries for WGBS. In brief, the method utilizes a hyperactive Tn5 transposase derivative to fragment genomic DNA and append adaptors in a single step, as previously characterized for the construction of DNA-seq libraries (Adey et al. 2010). In order for library molecules to withstand bisulfite treatment, the adaptors are methylated at all cytosine residues, and an oligonucleotide replacement strategy is employed to make each single-strand covalently flanked by adaptors. The high efficiency of the transposase and overall reduction in loss-associated steps permits construction of high-quality bisulfite sequencing libraries from as little as 10 ng that are comparable to ligation chemistry–based libraries generated from 100× more DNA, as well as useful sequence from 1 ng of input DNA. Additionally, the increased efficiency of transposase-mediated library construction may allow for preparation of WGBS libraries from poor-quality or degraded DNA samples.
Our results illustrate how derivatives of the transposase-based method for DNA-seq library preparation can enable key applications of next-generation sequencing where its advantages are perhaps even more relevant. The ability to generate such libraries from very low amounts of input genomic DNA substantially improves the practicality of whole methylome sequencing and removes a key advantage of less encompassing methods such as RRBS (Meissner et al. 2005; Harris et al. 2010). Specifically, low-input WGBS with Tn5mC-seq may make possible the comprehensive interrogation of methylation in many contexts where DNA quantity is a bottleneck, e.g., developing anatomical structures, microdissected tissues, or pathologies such as cancer, where the epigenetic landscape is of interest but tissue quantity limits high-resolution WGBS.
Methods
Tn5mC-seq library construction and sequencing
Transposome complexes were generated by incubating 2.5 μL of 10 μM Tn5mC-A1 (Tn5mC-A1top: 5′-GAT [5mC] TA [5mC] A[5mC] G [5mC] [5mC] T [5mC] [5mC] [5mC] T [5mC] G [5mC] G [5mC] [5mC] AT [5mC] AGAGATGTGTATAAGAGACAG-3′, IDT, annealed to Tn5mC-A1bot: 5′-[Phos]-CTGTCTCTTATACACA-3′, IDT, by incubating 10 μL of each oligo at 100 μM and 80 μL of EB [QIAGen] for 2 min at 95°C and then cooling to room temperature at 0.1°C/sec) with 2.5 μL 100% glycerol and 5 μL Ez-Tn5 transposase (Epicentre – Illumina) for 20 min at room temperature.
Genomic DNA prepared from GM20847 cell lines was used at respective input quantities with 4 μL Nextera HMW Buffer (Epicentre-Illumina), nuclease-free water (Ambion) to 17.5 μL and 2.5 μL prepared Tn5mC transposomes (regardless of the quantity of DNA used). Reactions were incubated for 9 min at 55°C in a thermocycler followed by SPRI bead cleanup (AMPure) using 36 μL of beads and the recommended protocol with elution in 14 μL nuclease-free water (Ambion). Adaptor 2 annealing was then carried out by adding 2 μL of 10× Ampligase Reaction Buffer (Epicentre-Illumina), 2 μL 10× dNTPs (2.5 mM each; Invitrogen), and 2 μL 10 μM Tn5mC-A2top (5′-/5Phos/ CTGTCTCTTATACACATCT [5mC] TGAG [5mC] GGG [5mC] TGG [5mC] AAGG [5mC] AGA [5mC] [5mC] GAT [5mC]-3′; IDT) to each reaction and incubating for 2 min at 50°C followed by 10 min at 45°C and cooling at 0.1°C/sec to 37°C and subsequent incubation for 10 min. Gap-repair was then performed by adding 3 μL of Ampligase at 5U/μL (Epicentre-Illumina) and 1 μL of either T4 DNA Polymerase (Tn5mC libraries A-G, NEB) or Sulfolobus DNA Polymerase IV (Tn5mC libraries H-J, NEB) and additional incubation for 30 min at 37°C. Reactions were then cleaned up using SPRI beads (AMPure) according to recommended protocol using 36 μL beads and elution in 50 μL nuclease-free water (Ambion). Bisulfite treatment was performed using an EZ DNA Methylation Kit (Zymo) according to recommended protocols with a 14-h 50°C incubation and 10 μL elution. Eluate was then used as the template for PCR using 12.5 μL Kapa 2G Robust HotStart ReadyMix (Kapa Biosystems), 1 μL 10 μM Tn5mC-P1 (5′-AATGATACGGCGACCACCGAGATCTACACGCCTCCCTCGCGCCATCAG-3′; IDT), 1 μL 10 μM barcoded P2 (from Adey et al. 2010), 0.15 μL 100× SYBR Green (Invitrogen), and 0.35 μL nuclease-free water (Ambion). Thermocycling was carried out on a BioRad Opticon Mini real-time machine with the following parameters: 5 min at 95°C; (15 sec at 95°C; 15 sec at 62°C; 40 sec at 72°C; Plate Read; 10 sec at 72°C) × 99. Reactions were monitored and removed from thermocycler as soon as plateau was reached (12–15 cycles).
Sequencing was carried out using either a full or partial lane on an Illumina HiSeq2000 using custom sequencing primers: read 1, Tn5mC-R1 (5′-GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGATAG-3′; IDT); index read, Tn5mC-Ix (5′-TTGTTTTTTATATATATTTCTGAGCGGGCTGGCAAGGC-3′; IDT); and read 2, Tn5mC-R2 (5′-GCCTTGCCAGCCCGCTCAGAAATATATATAAAAAACAA-3′; IDT). Read lengths were either single-read at 36 bp with a 9-bp index (SE36, libraries A and B, not included in Table 1) or 101 bp paired-end with a 9-bp index (PE101, libraries C–J). Libraries were only sequenced on runs that did not have lanes containing Nextera libraries as a precaution due to the similarity between sequencing primers.
Tn5mC-Seq 1.1 library preparation (Supplemental Fig. 1D) was carried out as previously described with several modifications: (1) Transposase recognition sequence reverse compliment is 3′ blocked to prevent nonspecific extension in final PCR. (2) Replacement oligo is methylated through the region complementary to the transposase recognition sequence to maintain complexity during bisulfite conversion and allow the use of standard Nextera sequencing primers. (3) Replacement oligo is 3′ blocked to prevent degradation by 3′→5′ exonuclease activity of gap-repair polymerase (replacement oligo: Tn5mC1.1-A2top 5′-/5Phos/ [5mC] TGT [5mC] T [5mC] TTATA [5mC] A [5mC] AT [5mC] T [5mC] TGAG [5mC] GGG [5mC] TGG [5mC] AAGG [5mC] AGA [5mC] [5mC] GA [inv dT]-3′, IDT; blocked transposase recognition sequence end: Tn5mC1.1-A1bot3block 5′-[Phos]-CTGTCTCTTATACA [ddC]-3′). Duplicate libraries were prepared from 100 ng, 10 ng, and 1 ng of starting material and were subject to PCR amplification using either Kapa HiFi U+ Hot Start Ready Mix, or Kapa 2G Robust Hot Start Ready Mix (Kapa Biosystems) and were sequenced on a single-end 36-bp read plus a 9-bp index read run on an Illumina GAIIx. Library characterization can be found in Supplemental Figure 5.
Ligation chemistry WGBS library construction and sequencing
We subjected 1000, 100, and 10 ng of genomic DNA prepared from GM18507 cell lines to ligation chemistry–based library preparation according to methods described by Lister et al. (2009) with several minor exceptions: (1) Bisulfite conversion was carried out using an EZ DNA Methylation Kit (Zymo), and (2) PCR was carried out using Kapa 2G Robust Hot Start Ready Mix (Kapa Biosystems). The change in PCR enzyme was due to several unpublished experiments demonstrating a much higher efficiency with Kapa Robust as opposed to PfuTurbo Cx used according to the method described by Lister et. al.(2009). Sequencing was performed on an Illumina MiSeq instrument using a single-end 100-bp sequence read run.
Read filtering and alignment
The hg19 reference genome was first bisulfite-converted in silico for both the top (C changed to T, C2T) and bottom (G changed to A, G2A) strands. Prior to alignment, reads were filtered based on the run metrics, as several libraries were run on lanes in which instrument valve failures resulted in poor quality or reads consisting primarily of N bases. Filtering was carried out by first calculating the base compositions as well as mean base quality scores at each position in the read. Many of the lanes had significantly reduced quality scores at the start and/or end of the read and were globally trimmed to remove any start or end positions that had a mean phed score of less than or equal to 10. The start and ends of the reads were additionally globally trimmed if a position within the first or last 25 bases of the read had a mean composition of 10% Ns, which generally corresponded to the quality-based trimming. Additionally, reads that contained three or more Ns were also removed. It is important to note that the reduced qualities in the runs were “flowcell-wide” regardless of the library that was run and not isolated to Tn5mC-seq libraries. Subsequent runs for the Tn5mC-seq 1.1 and polymerase testing experiments did not suffer instrument failures, and no trimming of the reads was necessary. Next, reads were aligned to both the C2T and G2A strands using BWA with default parameters. Reads that aligned to both strands were removed. Read pairs in which neither aligned to either strand were then pulled and trimmed to 76 bp (except for SE36 runs) and again aligned to both C2T and G2A strands. Duplicate reads (pairs sharing the same start positions for both reads 1 and 2) were removed and complexity determined. Reads with an alignment score less than 10 were then filtered out prior to secondary analysis. Total fold coverage was calculated using the total bases aligned from unique reads over the total alignable bases of the genome (slightly below 3 Gb per strand).
5mC calling
Methylated cytosines were called using a binomial distribution as in the method described by Lister et al. (2009), whereby a probability mass function is calculated for each methylation context (CpG, CHG, CHH) using the number of reads covering the position as the number of trials and reads maintaining cytosine status as successes with a probability of success based on the total error rates that were determined by the combined nonconversion rate and sequencing error rate. The total error rate was initially determined by unmethylated lambda DNA spike-ins; however, we found that the error rate estimation from the gap-repair portion of reads (as described in the main text) gave a more comprehensive estimate, which was slightly higher than that of the lambda estimate. Therefore to be conservative, we used the highest determined error rate at 0.009. If the probability was below the value of M, where M × (number of total unmethylated CpG) < 0.01 × (number of total methylated CpG), the position was called as being methylated, thus enforcing that no more than 1% of positions would be due to the error rate.
Data access
The sequence data presented in this study have been submitted to the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession no. SRP011746.
Acknowledgments
We thank Cholie (Charlie) Lee for performing all sequencing runs and the Shendure laboratory for helpful discussions. We also thank Nick Caruccio, Haiying Gruenwald, Brad Baas, and Igor Goryshin from Epicentre (Illumina) for help and ideas regarding transposase-based library preparation, as well as Eric Van Der Walt and colleagues at Kapa Biosystems for early access to reagents and protocols for library amplification. A.A. is funded by an NSF Graduate Research Fellowship. This work was supported in part by the Lowell Milken Prostate Cancer Foundation Young Investigator Award (J.S.).
Author contributions: A.A. performed experiments and data analysis. A.A. and J.S. designed experiments and wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.136242.111.
References
- Adey A, Morrison HG, Asan, Xun X, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, et al. 2010. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol 11: R119 doi: 10.1186/gb-2010-11-12-r119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM 2009. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol 27: 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SJ, Harrison J, Paul CL, Frommer M 1994. High sensitivity mapping of methylated cytosines. Nucleic Acids Res 22: 2990–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE 2008. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452: 215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Shoemaker R, Xie B, Gore A, LeProust EM, Antosiewicz-Bourget J, Egli D, Maherali N, Park IH, Yu J, et al. 2009. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol 27: 353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Graf S, Johnson N, Herrero J, Tomazou EM, et al. 2008. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol 26: 779–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz J, Varley KE, Davis NS, Baas BJ, Goryshin IY, Vaidyanathan R, Kuersten S, Myers RM 2012. Transposase mediated construction of RNA-seq libraries. Genome Res 22: 134–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunenwald H, Baas B, Goryshin I, Zhang B, Adey A, Hu S, Shendure J, Caruccio N, Maffitt M 2011. Nextera PCR-free DNA library preparation for next-generation sequencing. (Poster presentation, AGBT 2011) [Google Scholar]
- Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A 2011. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc 6: 468–481 [DOI] [PubMed] [Google Scholar]
- Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, et al. 2010. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol 28: 1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhu J, Tian G, Li N, Li Q, Ye M, Zheng H, Yu J, Wu H, Sun J, et al. 2010. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol 8: e1000533 doi: 10.1371/journal.pbio.1000533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R 2005. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 33: 5868–5877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D, Henikoff S 2007. Genome-wide analysis of DNA methylation patterns. Development 134: 3959–3965 [DOI] [PubMed] [Google Scholar]
- Ziller MJ, Muller F, Liao J, Zhang Y, Gu H, Bock C, Boyle P, Epstein CB, Bernstein BE, Lengauer T, et al. 2011. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet 7: e1002389 doi: 10.1371/journal.pgen.1002389 [DOI] [PMC free article] [PubMed] [Google Scholar]